
TLCrys: Transfer Learning Based Method for Protein Crystallization Prediction


Abstract
:1. Introduction
- As far as we know, this is the first time that transfer learning is applied to the protein crystallization prediction task based on the protein sequences. Compared with traditional machine learning models, our model is more interpretable and our predictions are more accurate.
- Since protein sequences generally contain complex spatial structure, we use global attention module and multi-task learning to pre-train the self-supervised model of protein sequences.
- In fine-tuning step, we apply a multi-head self-attention layer to extract the different levels of protein features from the global representation space during pre-training step.
2. Materials and Methods
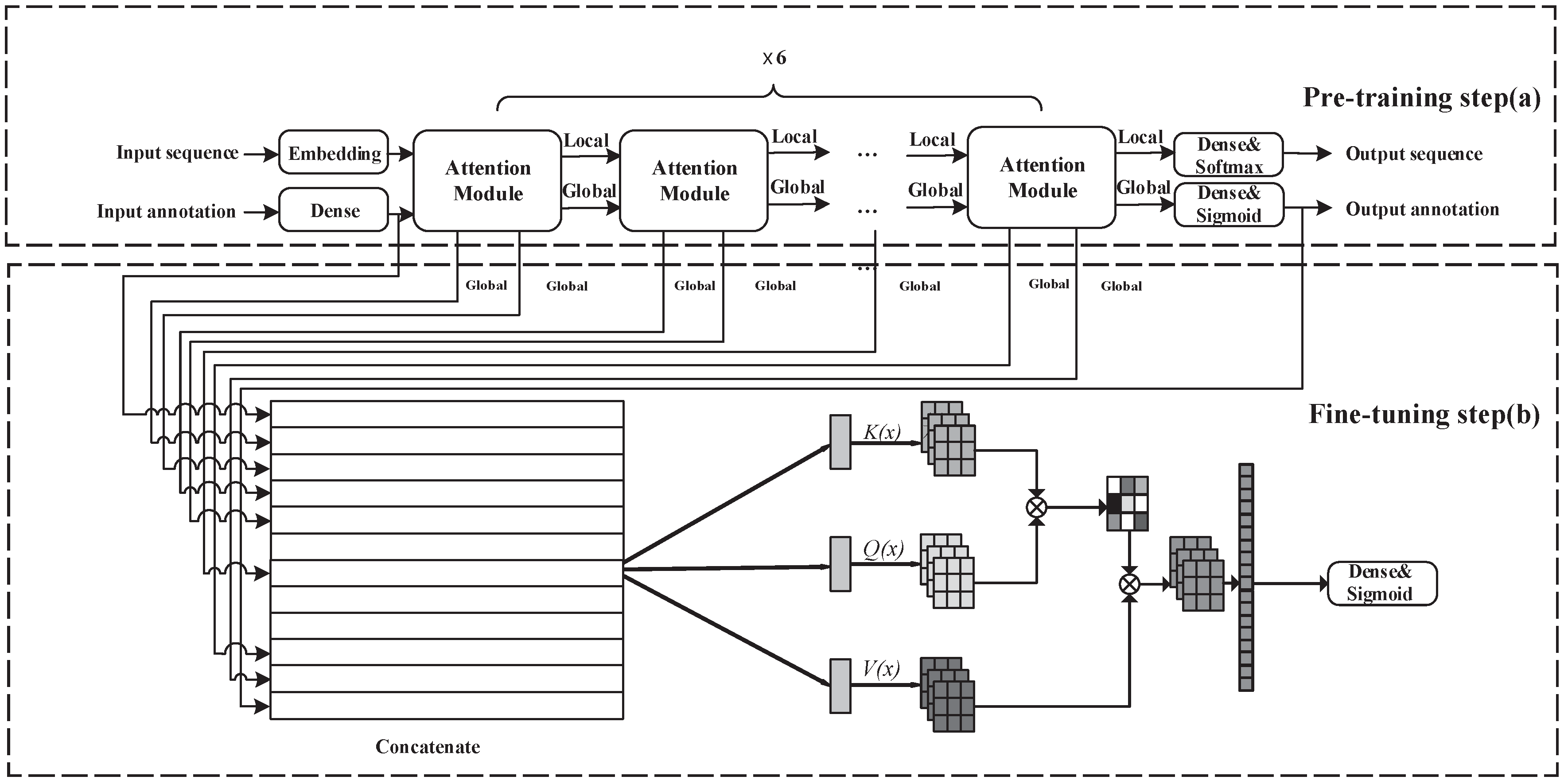
2.1. Overview of Model
- Learning task on source domain: self-supervised pre-training step for protein representation on protein sequences and Gene Ontology annotations.
- Learning task on target domain: supervised fine-tuning step on protein crystallization dataset with pre-trained parameters.
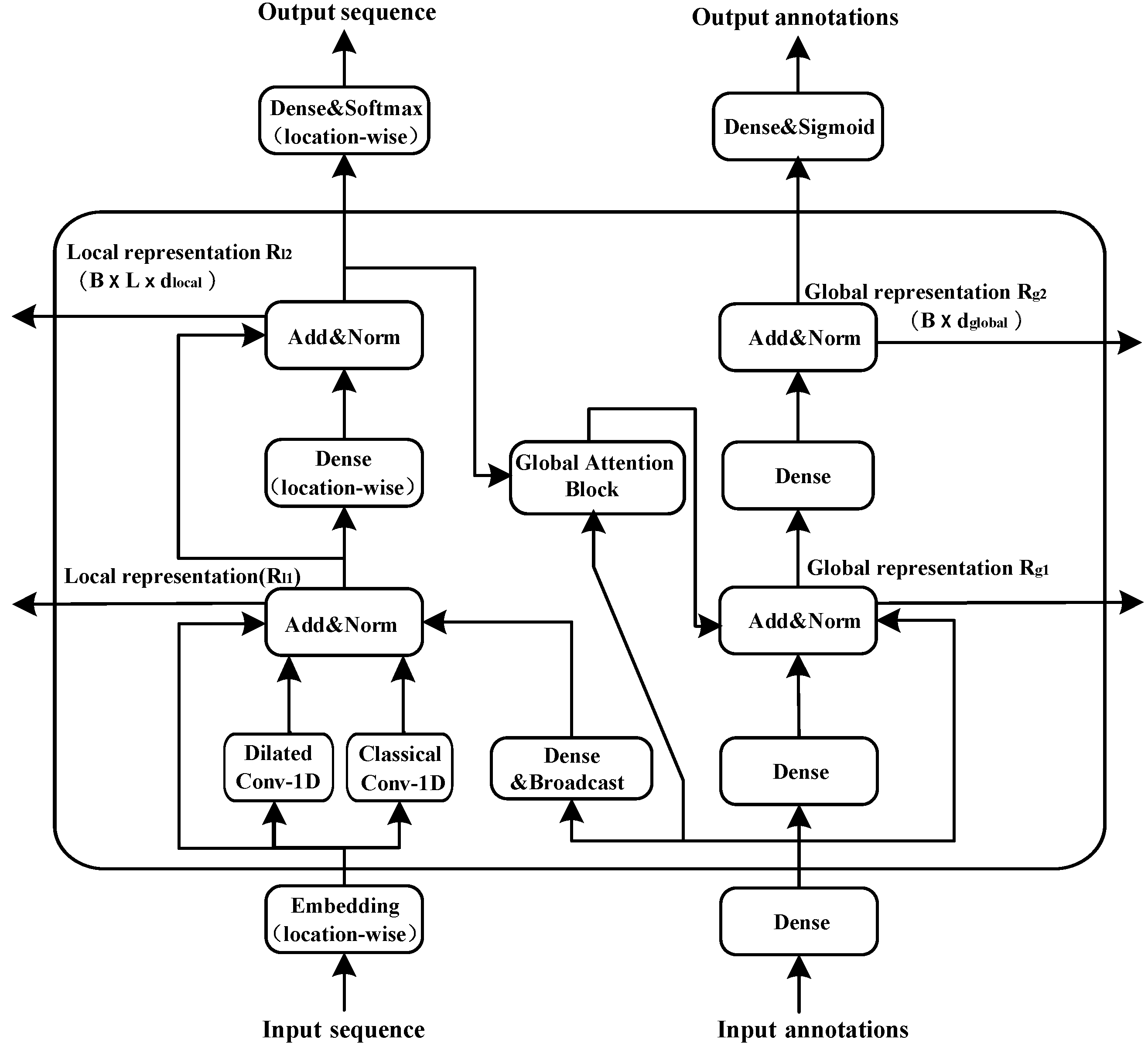
2.2. The Attention Module
- We use dilated convolution to expand the receptive field and capture multi-scale context information.
- Global attention regards global inputs as the key vector. Therefore, the number of computation parameters performed by the model grows only linearly with the sequence length, while the standard self-attention calculates a position-to-position consistency matrix and the amount of parameters grows quadratically. This linear growth is also applicable to the computation complexity and the memory consumption of the model.
- Owing to the informational interaction between the local and global representations, we can obtain not only the local features of adjacent amino acids, but also the global features of protein sequences
- The model, based on the convolutional and global attention layers, is more efficient and stable than that relying on recurrent layers.
2.3. Pre-Training
2.4. Fine-Tuning
2.5. Direct Learning
3. Results
3.1. Dataset and Environment
3.2. Metrics
3.3. Comparison with Other Methods
3.4. Ablation Experiments
3.5. Case Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Terwilliger, T.C. The success of structural genomics. J. Struct. Funct. Genom. 2011, 12, 43–44. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.D. High Resolution NMR: Theory and Chemical Applications; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Bradshaw, N.I.; Soares, D.C.; Zou, J.; Kennaway, C.K.; Porteous, D.J. 15:30 structural elucidation of disc1 pathway proteins using electron microscopy, chemical cross-linking and mass spectroscopy. Schizophr. Res. 2012, 136, S74. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Stuart, D.; Yokoyama, S. Lessons from structural genomics. Annu. Rev. Biophys. 2009, 38, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Service, R. Structural biology. Structural genomics, round 2. Science 2005, 307, 1554–1558. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, L.; Mizianty, M.J. Sequence-Based Protein Crystallization Propensity Prediction for Structural Genomics: Review and Comparative Analysis. Nat. Sci. 2009, 1, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Overton, I.M.; Barton, G.J. A normalised scale for structural genomics target ranking: The OB-Score. FEBS Lett. 2006, 580, 4005–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overton, I.M.; Padovani, G.; Girolami, M.A.; Barton, G.J. ParCrys: A Parzen window density estimation approach to protein crystallization propensity prediction. Bioinformatics 2008, 24, 901–907. [Google Scholar] [CrossRef]
- Kurgan, L.; Razib, A.A.; Aghakhani, S.; Dick, S.; Jahandideh, S. CRYSTALP2: Sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 2009, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Slabinski, L.; Jaroszewski, L.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. XtalPred: A web server for prediction of protein crystallizability. Bioinformatics 2007, 23, 3403–3405. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Kurgan, L. Sequence-based prediction of protein crystallization, purification and production propensity. Bioinformatics 2011, 27, i24–i33. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Shoombuatong, W.; Lee, H.C.; Chaijaruwanich, J.; Huang, H.L.; Ho, S.Y. SCMCRYS: Predicting protein crystallization using an ensemble scoring card method with estimating propensity scores of P-collocated amino acid pairs. PLoS ONE 2013, 8, e72368. [Google Scholar] [CrossRef]
- Kandaswamy, K.K.; Pugalenthi, G.; Suganthan, P.; Gangal, R. SVMCRYS: An SVM approach for the prediction of protein crystallization propensity from protein sequence. Protein Pept. Lett. 2010, 17, 423–430. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Tan, H.; Li, Y.; Zhang, Z.; Song, J. PredPPCrys: Accurate Prediction of Sequence Cloning, Protein Production, Purification and Crystallization Propensity from Protein Sequences Using Multi-Step Heterogeneous Feature Fusion and Selection. PLoS ONE 2014, 9, e105902. [Google Scholar] [CrossRef]
- Wang, H.; Feng, L.; Zhang, Z.; Webb, G.I.; Lin, D.; Song, J. Crysalis: An integrated server for computational analysis and design of protein crystallization. Sci. Rep. 2016, 6, 21383. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Zhang, H.; Jin, C.; Quan, X.; Yin, Y. A representation learning model based on variational inference and graph autoencoder for predicting lncRNA-disease associations. BMC Bioinform. 2021, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Shi, Z.; Zhang, H.; Yin, Y. Predicting lncRNA–protein interactions based on graph autoencoders and collaborative training. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021. [Google Scholar]
- Jin, C.; Shi, Z.; Lin, K.; Zhang, H. Predicting miRNA-Disease Association Based on Neural Inductive Matrix Completion with Graph Autoencoders and Self-Attention Mechanism. Biomolecules 2022, 12, 64. [Google Scholar] [CrossRef]
- Elbasir, A.; Moovarkumudalvan, B.; Kunji, K.; Kolatkar, P.R.; Mall, R.; Bensmail, H. DeepCrystal: A Deep Learning Framework for Sequence-based Protein Crystallization Prediction. Bioinformatics 2018, 35, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Gao, J.; Shi, Z.; Zhang, H. ATTCry: Attention-based neural network model for protein crystallization prediction. Neurocomputing 2021, 463, 265–274. [Google Scholar] [CrossRef]
- Pan, S.J.; Yang, Q. A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.U.; Polosukhin, I. Attention is All you Need. In Proceedings of the Advances in Neural Information Processing Systems 30, Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.08661. [Google Scholar]
- Cao, Y.; Shen, Y. TALE: Transformer-based protein function Annotation with joint sequence–Label Embedding. Bioinformatics 2021, 37, 2825–2833. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. In Proceedings of the International Conference on Learning Representations (ICLR), Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Han, K.; Li, Y.; Yang, J.Y.; Shen, H.B.; Yu, D.J. TargetCrys: Protein crystallization prediction by fusing multi-view features with two-layered SVM. Amino Acids 2016, 48, 2533–2547. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, S.; Moovarkumudalvan, B.; Lescar, J.; Kolatkar, P.R. Crystallization and X-ray diffraction analysis of the HMG domain of the chondrogenesis master regulator Sox9 in complex with a ChIP-Seq-identified DNA element. Acta Crystallogr. Sect. Struct. Biol. Commun. 2015, 71, 1437–1441. [Google Scholar] [CrossRef] [Green Version]
- Palasingam, P.; Jauch, R.; Ng, C.K.L.; Kolatkar, P.R. The structure of Sox17 bound to DNA reveals a conserved bending topology but selective protein interaction platforms. J. Mol. Biol. 2009, 388, 619–630. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Global Dim | Local Dim | Dilation Rate | Kernel Size |
| 512 | 128 | 5 | 9 |
| Stride Size | Key Dim | Value Dim | Head Number |
| 1 | 64 | 128 | 4 |
| Tasks | Dataset | Clone f. | Material Production f. | Purification f. | Crystallization f. | Crystallization |
|---|---|---|---|---|---|---|
| CLF | Train | N:9502 | P:14428 | |||
| Test | N:1939 | P:2852 | ||||
| MF | Train | N:17017 | P:6913 | |||
| Test | N:3347 | P:1444 | ||||
| PF | Train | - | N:2318 | P:4702 | ||
| Test | - | N:474 | P:932 | |||
| CF | Train | - | N:224 | P:631 | ||
| Test | - | N:35 | P:138 | |||
| CRYs | Train | N:19509 | P:4421 | |||
| Test | N:3892 | P:899 |
| Model | AUC | MCC | ACC (%) | SPEC (%) | SEN (%) | PRE (%) | F1 Score (%) | |
|---|---|---|---|---|---|---|---|---|
| CLF | PredPPCrys I | 0.711 | 0.296 | 65.33 | 63.58 | 66.50 | 73.16 | 69.67 |
| PredPPCrys II | 0.725 | 0.322 | 66.54 | 65.56 | 67.20 | 74.44 | 70.63 | |
| Crysalis I | 0.731 | 0.332 | 66.98 | 66.60 | 67.22 | 75.56 | 71.15 | |
| Crysalis II | 0.756 | 0.365 | 68.34 | 69.95 | 68.34 | 76.85 | 72.35 | |
| Direct learning | 0.701 | 0.326 | 64.65 | 76.14 | 56.84 | 77.81 | 65.69 | |
| TLCrys | 0.817 | 0.455 | 72.90 | 74.28 | 71.96 | 80.46 | 77.00 | |
| MF | PredPPCrys I | 0.772 | 0.380 | 69.93 | 68.21 | 72.88 | 49.95 | 59.27 |
| PredPPCrys II | 0.793 | 0.416 | 71.95 | 71.36 | 73.30 | 52.70 | 61.32 | |
| Crysalis I | 0.759 | 0.377 | 70.23 | 69.93 | 70.99 | 49.25 | 58.15 | |
| Crysalis II | 0.793 | 0.427 | 73.08 | 73.58 | 73.09 | 54.15 | 62.21 | |
| Direct learning | 0.745 | 0.307 | 73.31 | 88.67 | 37.74 | 58.98 | 46.03 | |
| TLCrys | 0.848 | 0.446 | 78.37 | 92.53 | 45.57 | 72.47 | 55.90 | |
| PF | PredPPCrys I | 0.800 | 0.460 | 74.83 | 70.52 | 77.02 | 83.77 | 80.25 |
| PredPPCrys II | 0.872 | 0.579 | 79.73 | 81.43 | 78.86 | 89.31 | 83.76 | |
| Crysalis I | 0.796 | 0.436 | 73.87 | 67.80 | 73.87 | 82.47 | 77.93 | |
| Crysalis II | 0.793 | 0.427 | 73.08 | 73.58 | 73.09 | 54.15 | 62.21 | |
| Direct learning | 0.778 | 0.505 | 78.52 | 60.97 | 73.09 | 54.15 | 62.21 | |
| TLCrys | 0.861 | 0.583 | 81.58 | 70.25 | 87.34 | 85.24 | 86.27 | |
| CF | PredPPCrys I | 0.712 | 0.280 | 67.05 | 67.65 | 66.91 | 89.42 | 76.54 |
| PredPPCrys II | 0.735 | 0.175 | 69.47 | 68.89 | 69.50 | 97.80 | 81.26 | |
| Crysalis I | 0.739 | 0.281 | 65.50 | 70.59 | 64.23 | 89.80 | 74.89 | |
| Crysalis II | 0.752 | 0.337 | 62.57 | 85.29 | 56.93 | 93.97 | 70.90 | |
| Direct learning | 0.694 | 0.123 | 71.10 | 31.43 | 81.16 | 82.35 | 81.75 | |
| TLCrys | 0.785 | 0.459 | 79.77 | 68.57 | 82.61 | 91.20 | 86.69 | |
| CRYs | ParCrys | 0.611 | 0.132 | 59.66 | 60.56 | 55.91 | 25.40 | 34.93 |
| OBScore | 0.638 | 0.184 | 59.28 | 57.78 | 65.49 | 27.14 | 38.38 | |
| CRYSTAP2 | 0.599 | 0.123 | 51.64 | 48.10 | 67.78 | 22.28 | 33.54 | |
| XtalPred | - | 0.224 | 65.04 | 65.61 | 62.51 | 29.31 | 39.91 | |
| SVMCRYs | - | 0.142 | 55.11 | 52.78 | 65.70 | 23.39 | 34.50 | |
| PPCPred | 0.704 | 0.254 | 63.63 | 62.09 | 70.67 | 29.03 | 41.15 | |
| XtalPred-RF | - | 0.205 | 60.94 | 59.67 | 66.41 | 27.56 | 38.95 | |
| SCMCRYS | - | 0.145 | 60.93 | 62.01 | 56.24 | 25.48 | 35.07 | |
| PredPPCrys I | 0.770 | 0.326 | 69.65 | 69.30 | 71.13 | 35.23 | 47.12 | |
| PredPPCrys II | 0.838 | 0.428 | 76.04 | 76.21 | 75.30 | 42.64 | 54.45 | |
| Crysalis I | 0.788 | 0.339 | 71.00 | 70.89 | 71.41 | 35.50 | 47.42 | |
| Crysalis II | 0.838 | 0.435 | 76.27 | 76.28 | 76.20 | 42.84 | 54.85 | |
| DeepCrystal | 0.858 | 0.477 | 77.83 | 77.43 | 79.51 | 45.90 | 58.20 | |
| Direct learning | 0.801 | 0.367 | 83.79 | 95.99 | 31.03 | 64.14 | 41.83 | |
| TLCrys | 0.879 | 0.546 | 87.24 | 94.96 | 53.84 | 71.18 | 61.30 |
| Models | AUC | MCC | ACC (%) | F1 Score (%) |
|---|---|---|---|---|
| without multi-head attention | 0.874 | 0.498 | 86.57 | 54.36 |
| 2 heads | 0.875 | 0.537 | 86.69 | 61.32 |
| 4 heads | 0.880 | 0.529 | 86.99 | 59.09 |
| TLCrys (6 heads) | 0.879 | 0.546 | 87.24 | 61.30 |
| 8 heads | 0.879 | 0.541 | 87.30 | 60.10 |
| Model | Sox9 FL (−) | Sox9 HMG (+) | Sox17 FL (−) | Sox17 HMG (+) | Sox17 EK-HMG (+) |
|---|---|---|---|---|---|
| TLCrys | 0.156 | 0.674 | 0.260 | 0.791 | 0.681 |
| DeepCrystal | 0.315 | 0.676 | 0.430 | 0.643 | 0.633 |
| TargetCrys | 0.032 | 0.045 | 0.037 | 0.029 | 0.031 |
| Crysalis II | 0.474 | 0.55 | 0.474 | 0.553 | 0.555 |
| Crysalis I | 0.438 | 0.482 | 0.487 | 0.567 | 0.557 |
| PPCPred | 0.039 | 0.658 | 0.089 | 0.462 | 0.523 |
| CrystalP2 | 0.327 | 0.459 | 0.470 | 0.436 | 0.402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, C.; Shi, Z.; Kang, C.; Lin, K.; Zhang, H. TLCrys: Transfer Learning Based Method for Protein Crystallization Prediction. Int. J. Mol. Sci. 2022, 23, 972. https://doi.org/10.3390/ijms23020972
Jin C, Shi Z, Kang C, Lin K, Zhang H. TLCrys: Transfer Learning Based Method for Protein Crystallization Prediction. International Journal of Molecular Sciences. 2022; 23(2):972. https://doi.org/10.3390/ijms23020972
Chicago/Turabian StyleJin, Chen, Zhuangwei Shi, Chuanze Kang, Ken Lin, and Han Zhang. 2022. "TLCrys: Transfer Learning Based Method for Protein Crystallization Prediction" International Journal of Molecular Sciences 23, no. 2: 972. https://doi.org/10.3390/ijms23020972