
Reactions of Recombinant Neuronal Nitric Oxide Synthase with Redox Cycling Xenobiotics: A Mechanistic Study


Abstract
:1. Introduction
2. Results
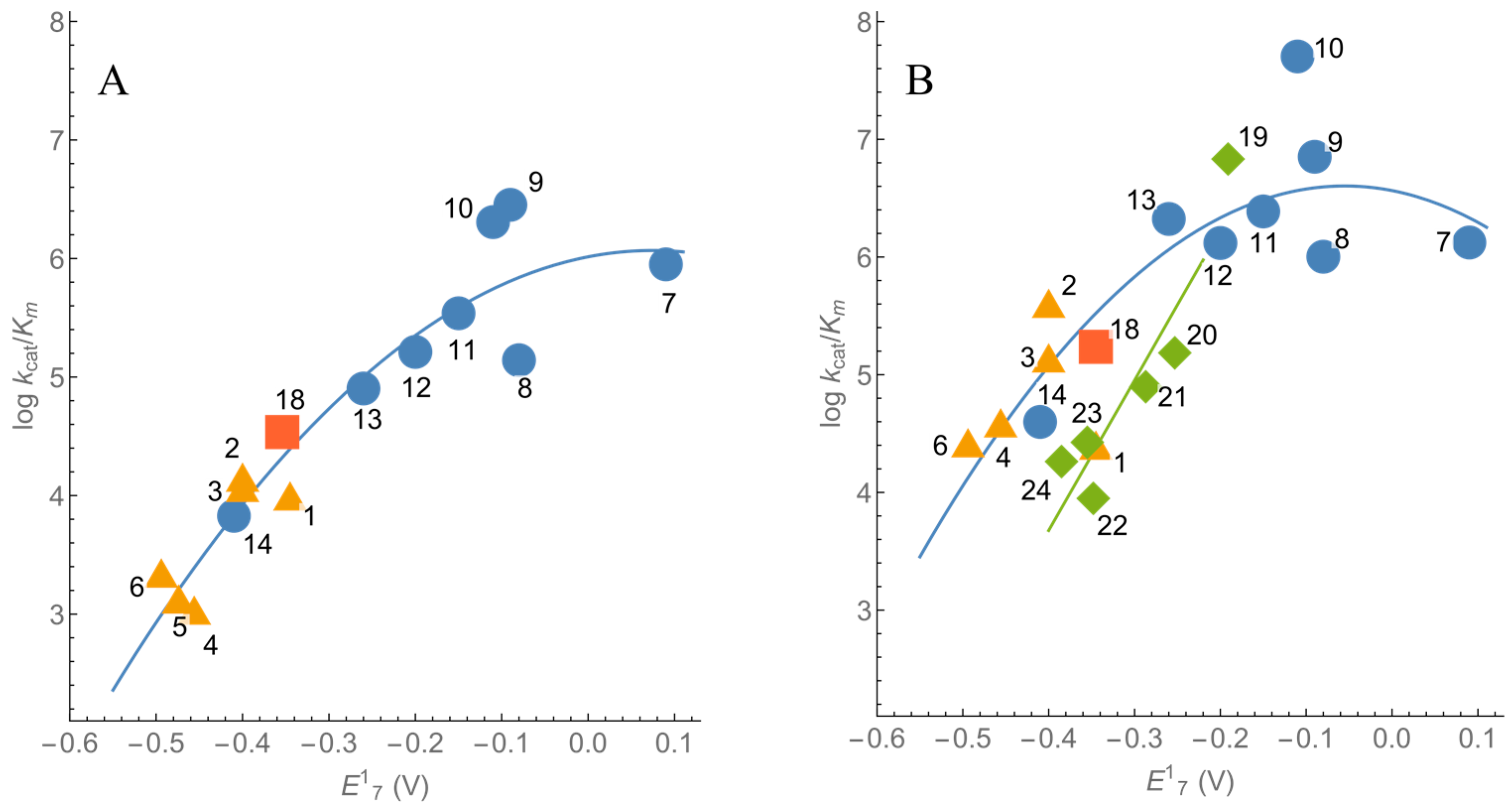
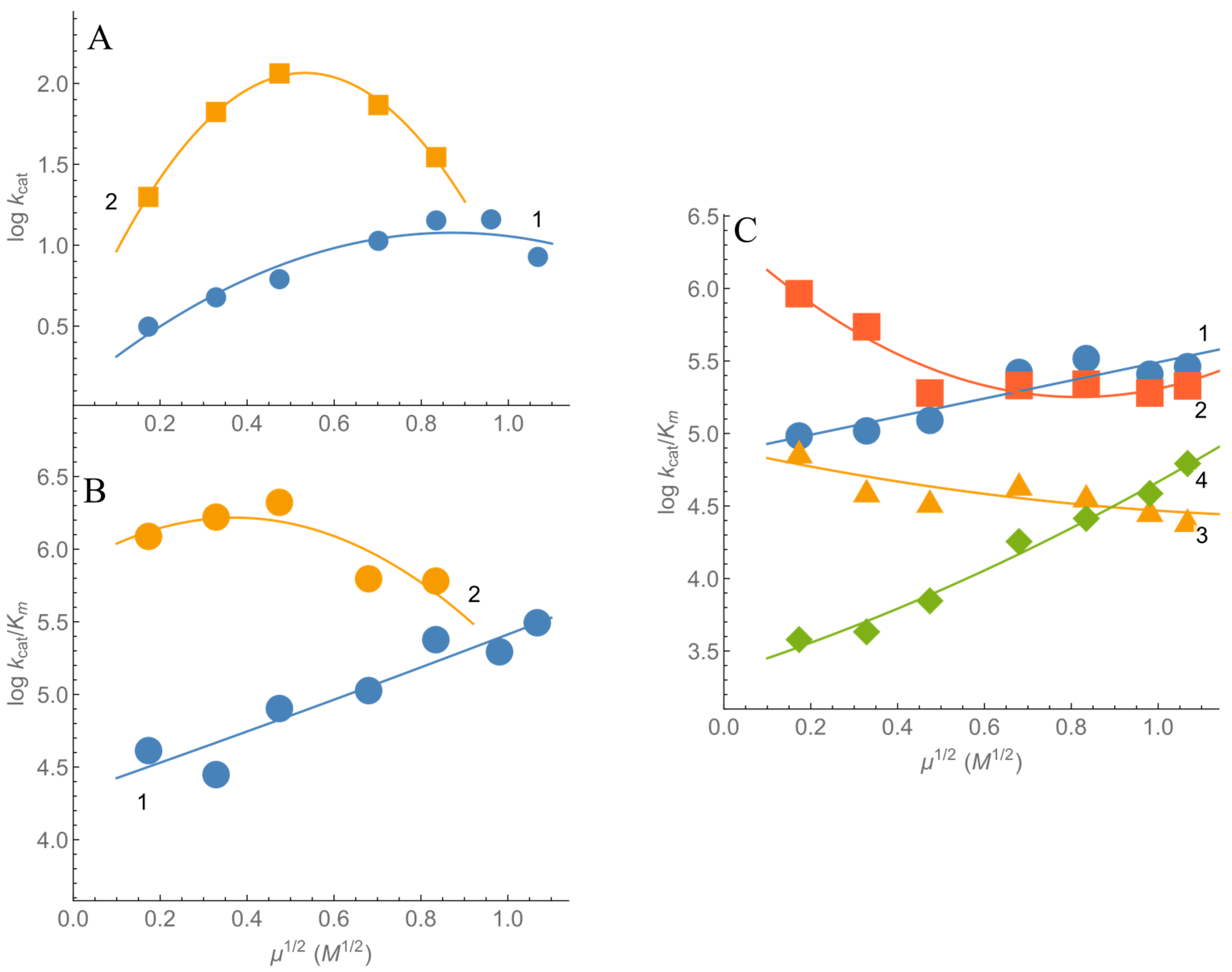
2.1. Steady-State Kinetics and Substrate Specificity of nNOS
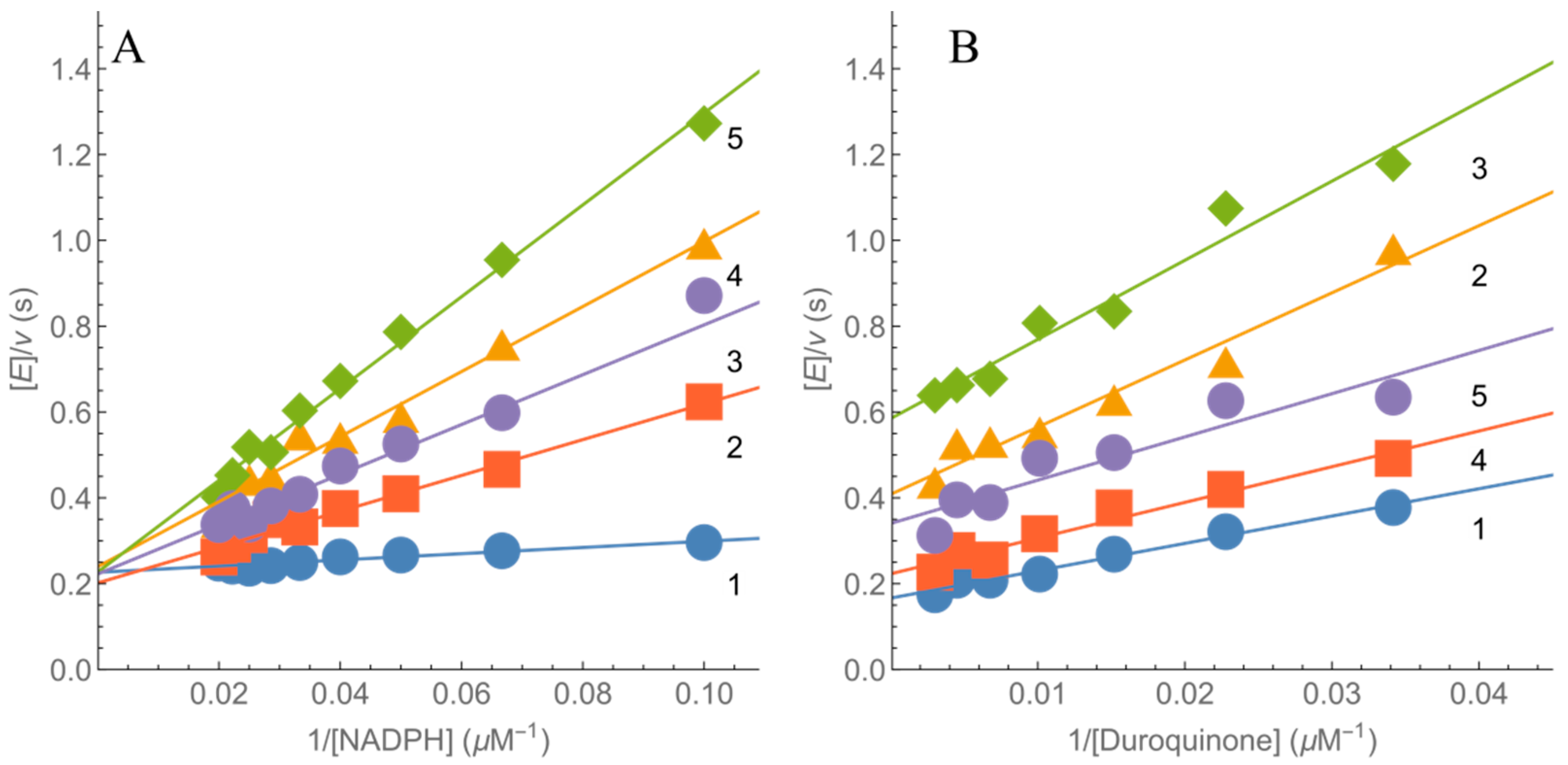
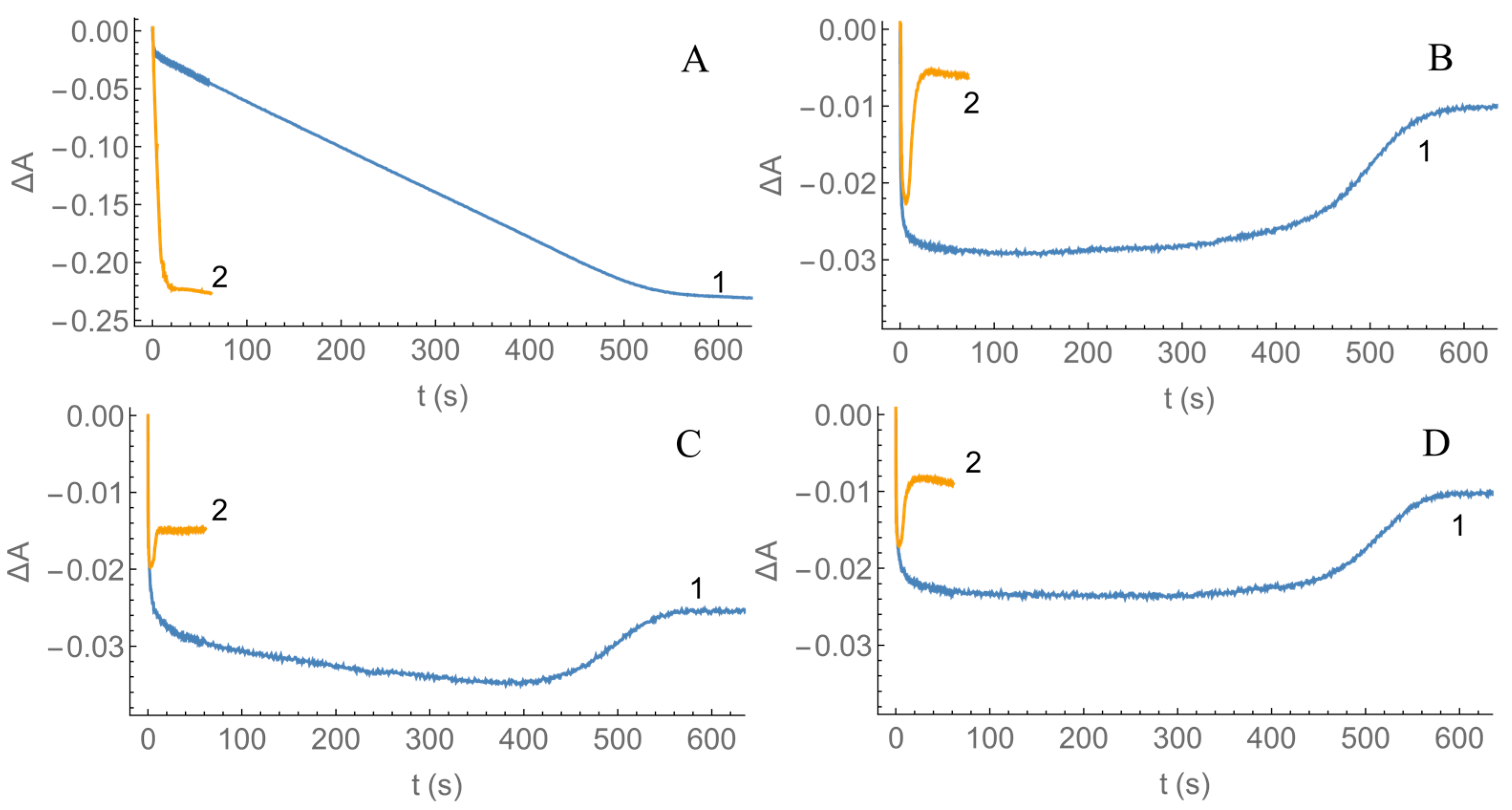
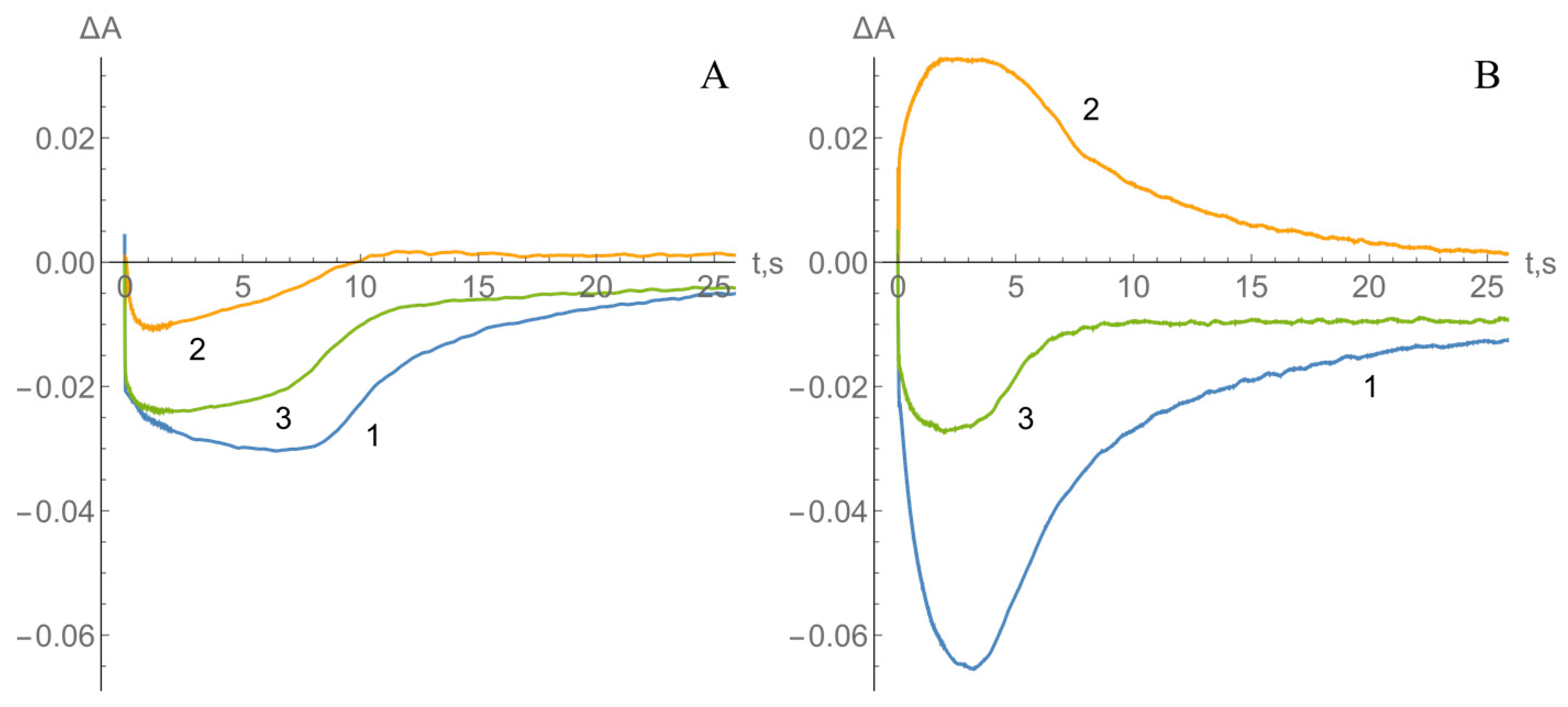
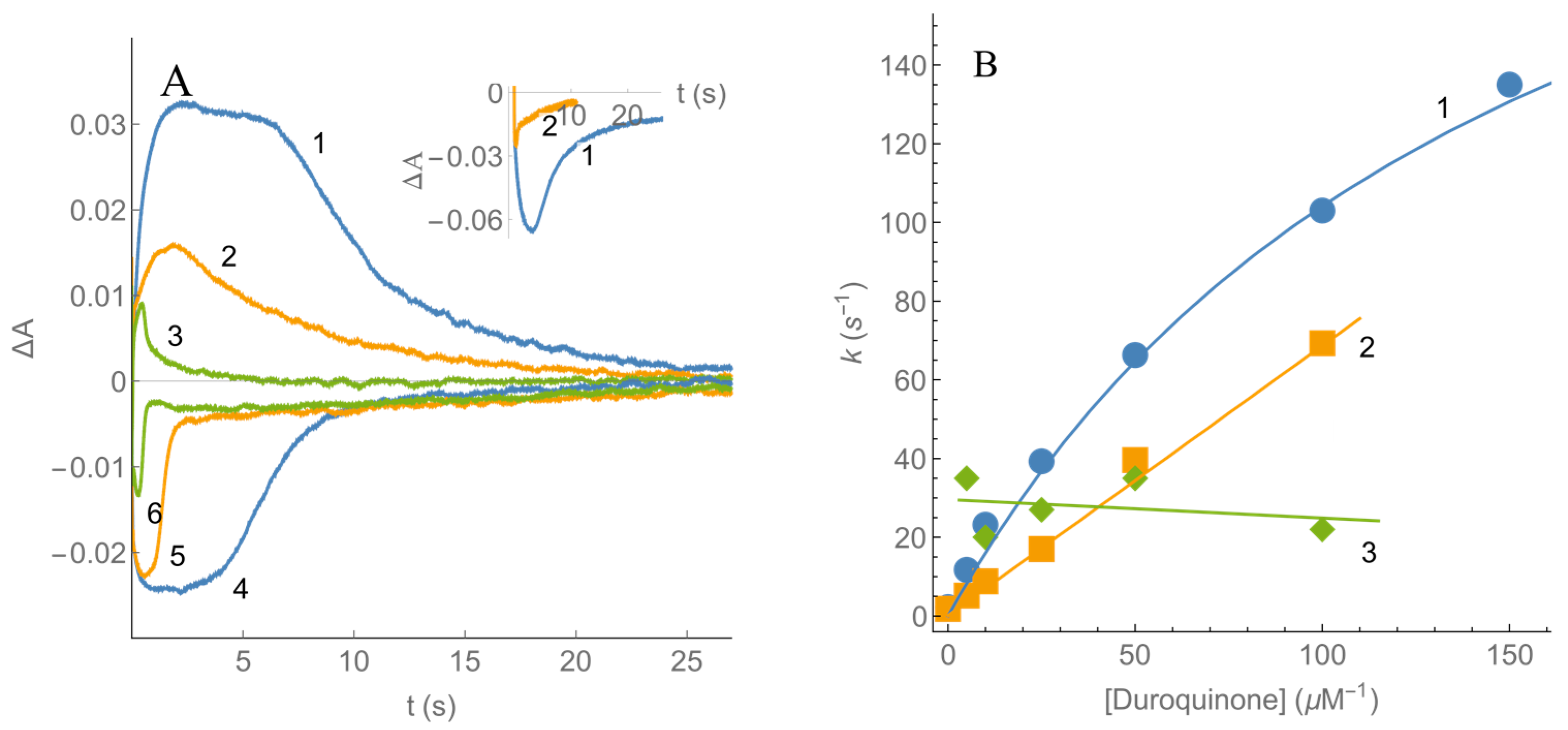
2.2. Kinetics of nNOS Oxidation under Multiple Turnover Conditions
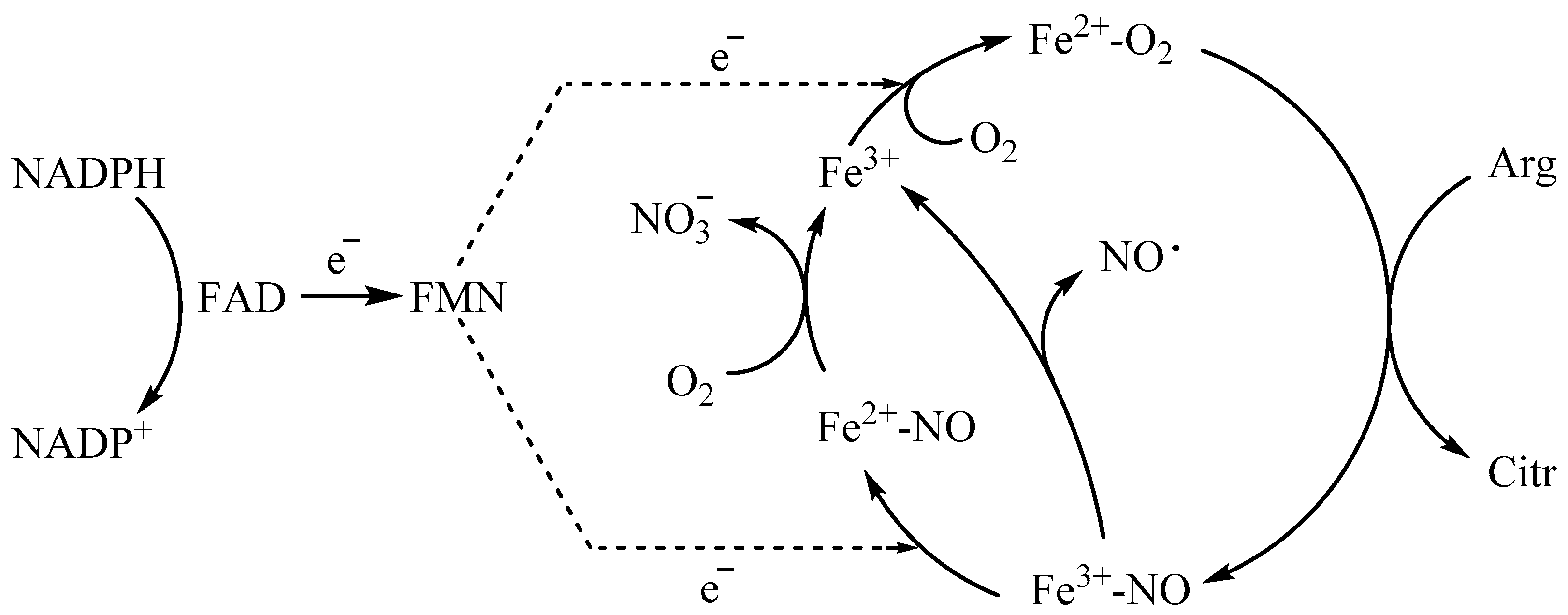
3. Discussion
4. Materials and Methods
4.1. Enzymes and Reagents
4.2. Steady-State Kinetic Studies
4.3. Presteady-State Kinetic Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

| Conditions | E1 (V) | |
|---|---|---|
| −CAM | +CAM | |
| Flavin reductase domain, pH 7.1 [55] | −0.274 (−0.268) | −0.267 (−0.261) |
| Holoenzyme, pH 7.0 [54] | −0.220 | −0.220 a |
| Flavin reductase domain, pH 7.5 [56] | −0.300 (−0.270) | |
| Flavin reductase domain, pH 7.4 [41] | −0.199 (−0.175) | −0.284 (−0.260) |
| Flavin reductase domain, pH 7.6 [39] | −0.276 (−0.240) | |
References
- Ignarro, L.J.; Freeman, B.A. (Eds.) Nitric Oxide. Biology and Pathobiology, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; 434p, ISBN 9780128042731. [Google Scholar]
- Stuehr, D.J.; Haque, M.M. Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. Br. J. Pharmacol. 2019, 176, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leferink, N.G.H.; Hay, S.; Rigby, S.E.J.; Scrutton, N.S. Towards the free energy landscape for catalysis in mammalian nitric oxide synthases. FEBS J. 2015, 282, 3016–3029. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.S.; Kim, J.-J.P. Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Martàsek, P.; Paschke, R.; Shea, T.; Masters, B.S.S.; Kim, J.-J.P. Crystal structure of the FAD/NADPH-binding domain of rat neuronal nitric-oxide synthase: Comparisons with NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem. 2001, 276, 37506–37513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcin, E.D.; Bruns, C.M.; Lloyd, S.J.; Hosfield, D.J.; Tiso, M.; Gachhui, R.; Stuehr, D.J.; Tainer, J.A.; Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 2004, 279, 37918–37927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, H.; Iyanagi, T. Calmodulin activates intramolecular electron transfer between the two flavins of neuronal nitric oxide synthase flavin domain. Biochim. Biophys. Acta Gen. Subj. 1999, 1473, 345–355. [Google Scholar] [CrossRef]
- Matsuda, H.; Kimura, S.; Iyanagi, T. One-electron reduction of quinones by the neuronal nitric-oxide synthase reductase domain. Biochim. Biophys. Acta Bioenerg. 2000, 1459, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.-W.; Kamatani, D.; Kimura, S.; Iyanagi, T. Mechanistic studies on the intramolecular one-electron transfer between the two flavins in the human neuronal nitric-oxide synthase and inducible nitric-oxide synthase flavin domains. J. Biol. Chem. 2003, 278, 30859–30868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Yamamoto, K.; Guan, Z.-W.; Kimura, S.; Iyanagi, T. Human neuronal nitric oxide synthase can catalyze one-electron reduction of adriamycin: Role of flavin domain. Arch. Biochem. Biophys. 2004, 427, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Wang, Z.Q.; Tejero, J.; Yang, Y.P.; Hemann, C.; Hille, R.; Stuehr, D.J. Catalytic reduction of a tetrahydrobiopterin radical within nitric-oxide synthase. J. Biol. Chem. 2008, 283, 11734–11742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Soud, H.M.; Wang, J.; Rousseau, D.L.; Fukuto, J.M.; Ignarro, L.J.; Stuehr, D.J. Neuronal nitric oxide synthase self-inactivates by forming a ferrous-nitrosyl complex during aerobic catalysis. J. Biol. Chem. 1995, 270, 22997–23006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, J.; Santolini, J.; Stuehr, D.J. Fast ferrous heme-NO oxidation in nitric oxide synthases. FEBS J. 2009, 276, 4505–4514. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Tejero, J.; Bayachou, M.; Wang, Z.-Q.; Fadlalla, M.; Stuehr, D.J. Thermodynamic characterization of five key kinetic parameters that define neuronal nitric oxide synthase catalysis. FEBS J. 2013, 280, 4439–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.H.; Chapman, S.K.; Daff, S. Calmodulin activates electron transfer through neuronal nitric-oxide synthase reductase domain by releasing an NADPH-dependent conformational lock. J. Biol. Chem. 2002, 277, 33987–33994. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Haque, M.M.; Stuehr, D.J.; Lu, H.P. Single-molecule spectroscopy reveals how calmodulin activates NO synthase by controlling its conformational fluctuation dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 11835–11840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.G.; Smith, B.C.; Potter, C.S.; Carragher, B.; Marletta, M.A. Molecular architecture of mammalian nitric oxide synthases. Proc. Natl. Acad. Sci. USA 2014, 111, E3614–E3623. [Google Scholar] [CrossRef] [Green Version]
- Garner, A.P.; Paine, M.J.I.; Rodriguez-Crespo, I.; Chinje, E.C.; Ortiz De Montellano, P.; Stratford, I.J.; Tew, D.G.; Wolf, C.R. Nitric oxide synthases catalyze the activation of redox cycling and bioreductive anticancer agents. Cancer Res. 1999, 59, 1929–1934. [Google Scholar] [PubMed]
- Ask, K.; Dijols, S.; Giroud, C.; Casse, L.; Frapart, Y.M.; Sari, M.A.; Kim, K.S.; Stuehr, D.J.; Mansuy, D.; Camus, P.; et al. Reduction of nilutamide by NO synthases: Implications for the adverse effects of this nitroaromatic antiandrogen drug. Chem. Res. Toxicol. 2003, 16, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Chandor, A.; Dijols, S.; Ramassamy, B.; Frapart, Y.; Mansuy, D.; Stuehr, D.; Helsby, N.; Boucher, J.-L. Metabolic activation of the antitumor drug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by NO synthases. Chem. Res. Toxicol. 2008, 21, 836–843. [Google Scholar] [CrossRef]
- Kumagai, Y.; Kikushima, M.; Nakai, Y.; Shimojo, N.; Kunimoto, M. Neuronal nitric oxide synthase (nNOS) catalyzes one-electron reduction of 2,4,6-trinitrotoluene, resulting in decreased nitric oxide production and increased nNOS gene expression: Implication for oxidative stress. Free Radic. Biol. Med. 2004, 37, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Anusevičius, Ž.; Nivinskas, H.; Šarlauskas, J.; Sari, M.-A.; Boucher, J.-L.; Čėnas, N. Single-electron reduction of quinone and nitroaromatic xenobiotics by recombinant rat neuronal nitric oxide synthase. Acta Biochim. Pol. 2013, 60, 217–222. [Google Scholar] [CrossRef]
- Lopes, M.Â.; Meisel, A.; Carvalho, F.D.; de Lourdes Bastos, M. Neuronal nitric oxide synthase is a key factor in doxorubicin-induced toxicity to rat-isolated cortical neurons. Neurotox. Res. 2011, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.T. Dinitrobenzene-mediated production of peroxynitrite by neuronal nitric oxide synthase. Chem. Res. Toxicol. 2002, 15, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Resende, F.F.B.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Function of nitric oxide synthase enzyme in temozolomide-induced damage of astrocytic tumor cells. Oncol. Lett. 2018, 15, 4891–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Guise, C.P.; Dachs, G.U.; Phung, Y.; Hsu, A.H.L.; Lambie, N.K.; Patterson, A.V.; Wilson, W.R. Identification of one-electron reductases that activate both the hypoxia prodrug SN30000 and diagnostic probe EF5. Biochem. Pharmacol. 2015, 91, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Nakajima, H.; Midorikawa, K.; Homma-Takeda, S.; Shimojo, N. Inhibition of nitric oxide formation by neuronal nitric oxide synthase by quinones: Nitric oxide synthase as a quinone reductase. Chem. Res. Toxicol. 1998, 11, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta Rev. Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Wardman, P.; Dennis, M.F.; Everett, S.A.; Patel, K.B.; Stratford, M.R.L.; Tracy, M. Radicals from one-electron reduction of nitro compounds, aromatic N-oxides and quinones: The kinetic basis for hypoxia-selective, bioreductive drugs. Biochem. Soc. Symp. 1995, 61, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Grampp, G.; Jaenicke, W. ESR-spectroscopic investigation of the parallel electron and proton exchange between quinones and their radicals: Part I. Measurements at 298 K. J. Electroanal. Chem. Interfacial Electrochem. 1987, 229, 297–303. [Google Scholar] [CrossRef]
- Wolthers, K.R.; Schimerlik, M.I. Reaction of neuronal nitric-oxide synthase with 2,6-dichloroindolphenol and cytochrome c3+: Influence of the electron acceptor and binding of Ca2+-activated calmodulin on the kinetic mechanism. Biochemistry 2001, 40, 4722–4737. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Reduction potentials of one electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Hay, M.P.; Gamage, S.A.; Kovacs, M.S.; Pruijn, F.B.; Anderson, R.F.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. Structure-activity relationships of 1,2,4-benzotriazine 1,4-dioxides as hypoxia-selective analogues of tirapazamine. J. Med. Chem. 2003, 46, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Nemeikaitė-Čėnienė, A.; Šarlauskas, J.; Jonušienė, V.; Marozienė, A.; Misevičienė, L.; Yantsevich, A.V.; Čėnas, N. Kinetics of flavoenzyme-catalyzed reduction of tirapazamine derivatives: Implications for their prooxidant cytotoxicity. Int. J. Mol. Sci. 2019, 20, 4602. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, J.S.; Narayanasami, R.; Miller, R.T.; Roman, L.J.; Panda, S.; Masters, B.S.S. The stimulatory effects of Hofmeister ions on the activities of neuronal nitric-oxide synthase. J. Biol. Chem. 1999, 274, 5399–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, K.; Haque, M.M.; Garcin-Hosfield, E.D.; Durra, D.; Getzoff, E.D.; Stuehr, D.J. Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J. Biol. Chem. 2006, 281, 36819–36827. [Google Scholar] [CrossRef] [Green Version]
- Welland, A.; Garnaud, P.E.; Kitamura, M.; Miles, C.S.; Daff, S. Importance of the domain−domain interface to the catalytic action of the NO synthase reductase domain. Biochemistry 2008, 47, 9771–9780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welland, A.; Daff, S. Conformation-dependent hydride transfer in neuronal nitric oxide synthase reductase domain. FEBS J. 2010, 277, 3833–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.M.; Tejero, J.; Bayachou, M.; Kenney, C.T.; Stuehr, D.J. A cross-domain charge interaction governs the activity of NO synthase. J. Biol. Chem. 2018, 293, 4545–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.T.; Martásek, P.; Omura, T.; Siler Masters, B.S. Rapid kinetic studies of electron transfer in the three isoforms of nitric oxide synthase. Biochem. Biophys. Res. Commun. 1999, 265, 184–188. [Google Scholar] [CrossRef]
- Dunford, A.J.; Rigby, S.E.J.; Hay, S.; Munro, A.W.; Scrutton, N.S. Conformational and thermodynamic control of electron transfer in neuronal nitric oxide synthase. Biochemistry 2007, 46, 5018–5029. [Google Scholar] [CrossRef] [Green Version]
- Chance, B. A simple relationship for a calculation of the “on” velocity constant in enzyme reactions. Arch. Biochem. Biophys. 1957, 71, 130–136. [Google Scholar] [CrossRef]
- Mauk, A.G.; Scott, R.A.; Gray, H.B. Distances of electron transfer to and from metalloprotein redox sites in reactions with inorganic complexes. J. Am. Chem. Soc. 1980, 102, 4360–4363. [Google Scholar] [CrossRef]
- Lesanavičius, M.; Aliverti, A.; Šarlauskas, J.; Čėnas, N. Reactions of Plasmodium falciparum ferredoxin:NADP+ oxidoreductase with redox cycling xenobiotics: A mechanistic study. Int. J. Mol. Sci. 2020, 21, 3234. [Google Scholar] [CrossRef]
- Čėnas, N.; Anusevičius, Ž.; Bironaitė, D.; Bachmanova, G.I.; Archakov, A.I.; Ollinger, K. The electron-transfer reactions of NADPH-cytochrome P450 reductase with nonphysiological oxidants. Arch. Biochem. Biophys. 1994, 315, 400–406. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Feldman, P.L.; Clark, P.; Stuehr, D.J. Electron transfer in the nitric-oxide synthases. Characterization of L-arginine analogs that block heme iron reduction. J. Biol. Chem. 1994, 269, 32318–32326. [Google Scholar] [CrossRef]
- Moussaoui, M.; Misevičienė, L.; Anusevičius, Ž.; Marozienė, A.; Lederer, F.; Baciou, L.; Čėnas, N. Quinones and nitroaromatic compounds as subversive substrates of Staphylococcus aureus flavohemoglobin. Free Radic. Biol. Med. 2018, 123, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Nishida, C.R.; Ortiz de Montellano, P.R. Reductive heme-dependent activation of the N-oxide prodrug AQ4N by nitric oxide synthase. J. Med. Chem. 2008, 51, 5118–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavaggi, M.L.; Cabrera, M.; González, M.; Cerecetto, H. Differential enzymatic reductions governing the differential hypoxia-selective cytotoxicities of phenazine 5,10-dioxides. Chem. Res. Toxicol. 2008, 21, 1900–1906. [Google Scholar] [CrossRef] [PubMed]
- Goeptar, A.R.; Scheerens, H.; Vermeulen, N.P.E. Oxygen and xenobiotic reductase activities of cytochrome P450. Crit. Rev. Toxicol. 1995, 25, 25–65. [Google Scholar] [CrossRef] [PubMed]
- Pochapsky, T.C.; Wong, N.; Zhuang, Y.; Futcher, J.; Pandelia, M.-E.; Teitz, D.R.; Colthart, A.M. NADH reduction of nitroaromatics as a probe for residual ferric form high-spin in a cytochrome P450. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Voegtle, H.L.; Sono, M.; Adak, S.; Pond, A.E.; Tomita, T.; Perera, R.; Goodin, D.B.; Ikeda-Saito, M.; Stuehr, D.J.; Dawson, J.H. Spectroscopic characterization of five- and six-coordinate ferrous−NO heme complexes. evidence for heme Fe−proximal cysteinate bond cleavage in the ferrous−NO adducts of the Trp-409Tyr/Phe proximal environment mutants of neuronal nitric oxide synthase. Biochemistry 2003, 42, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Miškinienė, V.; Sergedienė, E.; Nemeikaitė, A.; Segura-Aguilar, J.; Čėnas, N. Role of redox cycling and activation by DT-diaphorase in the cytotoxicity of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB-1954) and its analogs. Cancer Lett. 1999, 146, 217–222. [Google Scholar] [CrossRef]
- Gao, Y.T.; Smith, S.M.E.; Weinberg, J.B.; Montgomery, H.J.; Newman, E.; Guillemette, J.G.; Ghosh, D.K.; Roman, L.J.; Martasek, P.; Salerno, J.C. Thermodynamics of oxidation-reduction reactions in mammalian nitric-oxide synthase isoforms. J. Biol. Chem. 2004, 279, 18759–18766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, M.A.; Munro, A.W.; Rivers, S.L.; Robledo, L.; Daff, S.N.; Yellowlees, L.J.; Shimizu, T.; Sagami, I.; Guillemette, J.G.; Chapman, S.K. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry 1999, 38, 16413–16418. [Google Scholar] [CrossRef] [PubMed]
- Garnaud, P.E.; Koetsier, M.; Ost, T.W.B.; Daff, S. Redox properties of the isolated flavin mononucleotide- and flavin adenine dinucleotide-binding domains of neuronal nitric oxide synthase. Biochemistry 2004, 43, 11035–11044. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






| No. | Compound | E17 (V) [32,33] | −CAM | +CAM | ||
|---|---|---|---|---|---|---|
| kcat (s−1) | kcat/Km (M−1s−1) | kcat (s−1) | kcat/Km (M−1s−1) | |||
| Aromatic N-oxides | ||||||
| 1. | 7-CF3-tirapazamine | −0.345 | 3.98 ± 0.38 | 9.9 ± 1.2 × 103 | 16.3 ± 1.8 | 2.5 ± 0.2 × 104 |
| 2. | 7-Cl-tirapazamine | −0.400 | 0.84 ± 0.08 | 1.4 ± 0.1 × 104 | 14.8 ± 1.7 | 4.0 ± 0.6 × 105 |
| 3. | 7-F-tirapazamine | −0.400 | 2.84 ± 0.23 | 1.2 ± 0.1 × 104 | 11.1 ± 1.1 | 1.4 ± 0.1 × 105 |
| 4. | 3-Amino-1,2,4-benzotriazine-1,4- dioxide (tirapazamine) | −0.456 | 0.69 ± 0.09 | 1.1 ± 0.1 × 103 | 5.22 ± 0.66 | 3.9 ± 0.2 × 104 |
| 5. | 7-CH3-tirapazamine | −0.474 | 0.60 ± 0.09 | 1.3 ± 0.2 × 103 | ND | ND |
| 6. | 7-C2H5O-tirapazamine | −0.494 | 0.67 ± 0.09 | 2.2 ± 0.2 × 103 | 2.94 ± 0.71 | 2.6 ± 0.2 × 104 |
| Quinones | ||||||
| 7. | 1,4-Benzoquinone | 0.090 | 12.9 ± 1.5 | 5.1 ± 0.7 × 105 | 61.4 ± 7.2 | 1.3 ± 0.2 × 106 |
| 8. | 2,6-(CH3)2-1,4-benzoquinone | −0.080 | 6.84 ± 0.42 | 1.4 ± 0.2 × 105 | 55.8 ± 3.4 | 1.0 ± 0.1 × 106 |
| 9. | 5-OH-1,4-naphthoquinone | −0.090 | 23.3 ± 1.9 | 2.8 ± 0.4 × 106 | 42.2 ± 1.8 | 7.0 ± 0.6 × 106 |
| 10. | 5,8-(OH)2-1,4-naphthoquinone | −0.110 | 11.5 ± 1.2 | 1.8 ± 0.2 × 106 | 24.7 ± 4.8 | 5.0 ± 0.5 × 107 |
| 11. | 1,4-Naphthoquinone | −0.150 | 7.98 ± 0.40 | 3.4 ± 0.7 × 105 | 38.2 ± 2.6 | 2.4 ± 0.3 × 106 |
| 12. | 2-CH3-1,4-naphthoquinone | −0.200 | 8.50 ± 0.80 | 1.6 ± 0.3 × 105 | 37.9 ± 2.5 | 1.3 ± 0.2 × 106 |
| 13. | (CH3)4-1,4-benzoquinone (duroquinone) | −0.260 | 4.71 ± 0.34 | 8.0 ± 1.0 × 104 | 115.0 ± 7.0 112.2 ± 4.3 a 84.3 ± 5.1 b | 2.1 ± 0.2 × 106 1.2 ± 0.1 × 106 a 1.1 ± 0.1 × 106 b |
| 14. | 2-OH-1,4-naphthoquinone | −0.410 | 0.39 ± 0.05 | 4.4 ± 0.4 × 103 | 3.11 ± 0.35 | 4.0 ± 0.3 × 104 |
| Single electron acceptors | ||||||
| 15. | Ferricyanide | 0.410 | 30.4 ± 3.5 | 1.2 ± 0.2 × 105 | 102.4 ± 5.9 | 3.3 ± 0.3 × 106 |
| 16. | Cytochrome c | 0.250 | 9.66 ± 0.35 | 1.9 ± 0.2 × 105 | 104.1 ± 2.2 | 5.3 ± 0.3 × 106 |
| 17. | Fe(EDTA)− | 0.120 | 7.30 ± 1.30 | 7.0 ± 1.0 × 103 | 11.0 ± 1.2 | 2.8 ± 0.2 × 104 |
| 18. | Benzylviologen | −0.354 | 6.60 ± 0.40 | 3.4 ± 0.2 × 104 | 23.5 ± 1.4 | 1.7 ± 0.1 × 105 |
| Nitroaromatic compounds | ||||||
| 19. | 2,4,6-Trinitrophenyl-N-methyl- nitramine (tetryl) | −0.191 | ND | ND | 52.0 ± 2.6 | 6.8 ± 0.5 × 106 |
| 20. | 2,4,6-Trinitrotoluene | −0.253 | ND | ND | 46.4 ± 5.2 | 1.5 ± 0.1 × 105 |
| 21. | o-Dinitrobenzene | −0.287 | ND | ND | 18.9 ± 3.0 | 8.0 ± 0.7 × 104 |
| 22. | m-Dinitrobenzene | −0.348 | ND | ND | 10.1 ± 0.9 | 8.9 ± 0.6 × 103 |
| 23. | p-Nitroacetophenone | −0.355 | ND | ND | 4.67 ± 1.40 | 2.7 ± 0.3 × 104 |
| 24. | 5-(1-Aziridinyl)-2,4-dinitro- benzamide (CB-1954) | −0.385 | ND | ND | 8.83 ± 0.80 | 2.7 ± 0.2 × 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lesanavičius, M.; Boucher, J.-L.; Čėnas, N. Reactions of Recombinant Neuronal Nitric Oxide Synthase with Redox Cycling Xenobiotics: A Mechanistic Study. Int. J. Mol. Sci. 2022, 23, 980. https://doi.org/10.3390/ijms23020980
Lesanavičius M, Boucher J-L, Čėnas N. Reactions of Recombinant Neuronal Nitric Oxide Synthase with Redox Cycling Xenobiotics: A Mechanistic Study. International Journal of Molecular Sciences. 2022; 23(2):980. https://doi.org/10.3390/ijms23020980
Chicago/Turabian StyleLesanavičius, Mindaugas, Jean-Luc Boucher, and Narimantas Čėnas. 2022. "Reactions of Recombinant Neuronal Nitric Oxide Synthase with Redox Cycling Xenobiotics: A Mechanistic Study" International Journal of Molecular Sciences 23, no. 2: 980. https://doi.org/10.3390/ijms23020980