
Caffeic Acid/Eu(III) Complexes: Solution Equilibrium Studies, Structure Characterization and Biological Activity

 , , , , , , , , , , , ,
, , , , , , , , , , , ,
Abstract
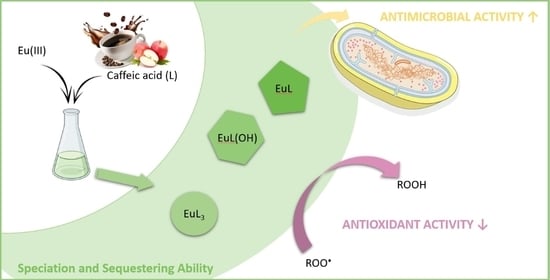
:
1. Introduction
2. Results and Discussion
2.1. Molecular Structure of [Eu(CFA)3]
2.2. Solution Equilibrium Studies
2.2.1. Acid–Base Properties of CFA and Eu3+
2.2.2. Formation and Stability of Eu(III)-CFA Species
2.2.3. Chemical Speciation and Sequestering Ability
2.3. Biological Studies
2.3.1. Antimicrobial Activity
2.3.2. Microelectrophoretic Mobility Measurements
2.3.3. Antioxidant Activity
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis and Characterization of Eu(III) Metal Complexes
3.3. Potentiometric Measurements
3.4. UV-Vis Spectrophotometric Measurements
3.5. H-NMR Measurements
3.6. ESI-MS Measurements
3.7. Thermodynamic Calculations
Evaluation of the Sequestering Ability
3.8. Antimicrobial Studies
3.9. Microelectrophoretic Mobility Measurements
3.10. Anti-Oxidative Studies
3.10.1. DPPH Assay
3.10.2. ABTS Assay
3.10.3. FRAP Assay
3.10.4. CUPRAC Assay
3.10.5. Inhibition of the Lipid Peroxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Godlewska-Żyłkiewicz, B.; Świsłocka, R.; Kalinowska, M.; Golonko, A.; Świderski, G.; Arciszewska, Ż.; Nalewajko-Sieliwoniuk, E.; Naumowicz, M.; Lewandowski, W. Biologically Active Compounds of Plants: Structure-Related Antioxidant, Microbiological and Cytotoxic Activity of Selected Carboxylic Acids. Materials 2020, 13, 4454. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Cheng, K.; Zhang, Z.-M.; Fang, R.-Q.; Zhu, H.-L. Synthesis, structure and structure–activity relationship analysis of caffeic acid amides as potential antimicrobials. Eur. J. Med. Chem. 2010, 45, 2638–2643. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, L.; Chochkova, M.; Totseva, I.; Seizova, K.; Marinova, E.; Ivanova, G.; Ninova, M.; Najdenski, H.; Milkova, T. Anti-tyrosinase, antioxidant and antimicrobial activities of hydroxycinnamoylamides. Med. Chem. Res. 2013, 22, 4173–4182. [Google Scholar] [CrossRef]
- Lima, V.N.; Oliveira-Tintino, C.D.; Santos, E.S.; Morais, L.P.; Tintino, S.R.; Freitas, T.S.; Geraldo, Y.S.; Pereira, R.L.; Cruz, R.P.; Menezes, I.R.; et al. Antimicrobial and enhancement of the antibiotic activity by phenolic compounds: Gallic acid, caffeic acid and pyrogallol. Microb. Pathog. 2016, 99, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Kyselka, J.; Rabiej, D.; Dragoun, M.; Kreps, F.; Burčová, Z.; Němečková, I.; Smolová, J.; Bjelková, M.; Szydłowska-Czerniak, A.; Schmidt, Š.; et al. Antioxidant and antimicrobial activity of linseed lignans and phenolic acids. Eur. Food Res.Tech. 2017, 243, 1633–1644. [Google Scholar] [CrossRef]
- Kępa, M.; Miklasińska-Majdanik, M.; Wojtyczka, R.D.; Idzik, D.; Korzeniowski, K.; Smoleń-Dzirba, J.; Wąsik, T.J. Antimicrobial Potential of Caffeic Acid against Staphylococcus aureus: Clinical Strains. BioMed Res. Int. 2018, 2018, 7413504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.; Bamunuarachchi, N.I.; Tabassum, N.; Kim, Y.-M. Caffeic Acid and Its Derivatives: Antimicrobial Drugs toward Microbial Pathogens. J. Agric. Food Chem. 2021, 69, 2979–3004. [Google Scholar] [CrossRef]
- Damasceno, S.; Dantas, B.; Ribeiro-Filho, J.; Antônio, M.; Galberto, M. Chemical Properties of Caffeic and Ferulic Acids in Biological System: Implications in Cancer Therapy. A Review. Curr. Pharm. Des. 2017, 23, 3015–3023. [Google Scholar] [CrossRef]
- Świsłocka, R. Spectroscopic (FT-IR, FT-Raman, UV absorption, 1H and 13C NMR) and theoretical (in B3LYP/6-311++G** level) studies on alkali metal salts of caffeic acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 100, 21–30. [Google Scholar] [CrossRef]
- Matejczyk, M.; Świsłocka, R.; Golonko, A.; Lewandowski, W.; Hawrylik, E. Cytotoxic, genotoxic and antimicrobial activity of caffeic and rosmarinic acids and their lithium, sodium and potassium salts as potential anticancer compounds. Adv. Med. Sci. 2018, 63, 14–21. [Google Scholar] [CrossRef]
- Cornard, J.P.; Lapouge, C. Theoretical and Spectroscopic Investigations of a Complex of Al(III) with Caffeic Acid. J. Phys. Chem. A 2004, 108, 4470–4478. [Google Scholar] [CrossRef]
- Cornard, J.-P.; Caudron, A.; Merlin, J.-C. UV–visible and synchronous fluorescence spectroscopic investigations of the complexation of Al(III) with caffeic acid, in aqueous low acidic medium. Polyhedron 2006, 25, 2215–2222. [Google Scholar] [CrossRef]
- Thoma, V.; Tampouris, K.; Petrou, A.L. Kinetics and mechanism of the reaction between chromium(III) and 3,4-dihydroxy-phenyl-propenoic acid (caffeic acid) in weak acidic aqueous solutions. Bioinorg. Chem. Appl. 2008, 2008, 624583. [Google Scholar] [CrossRef] [Green Version]
- Boilet, L.; Cornard, J.P.; Lapouge, C. Determination of the Chelating Site Preferentially Involved in the Complex of Lead(II) with Caffeic Acid: A Spectroscopic and Structural Study. J. Phys. Chem. A 2005, 109, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, C.; Foti, C.; Giuffrè, O.; Sammartano, S. Acid–base and UV behavior of 3-(3,4-dihydroxyphenyl)-propenoic acid (caffeic acid) and complexing ability towards different divalent metal cations in aqueous solution. J. Mol. Liq. 2014, 195, 9–16. [Google Scholar] [CrossRef]
- Živanović, S.C.; Veselinović, A.M.; Mitić, Ž.J.; Nikolić, G.M. The study of the influence of Mg(II) and Ca(II) ions on caffeic acid autoxidation in weakly alkaline aqueous solution using MCR-ALS analysis of spectrophotometric data. New J. Chem. 2018, 42, 6256–6263. [Google Scholar] [CrossRef]
- Liddle, S.T.; Mills, D.P.; Natrajan, L.S. The Lanthanides and Actinides; World Scientific Publishing Europe Ltd.: London, UK, 2021. [Google Scholar]
- Crea, F.; De Stefano, C.; Milea, D.; Sammartano, S. Thermodynamic data for lanthanoid(III) sequestration by phytate at different temperatures. Monatsh. Chem. 2010, 141, 511–520. [Google Scholar] [CrossRef]
- Cigala, R.M.; De Stefano, C.; Irto, A.; Milea, D.; Sammartano, S. Thermodynamic Data for the Modeling of Lanthanoid(III) Sequestration by Reduced Glutathione in Aqueous Solution. J. Chem. Eng. Data 2015, 60, 192–201. [Google Scholar] [CrossRef]
- Cotruvo, J.A., Jr. The Chemistry of Lanthanides in Biology: Recent Discoveries, Emerging Principles, and Technological Applications. ACS Cent. Sci. 2019, 5, 1496–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostova, I.; Traykova, M. Cerium(III) and neodymium(III) complexes as scavengers of X/XO-derived superoxide radical. Med. Chem. 2006, 2, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I.; Peica, N.; Kiefer, W. Theoretical and spectroscopic studies of 5-aminoorotic acid and its new lanthanide(III) complexes. J. Raman Spectrosc. 2007, 38, 205–216. [Google Scholar] [CrossRef]
- Kostova, I.; Momekov, G. New cerium(III) complexes of coumarins—Synthesis, characterization and cytotoxicity evaluation. Eur. J. Med. Chem. 2008, 43, 178–188. [Google Scholar] [CrossRef]
- Kostova, I.; Valcheva-Traykova, M. New samarium(III) complex of 5-aminoorotic acid with antioxidant activity. Appl. Organometal. Chem. 2015, 29, 815–824. [Google Scholar] [CrossRef]
- Ajlouni, A.M.; Abu-Salem, Q.; Taha, Z.A.; Hijazi, A.K.; Al Momani, W. Synthesis, characterization, biological activities and luminescent properties of lanthanide complexes with [2-thiophenecarboxylic acid, 2-(2-pyridinylmethylene)hydrazide] Schiff bases ligand. J. Rare Earths 2016, 34, 986–993. [Google Scholar] [CrossRef]
- Kaczmarek, M.T.; Zabiszak, M.; Nowak, M.; Jastrzab, R. Lanthanides: Schiff base complexes, applications in cancer diagnosis, therapy, and antibacterial activity. Coord. Chem. Rev. 2018, 370, 42–54. [Google Scholar] [CrossRef]
- Cota, I.; Marturano, V.; Tylkowski, B. Ln complexes as double faced agents: Study of antibacterial and antifungal activity. Coord. Chem. Rev. 2019, 396, 49–71. [Google Scholar] [CrossRef]
- Kostova, I. Lanthanides as anticancer agents. Curr. Med. Chem. Anticancer Agents 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Boldyrev, I.A.; Gaenko, G.P.; Moiseeva, E.V.; Deligeorgiev, T.; Kaloyanova, S.; Lesev, N.; Vasilev, A.; Molotkovsky, J.G. Europium complexes of 1,10-phenanthrolines: Their inclusion in liposomes and cytotoxicity. Russ. J. Bioorg. Chem. 2011, 37, 364–368. [Google Scholar] [CrossRef]
- Trusova, V.; Yudintsev, A.; Limanskaya, L.; Gorbenko, G.; Deligeorgiev, T. Europium coordination complexes as potential anticancer drugs: Their partitioning and permeation into lipid bilayers as revealed by pyrene fluorescence quenching. J. Fluoresc. 2013, 23, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Wydro, U.; Wołejko, E.; Świderski, G.; Lewandowski, W. Biological Activity of New Cichoric Acid-Metal Complexes in Bacterial Strains, Yeast-Like Fungi, and Human Cell Cultures In Vitro. Nutrients 2020, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- Kalinowska, M.; Sienkiewicz-Gromiuk, J.; Świderski, G.; Pietryczuk, A.; Cudowski, A.; Lewandowski, W. Zn(II) Complex of Plant Phenolic Chlorogenic Acid: Antioxidant, Antimicrobial and Structural Studies. Materials 2020, 13, 3745. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, W.; Lewandowska, H.; Golonko, A.; Świderski, G.; Świsłocka, R.; Kalinowska, M. Correlations between molecular structure and biological activity in “logical series” of dietary chromone derivatives. PLoS ONE 2020, 15, e0229477. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, M.; Gołębiewska, E.; Mazur, L.; Lewandowska, H.; Pruszyński, M.; Świderski, G.; Wyrwas, M.; Pawluczuk, N.; Lewandowski, W. Crystal Structure, Spectroscopic Characterization, Antioxidant and Cytotoxic Activity of New Mg(II) and Mn(II)/Na(I) Complexes of Isoferulic Acid. Materials 2021, 14, 3236. [Google Scholar] [CrossRef]
- Arena, K.; Brancato, G.; Cacciola, F.; Crea, F.; Cataldo, S.; De Stefano, C.; Gama, S.; Lando, G.; Milea, D.; Mondello, L.; et al. 8-Hydroxyquinoline-2-Carboxylic Acid as Possible Molybdophore: A Multi-Technique Approach to Define Its Chemical Speciation, Coordination and Sequestering Ability in Aqueous Solution. Biomolecules 2020, 10, 930. [Google Scholar] [CrossRef]
- Cigala, R.M.; Cordaro, M.; Crea, F.; De Stefano, C.; Fracassetti, V.; Marchesi, M.; Milea, D.; Sammartano, S. Acid–Base Properties and Alkali and Alkaline Earth Metal Complex Formation in Aqueous Solution of Diethylenetriamine-N,N,N′,N″,N″-pentakis(methylenephosphonic acid) Obtained by an Efficient Synthetic Procedure. Ind. Eng. Chem. Res. 2014, 53, 9544–9553. [Google Scholar] [CrossRef]
- Palma, E.; Mendes, F.; Morais, G.R.; Rodrigues, I.; Santos, I.C.; Campello, M.P.C.; Raposinho, P.; Correia, I.; Gama, S.; Belo, D.; et al. Biophysical characterization and antineoplastic activity of new bis(thiosemicarbazonato) Cu(II) complexes. J. Inorg. Biochem. 2017, 167, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Gabano, E.; Gama, S.; Mendes, F.; Gariboldi, M.B.; Monti, E.; Bombard, S.; Bianco, S.; Ravera, M. Study of the synthesis, antiproliferative properties, and interaction with DNA and polynucleotides of cisplatin-like Pt(II) complexes containing carcinogenic polyaromatic amines. J. Biol. Inorg. Chem. 2013, 18, 791–801. [Google Scholar] [CrossRef]
- Kalinowska, M.; Piekut, J.; Bruss, A.; Follet, C.; Sienkiewicz-Gromiuk, J.; Świsłocka, R.; Rzączyńska, Z.; Lewandowski, W. Spectroscopic (FT-IR, FT-Raman, 1H, 13C NMR, UV/VIS), thermogravimetric and antimicrobial studies of Ca(II), Mn(II), Cu(II), Zn(II) and Cd(II) complexes of ferulic acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 122, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Świderski, G.; Łyszczek, R.; Wojtulewski, S.; Kalinowska, M.; Świsłocka, R.; Lewandowski, W. Comparison of structural, spectroscopic, theoretical and thermal properties of metal complexes (Zn(II), Mn(II), Cu(II), Ni(II) and Co(II)) of pyridazine-3-carboxylic acid and pyridazine-4-carboxylic acids. Inorg. Chim. Acta 2020, 512, 119865. [Google Scholar] [CrossRef]
- Varsányi, G.; Kovner, M.A.; Láng, L. Assignments for Vibrational Spectra of 700 Benzene Derivatives; Akademiai Kiadó: Budapest, Hungary, 1973. [Google Scholar]
- Lewandowski, W.; Fuks, L.; Kalinowska, M.; Koczoń, P. The influence of selected metals on the electronic system of biologically important ligands. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2003, 59, 3411–3420. [Google Scholar] [CrossRef]
- Lewandowski, W.; Kalinowska, M.; Lewandowska, H. The influence of metals on the electronic system of biologically important ligands. Spectroscopic study of benzoates, salicylates, nicotinates and isoorotates. J. Inorg. Biochem. 2005, 99, 1407–1423. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; McCarthy, P.J. Spectroscopy and Structure of Metal Chelate Compounds; Wiley: New York, NY, USA, 1968. [Google Scholar]
- Kalinowska, M.; Świsłocka, R.; Lewandowski, W. The spectroscopic (FT-IR, FT-Raman and 1H, 13C NMR) and theoretical studies of cinnamic acid and alkali metal cinnamates. J. Mol. Struct. 2007, 834, 572–580. [Google Scholar] [CrossRef]
- Swislocka, R.; Kowczyk-Sadowy, M.; Kalinowska, M.; Lewandowski, W. Spectroscopic (FT-IR, FT-Raman, 1H and 13C NMR) and theoretical studies of p-coumaric acid and alkali metal p-coumarates. J. Spectrosc. 2012, 27, 546146. [Google Scholar] [CrossRef]
- Lin, X.; Ning, E.; Li, X.; Li, Q. Construction of mixed carboxylate and pyrogallate building units for luminescent metal–organic frameworks. Chin. Chem. Lett. 2020, 31, 813–817. [Google Scholar] [CrossRef]
- Dan, M.; Cheetham, A.K.; Rao, C.N.R. Diverse Structures and Dimensionalities in Hybrid Frameworks of Strontium and Lanthanum with Isomeric Dihydroxybenzoates. Inorg. Chem. 2006, 45, 8227–8238. [Google Scholar] [CrossRef]
- Bizri, Y.; Cromer, M.; Lamy, I.; Scharff, J.P. Complexation dans les systèmes organo-minéraux modèles (acide cafféique et tiron) et naturels (substances humiques). Analusis 1985, 13, 128–133. [Google Scholar]
- Gans, P.; Sabatini, A.; Vacca, A. HypSpec. Available online: http://www.hyperquad.co.uk (accessed on 15 October 2021).
- Frassineti, C.; Ghelli, S.; Gans, P.; Sabatini, A.; Moruzzi, M.S.; Vacca, A. Nuclear Magnetic Resonance as a Tool for Determining Protonation Constants of Natural Polyprotic Bases in Solution. Anal. Biochem. 1995, 231, 374–382. [Google Scholar] [CrossRef]
- Crea, F.; Milea, D.; Sammartano, S. Enhancement of hydrolysis through the formation of mixed hetero-metal species. Talanta 2005, 65, 229–238. [Google Scholar] [CrossRef]
- Crea, F.; Milea, D.; Sammartano, S. Enhancement of Hydrolysis through the Formation of Mixed Hetero-Metal Species: Dioxouranium(VI)–Cadmium(II) Mixtures. Ann. Chim. 2005, 95, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, C.; Brown, P.L. Hydrolysis of Metal Ions; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016. [Google Scholar]
- Klungness, G.D.; Byrne, R.H. Comparative hydrolysis behavior of the rare earths and yttrium: The influence of temperature and ionic strength. Polyhedron 2000, 19, 99–107. [Google Scholar] [CrossRef]
- Kiss, T.; Nagy, G.; Pécsi, M.; Kozlowski, H.; Micera, G.; Erre, L.S. Complexes of 3,4-dihydroxyphenyl derivatives—X. Copper(II) complexes of chlorogenic acid and related compounds. Polyhedron 1989, 8, 2345–2349. [Google Scholar] [CrossRef]
- Sahoo, S.; Bera, R.K.; Baral, M.; Kanungo, B.K.A.C.S. Spectroscopic and Potentiometric Study of 2,3-Dihydroxybenzoic Acid and its Complexation with La(III) Ion. Acta Chim. Slov. 2008, 55, 243–247. [Google Scholar]
- Cocks, S.; Under, P.W.; Voyé, A. Potentiometric Investigations of Equilibria Between Caffeic Acid and Manganese(II), Cobalt(II), Nickel(II) and Cadmium(II) Ions in Aqueous Solution. J. Coord. Chem. 1992, 25, 211–220. [Google Scholar] [CrossRef]
- Adams, M.L.; O’Sullivan, B.; Downard, A.J.; Powell, K.J. Stability Constants for Aluminum(III) Complexes with the 1,2-Dihydroxyaryl Ligands Caffeic Acid, Chlorogenic Acid, DHB, and DASA in Aqueous Solution. J. Chem. Eng. Data 2002, 47, 289–296. [Google Scholar] [CrossRef]
- Beneduci, A.; Furia, E.; Russo, N.; Marino, T. Complexation behaviour of caffeic, ferulic and p-coumaric acids towards aluminium cations: A combined experimental and theoretical approach. New J. Chem. 2017, 41, 5182–5190. [Google Scholar] [CrossRef]
- Malacaria, L.; Corrente, G.A.; Beneduci, A.; Furia, E.; Marino, T.; Mazzone, G. A Review on Coordination Properties of Al(III) and Fe(III) toward Natural Antioxidant Molecules: Experimental and Theoretical Insights. Molecules 2021, 26, 2603. [Google Scholar] [CrossRef]
- Zhu, D.-H.; Kappel, M.J.; Raymond, K.N. Coordination chemistry of lanthanide catecholates. Inorg. Chim. Acta 1988, 147, 115–121. [Google Scholar] [CrossRef]
- Taha, M.; Khan, I.; Coutinho, J.A. Complexation and molecular modeling studies of europium(III)-gallic acid-amino acid complexes. J. Inorg. Biochem. 2016, 157, 25–33. [Google Scholar] [CrossRef]
- Gama, S.; Frontauria, M.; Ueberschaar, N.; Brancato, G.; Milea, D.; Sammartano, S.; Plass, W. Thermodynamic study on 8-hydroxyquinoline-2-carboxylic acid as a chelating agent for iron found in the gut of Noctuid larvae. New J. Chem. 2018, 42, 8062–8073. [Google Scholar] [CrossRef]
- Gama, S.; Hermenau, R.; Frontauria, M.; Milea, D.; Sammartano, S.; Hertweck, C.; Plass, W. Iron Coordination Properties of Gramibactin as Model for the New Class of Diazeniumdiolate Based Siderophores. Chem. Eur. J. 2021, 27, 2724–2733. [Google Scholar] [CrossRef]
- Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Clares, M.P.; García-España, E. Addressing selectivity criteria in binding equilibria. Coord. Chem. Rev. 2012, 256, 13–27. [Google Scholar] [CrossRef]
- Crea, F.; De Stefano, C.; Foti, C.; Milea, D.; Sammartano, S. Chelating agents for the sequestration of mercury(II) and monomethyl mercury(II). Curr. Med. Chem. 2014, 21, 3819–3836. [Google Scholar] [CrossRef]
- Harris, W.R.; Carrano, C.J.; Cooper, S.R.; Sofen, S.R.; Avdeef, A.E.; McArdle, J.V.; Raymond, K.N. Coordination chemistry of microbial iron transport compounds. 19. Stability constants and electrochemical behavior of ferric enterobactin and model complexes. J. Am. Chem. Soc. 1979, 101, 6097–6104. [Google Scholar] [CrossRef]
- Overton, E. Studien über d. Narkose Zugleich ein Beitrag zur Allgemeinen Pharmakologie; Gustav Fisher: Jena, Germany, 1901; p. 195. [Google Scholar]
- Tweedy, B.G. Plant Extracts with Metal Ions as Potential Antimicrobial Agents. Phytopatology 1964, 55, 910–918. [Google Scholar]
- Zong, G.-C.; Huo, J.-X.; Ren, N.; Zhang, J.-J.; Qi, X.-X.; Gao, J.; Geng, L.-N.; Wang, S.-P.; Shi, S.-K. Preparation, characterization and properties of four new trivalent lanthanide complexes constructed using 2-bromine-5-methoxybenzoic acid and 1,10-phenanthroline. Dalton Trans. 2015, 44, 14877–14886. [Google Scholar] [CrossRef]
- Mohanan, K.; Devi, S.N. Synthesis, characterization, thermal stability, reactivity, and antimicrobial properties of some novel lanthanide(III) complexes of 2-(N-salicylideneamino)-3-carboxyethyl-4,5,6,7-tetrahydrobenzo[b]thiophene. Russ. J. Coord. Chem. 2006, 32, 600–609. [Google Scholar] [CrossRef]
- Ivanova, E.; Mitik-Dineva, N.; Mocanasu, R.; Murphy, S.; Wang, J.; Riessen, G.v.; Crawford, R. Vibrio fischeri and Escherichia coli adhesion tendencies towards photolithographically modified nanosmooth poly (tert-butyl methacrylate) polymer surfaces. Nanotechnol. Sci. Appl. 2008, 1, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Schott, H.; Young, C.Y. Electrokinetic Studies of Bacteria III: Effect of Polyvalent Metal Ions on Electrophoretic Mobility and Growth of Streptococcus faecalis. J. Pharm. Sci. 1973, 62, 1797–1801. [Google Scholar] [CrossRef]
- Collins, Y.E.; Stotzky, G. Heavy metals alter the electrokinetic properties of bacteria, yeasts, and clay minerals. Appl. Environ. Microbiol. 1992, 58, 1592–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, M.; Kapusta, K.; Kołodziejczyk, W.; Saloni, J.; Żbikowska, B.; Hill, G.A.; Sroka, Z. Antioxidant Activity of Selected Phenolic Acids–Ferric Reducing Antioxidant Power Assay and QSAR Analysis of the Structural Features. Molecules 2020, 25, 3088. [Google Scholar] [CrossRef]
- Chen, J.; Yang, J.; Ma, L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci. Rep. 2020, 10, 2611. [Google Scholar] [CrossRef] [PubMed]
- Santos-Sánchez, F.; Villanueva-Cañongo, C.; Hernández-Carlos, B. Antioxidant compounds and their antioxidant mechanism in antioxidants. In Antioxidants; Shalaby, E., Ed.; IntechOpen: London, UK, 2019; pp. 1–28. [Google Scholar]
- Litwinienko, G.; Ingold, K.U. Abnormal solvent effects on hydrogen atom abstraction. 2. Resolution of the curcumin antioxidant controversy. The role of sequential proton loss electron transfer. J. Org. Chem. 2004, 69, 5888–5896. [Google Scholar] [CrossRef] [PubMed]
- Krȩżel, A.; Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, C.; Sammartano, S.; Mineo, P.; Rigano, C. Computer tools for the speciation of natural fluids. In Marine Chemistry—An Environmental Analytical Chemistry Approach; Gianguzza, A., Pelizzetti, E., Sammartano, S., Eds.; Kluwer Academic Publishers: Amsterdam, The Netherlands, 1997; pp. 71–83. [Google Scholar]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Kruszewski, M.; Kusaczuk, M.; Kotyńska, J.; Gál, M.; Krętowski, R.; Cechowska-Pasko, M.; Naumowicz, M. The effect of quercetin on the electrical properties of model lipid membranes and human glioblastoma cells. Bioelectrochemistry 2018, 124, 133–141. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Diplock, A.T.; Symons, M.C.R.; Rice-Evans, C.A. Techniques in Free Radical Research; Elsevier Science: Amsterdam, The Netherlands, 1991; Volume 22. [Google Scholar]
- Apak, R.; Güçlü, K.; Özyürek, M.; Karademir, S.E. Novel Total Antioxidant Capacity Index for Dietary Polyphenols and Vitamins C and E, Using Their Cupric Ion Reducing Capability in the Presence of Neocuproine: CUPRAC Method. J. Agric. Food Chem. 2004, 52, 7970–7981. [Google Scholar] [CrossRef]
- Kikuzaki, H.; Nakatani, N. Antioxidant Effects of Some Ginger Constituents. J. Food Sci. 1993, 58, 1407–1410. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CFA | Eu(III)-Caffeinate | Assignment | Varsanyi’s Designation [41] | |||
|---|---|---|---|---|---|---|
| IRKBr | IRATR | Raman | IRKBr | IRATR | ||
| 3431 s a | 3399 m | 3421 vs | 3396 m | ν(OH)ar b | ||
| 3235 m | 3218 m | - | - | ν(OH)ar | ||
| 3000 w | 3056 m | 3032 vw | 2963 w | ν(CH), ν(CH)C=C | 20b | |
| 2839–2569 w | ν(OH) | |||||
| 1647 vs | 1634 s | 1641 m | 1630 s | 1629 sh | ν(C=O), ν(CH)C=C | |
| 1611 vs | 1602 vs | 1613 vs | - | ν(CC) | 8a | |
| 1526 w | 1526 m | 1532 vw | ν(CC) | 8b | ||
| 1501 m | 1499 vs | νas(COO) | ||||
| 1505 vs | νas(COO) | |||||
| 1450 vs | 1442 vs | 1453 vw | ν(CC) | 19b | ||
| 1409 s | 1409 s | νs(COO) | ||||
| 1364 w | 1362 w | 1353 w | ν(CC), β(OH) | 14 | ||
| 1299 s | β(OH) | |||||
| 1283 vs | 1271 s | 1271 s | 1271 vs | ν(C-OH) | ||
| 1215 s | 1209 s | β(OH), β(CH)C=C | ||||
| 1174 m | - | 1186 w | 1169 w | 1170 m | β(CH) | 18a |
| 1115 m | 1110 s | 1108 vw | 1119 m | 1120 m | β(CH) | 18b |
| 982 w | 980 w | βs(COO) | ||||
| 972 w | 965 m | 974 vw | 961 w | 977 w | γ(CH)C=C, γ(CH) | 17b |
| 936 w | 939 w | 932 w | γ(CH) | 17a | ||
| 901 w | 892 s | ν(CCO) | ||||
| 856 w | 853 m | 852 vw | 861 w | 853 vw | γ(CH) | 5 |
| 812 w | β(C=O) | |||||
| 816 w | 823 w | α(CCC) | 1 | |||
| 801 w | 806 s | 803 vw | 793 m | γ(CH) | 10a | |
| 787 w | 788 m | 779 vw | 756 sh | α(CCC) | 12 | |
| 701 w | 695 m | γs(COO) | ||||
| 699 w | 695 w | 686 vw | γ(C=O) | |||
| 645 w | 642 m | 664 vw | φ(CC) | 16a | ||
| 603 w | 603 vw | 603 w | 595 s | α(CCC) | 6a | |
| 575 m | 534 vw | 535 w | γ(OH) | |||
| 459 w | 459 vw | 547 vw | 547 s | α(CCC) | 6b | |
| 409 w | 446 vw | 407 vw | φ(CC) | 16b | ||
| IRKBr | IRATR | IRKBr | IRATR | Assignment | Varsanyi’s Designation [41] |
|---|---|---|---|---|---|
| Cinnamic acid | Eu(III)-cinnamate | ||||
| 1684 vs | 1667 vs | - | - | ν(C=O) | |
| - | - | 1536 s | 1520 s | νas(COO−) | |
| 1494 w | 1492 m | - | 1507 s | β (CH)aro. | 19 a |
| 1448 m | 1449 m | 1448 s | 1446 m | ν(CC)aro+ β(CH)aro. | 19 b |
| - | - | 1412 vs | 1399 s | νs(COO−) | |
| 1073 w | 1071 m | 1069 w | 1071 vw | ν(CC)aro+ β(CH)aro. | 18b |
| 1025 w | 1030 w | 1029 w | 1026 vw | ν(CC)aro+ β(CH)aro. | 18a |
| p-coumaric acid | Eu(III)-p-coumarate | ||||
| 3422 vs | 3330 w | 3423 vs | - | ν(OH)aro. | |
| 2924 w- 2580 w | 2944 w-2379 m | 2924 w- 2613 vw | 2804 m | ν(OH)aro. | |
| 1671 s | 1664 vs | - | - | ν(C=O) | |
| - | - | 1559 m | 1559 m | νas(COO−) | |
| 1511 m | 1503 m | 1512 s | 1502 vs | ν(CC)aro | 19a |
| 1448 m | 1440 s | 1439 m | 1442 m | ν(CC)aro | 19b |
| - | - | 1409 s | - | νs(COO−) | |
| 1106 w | 1105 m | 1104 w | 1107 m | β(CH) | 18b |
| 1063 w | - | - | - | β(CH) | 18a |
| Species | (p:q:r) | log βpqr a (log K01r) b |
|---|---|---|
| LH | 0:1:1 | 11.93 ± 0.08 |
| LH2 | 0:1:2 | 20.59 ± 0.08 (8.66) (20.47 ± 0.06) c (20.9 ± 0.3) d |
| LH3 | 0:1:3 | 24.92 ± 0.01 (4.33) (24.97 ± 0.03) c (25.3 ± 0.3) d |
| Nucleus | δ/ppm | |||
|---|---|---|---|---|
| L | LH | LH2 | LH3 | |
| Hb | 7.2419 | 7.2429 | 7.2612 | 7.6202 |
| Hc | 6.9927 | 7.035 | 7.1463 | 7.1989 |
| He | 6.8656 | 6.918 | 7.0582 | 7.1294 |
| Hd | 6.6373 | 6.701 | 6.9168 | 6.9433 |
| Ha | 6.1865 | 6.2177 | 6.3258 | 6.3534 |
| Species | (p:q:r) | log βpqr a |
|---|---|---|
| EuL | 1:1:0 | 10.52 ± 0.02 (10.57 ± 0.03) b |
| [EuL(OH)]− | 1:1:−1 | -- (0.03 ± 0.05) b |
| Microorganism | MIC | |||
|---|---|---|---|---|
| mg·cm−3 (mmol·dm−3) | ||||
| CFA | Eu-Caffeinate | EuCl3 | Positive Control | |
| Escherichia coli Gram(−) bacteria | 13 (72.2) | 4 (5.1) | 10 (27.3) | Gentamicin 0.0005 (0.001) |
| Bacillus subtilis Gram(+) bacteria | 18 (99.9) | 4 (5.1) | 12 (32.8) | Gentamicin 0.005 (0.01) |
| Candida albicans (fungi) | 29 (161.0) | >4 (>5.1) | 6 (16.4) | Fluconazole 0.00025 (0.00082) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arciszewska, Ż.; Gama, S.; Kalinowska, M.; Świderski, G.; Świsłocka, R.; Gołębiewska, E.; Naumowicz, M.; Worobiczuk, M.; Cudowski, A.; Pietryczuk, A.; et al. Caffeic Acid/Eu(III) Complexes: Solution Equilibrium Studies, Structure Characterization and Biological Activity. Int. J. Mol. Sci. 2022, 23, 888. https://doi.org/10.3390/ijms23020888
Arciszewska Ż, Gama S, Kalinowska M, Świderski G, Świsłocka R, Gołębiewska E, Naumowicz M, Worobiczuk M, Cudowski A, Pietryczuk A, et al. Caffeic Acid/Eu(III) Complexes: Solution Equilibrium Studies, Structure Characterization and Biological Activity. International Journal of Molecular Sciences. 2022; 23(2):888. https://doi.org/10.3390/ijms23020888
Chicago/Turabian StyleArciszewska, Żaneta, Sofia Gama, Monika Kalinowska, Grzegorz Świderski, Renata Świsłocka, Ewelina Gołębiewska, Monika Naumowicz, Mateusz Worobiczuk, Adam Cudowski, Anna Pietryczuk, and et al. 2022. "Caffeic Acid/Eu(III) Complexes: Solution Equilibrium Studies, Structure Characterization and Biological Activity" International Journal of Molecular Sciences 23, no. 2: 888. https://doi.org/10.3390/ijms23020888