
Inflammatory Burden and Immunomodulative Therapeutics of Cardiovascular Diseases


Abstract
:1. Background
2. Inflammation and Cardiovascular Diseases
2.1. Atherosclerosis
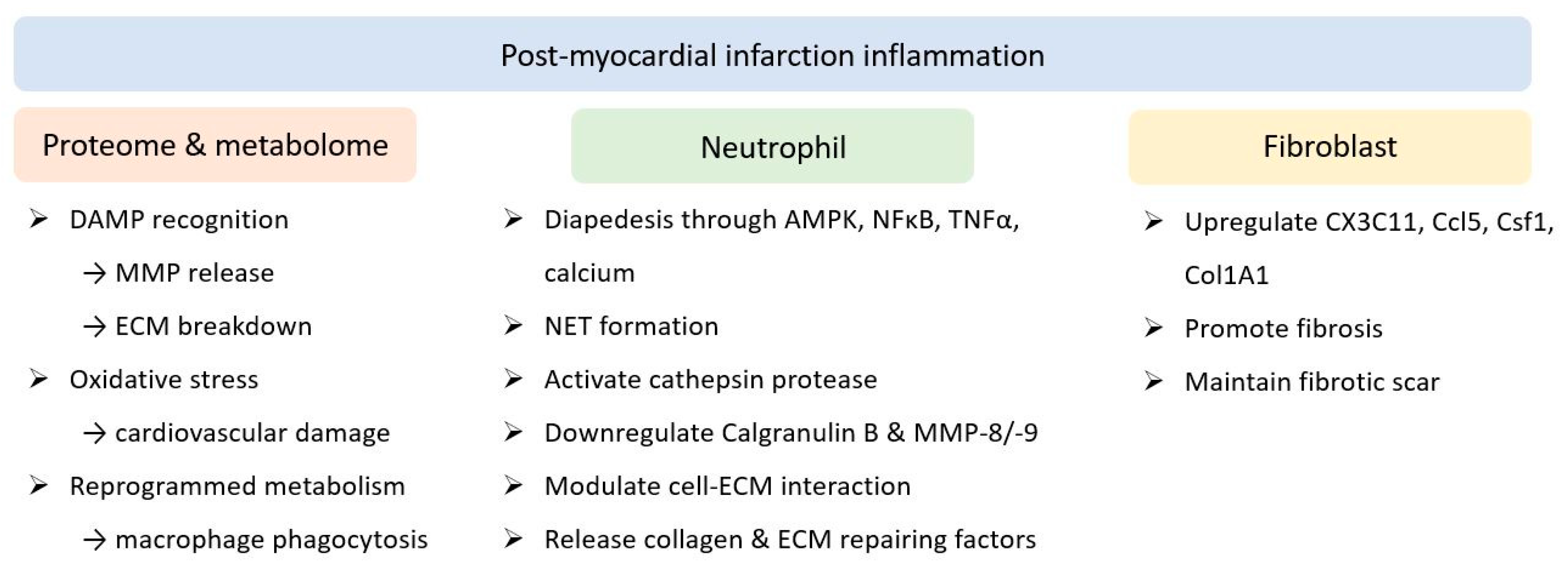
2.2. Myocardial Infarction
3. Molecular Mechanism Underlying Inflammation
4. Pharmaceutical Advancements
4.1. Colchicine
4.2. Interleukin-1 Antagonist
4.3. Interleukin-6 Antagonist
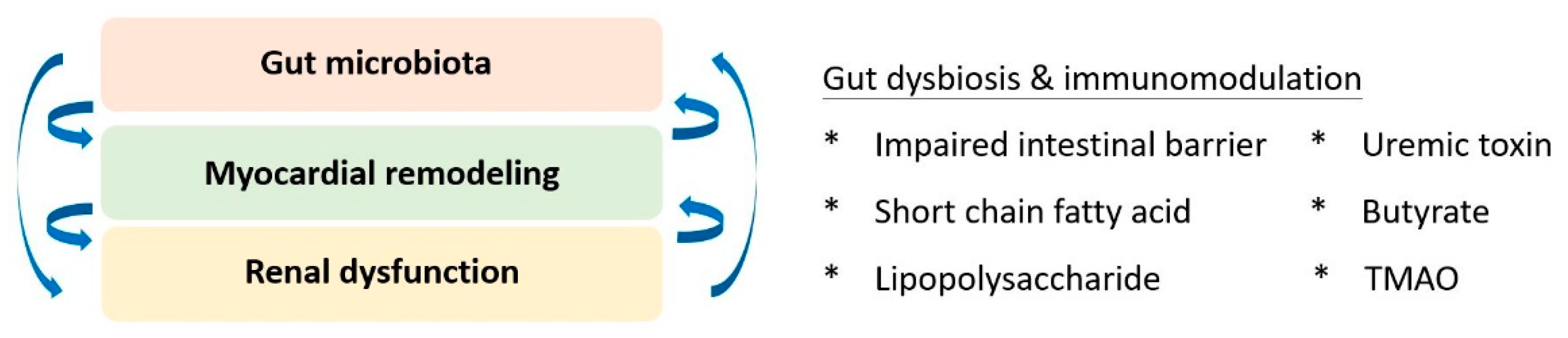
5. The Role of Gut Microbiota
6. Gut-Cardio-Renal Triplet
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friuli, M.; Eramo, B.; Valenza, M.; Scuderi, C.; Provensi, G.; Romano, A. Targeting the Oxytocinergic System: A Possible Pharmacological Strategy for the Treatment of Inflammation Occurring in Different Chronic Diseases. Int. J. Mol. Sci. 2021, 22, 10250. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.-L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; De Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Melnichenko, A.A.; Grechko, A.V.; Myasoedova, V.A.; Orekhov, A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp. Mol. Pathol. 2018, 104, 114–124. [Google Scholar] [CrossRef]
- Zeng, X.; Guo, R.; Dong, M.; Zheng, J.; Lin, H.; Lu, H. Contribution of TLR4 signaling in intermittent hypoxia-mediated atherosclerosis progression. J. Transl. Med. 2018, 16, 106. [Google Scholar] [CrossRef] [Green Version]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daseke, M.J., 2nd; Valerio, F.M.; Kalusche, W.J.; Ma, Y.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res. Cardiol. 2019, 114, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.Y.; Chan, C.K.; Braun, K.R.; Green, P.S.; O’Brien, K.D.; Chait, A.; Day, A.J.; Wight, T.N. Mono-cyte-to-macrophage differentiation: Synthesis and secretion of a complex extracellular matrix. J. Biol. Chem. 2012, 287, 14122–14135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef]
- Meijers, W.C.; van der Velde, A.R.; Pascual-Figal, D.A.; de Boer, R.A. Galectin-3 and post-myocardial infarction cardiac remodeling. Eur. J. Pharmacol. 2015, 763, 115–121. [Google Scholar] [CrossRef]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J., 2nd; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef] [Green Version]
- Daseke, M.J., 2nd; Tenkorang-Impraim, M.A.A.; Ma, Y.; Chalise, U.; Konfrst, S.R.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. J. Mol. Cell. Cardiol. 2020, 145, 112–121. [Google Scholar] [CrossRef]
- Alvarez-Argote, S.; O’Meara, C.C. The evolving roles of cardiac macrophages in homeostasis, regeneration, and repair. Int. J. Mol. Sci. 2021, 22, 7923. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.P.; Patterson, N.L.; Zouein, F.A.; Ma, Y.; Dive, V.; de Castro Brás, L.E.; Lindsey, M.L. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int. J. Cardiol. 2015, 185, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat. Rev. Cardiol. 2018, 15, 471–479. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Mann, D.L.; Topkara, V.K.; Evans, S.; Barger, P.M. Innate Immunity in the Adult Mammalian Heart: For Whom the Cell Tolls. Trans. Am. Clin. Clim. Assoc. 2010, 121, 34–50. [Google Scholar]
- Ridker, P.M. From C-reactive protein to interleukin-6 to interleukin-1: Moving upstream to identify novel targets for atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M. Anticytokine agents: Targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ. Res. 2019, 124, 437–450. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yuan, W.; Hu, A.; Lin, J.; Xia, Z.; Yang, C.F.; Li, Y.; Zhang, Z. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 2020, 22, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.M.; Moye, L.A.; Goldman, S.; Flaker, G.C.; Braunwald, E. Inflammation, Pravastatin, and the Risk of Coronary Events After Myocardial Infarction in Patients with Average Cholesterol Levels. Circulation 1998, 98, 839–844. [Google Scholar] [CrossRef]
- De Lemos, J.A.; Blazing, M.A.; Wiviott, S.D.; Lewis, E.F.; Fox, K.A.; White, H.D.; Rouleau, J.L.; Pedersen, T.R.; Gardner, L.H.; Mukherjee, R.; et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: Phase Z of the A to Z trial. JAMA 2004, 292, 1307–1316. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Brown, B.G.; Ganz, P.; Vogel, R.A.; Crowe, T.; Howard, G.; Cooper, C.J.; Brodie, B.; et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: A randomized controlled trial. JAMA 2004, 291, 1071–1080. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–21207. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E.; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Giugliano, R.P.; Cannon, C.P.; Zhou, J.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Blazing, M.A.; Braunwald, E. Achievement of Dual Low-Density Lipoprotein Cholesterol and High-Sensitivity C-Reactive Protein Targets More Frequent with the Addition of Ezetimibe to Simvastatin and Associated with Better Outcomes in IMPROVE-IT. Circulation 2015, 132, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhou, S.; Dreyer, R.P.; Spatz, E.S.; Geda, M.; Lorenze, N.P.; D’Onofrio, G.; Lichtman, J.H.; Spertus, J.A.; Ridker, P.M.; et al. Sex differences in inflammatory markers and health status among young adults with acute myocardial infarction: Results from the VIRGO (Variation in Recovery: Role of Gender on Outcomes of Young Acute Myocardial Infarction Patients) study. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003470. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, D.N.; Aquino, M.; Claessen, B.E.; Baber, U.; Guedeney, P.; Sorrentino, S.; Vogel, B.; De Winter, R.J.; Sweeny, J.; Kovacic, J.C.; et al. Residual inflammatory risk and the impact on clinical outcomes in patients after percutaneous coronary interventions. Eur. Heart J. 2018, 39, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Alexopoulos, N.; Filippatos, G.; Papoutsidakis, N.; Sianos, G.; Goudevenos, J.; Alexopoulos, D.; Pyrgakis, V.; et al. Anti-inflammatory treatment with colchicine in acute myocardial infarction: A pilot study. Circulation 2015, 132, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, T.; Soh, L.; Bowman, M.; Kurup, R.; Schultz, C.; Patel, S.; Hillis, G.S. The low dose colchicine after myocardial infarction (LoDoCo-MI) study: A pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am. Heart J. 2019, 215, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Akodad, M.; Lattuca, B.; Nagot, N.; Georgescu, V.; Buisson, M.; Cristol, J.-P.; Leclercq, F.; Macia, J.-C.; Gervasoni, R.; Cung, T.-T.; et al. COLIN trial: Value of colchicine in the treatment of patients with acute myocardial infarction and inflammatory response. Arch. Cardiovasc. Dis. 2017, 110, 395–402. [Google Scholar] [CrossRef]
- Shah, B.; Pillinger, M.; Zhong, H.; Cronstein, B.; Xia, Y.; Lorin, J.D.; Smilowitz, N.R.; Feit, F.; Ratnapala, N.; Keller, N.M.; et al. Effects of Acute Colchicine Administration Prior to Percutaneous Coronary Intervention: COLCHICINE-PCI Randomized Trial. Circ. Cardiovasc. Interv. 2020, 13, e008717. [Google Scholar] [CrossRef]
- Colchicine and Spironolactone in Patients with MI/SYNERGY Stent Registry (CLEAR SYNERGY). Available online: www.ClinicalTrials.gov (accessed on 6 January 2022).
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 Blockade with Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Morton, A.C.; Rothman, A.M.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.X.; Ur-Rahman, M.A.; Sage, A.P.; Victor, S.; Kurian, R.; Fielding, S.; Ait-Oufella, H.; Chiu, Y.D.; Binder, C.J.; Mckie, M.; et al. Rituximab in patients with acute ST-elevation myocardial infarction (RITA-MI): An experimental medicine safety study. Cardiovasc. Res. 2021, cvab113. [Google Scholar] [CrossRef]
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium; Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A. The Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Georgakis, M.K.; Malik, R.; Gill, D.; Franceschini, N.; Sudlow, C.L.M.; Dichgans, M.; INVENT Consortium, CHARGE Inflammation Working Group. Interleukin-6 signaling effects on ischemic stroke and other cardiovascular outcomes: A mendelian randomization study. Circ. Genom. Precis Med. 2020, 13, e002872. [Google Scholar] [CrossRef]
- Rosa, M.; Chignon, A.; Li, Z.; Boulanger, M.C.; Arsenault, B.J.; Bossé, Y.; Thériault, S.; Mathieu, P. A Mendelian randomization study of IL6 signaling in cardiovascular diseases, immune-related disorders and longevity. NPJ Genom. Med. 2019, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Akita, K.; Isoda, K.; Sato-Okabayashi, Y.; Kadoguchi, T.; Kitamura, K.; Ohtomo, F.; Shimada, K.; Daida, H. An Interleukin-6 Receptor Antibody Suppresses Atherosclerosis in Atherogenic Mice. Front. Cardiovasc. Med. 2017, 4, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and inter-leukin-6 after successful interleukin-1β inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- A Research Study to Look at How Ziltivekimab Works Compared to Placebo in People with Cardiovascular Disease, Chronic Kidney Disease and Inflammation (ZEUS). Available online: www.ClinicalTrials.gov (accessed on 6 January 2022).
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.S.; Obeid, S.; Wang, Z.; Hazen, B.J.; Li, L.; Wu, Y.; Hurd, A.G.; Gu, X.; Pratt, A.; Levison, B.S.; et al. Trimethyllysine, a trimethylamine N-oxide precursor, provides near- and long-term prognostic value in patients presenting with acute coronary syndromes. Eur. Heart J. 2019, 40, 2700–2709. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Kao, T.-W.; Huang, C.-C. Recent Progress in Metabolic Syndrome Research and Therapeutics. Int. J. Mol. Sci. 2021, 22, 6862. [Google Scholar] [CrossRef]
- Luedde, M.; Winkler, T.; Heinsen, F.-A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F. Pathogenic gut flora in patients with chronic heart failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lip, G.Y.; Apostolakis, S. Inflammation in Atrial Fibrillation. J. Am. Coll. Cardiol. 2012, 60, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Svingen, G.F.T.; Zuo, H.; Ueland, P.M.; Seifert, R.; Løland, K.H.; Pedersen, E.R.; Schuster, P.M.; Karlsson, T.; Tell, G.S.; Schartum-Hansen, H.; et al. Increased plasma trimethylamine-N-oxide is associated with incident atrial fibrillation. Int. J. Cardiol. 2018, 267, 100–106. [Google Scholar] [CrossRef]
- Jacob, K.A.; Nathoe, H.M.; Dieleman, J.M.; van Osch, D.; Kluin, J.; Van Dijk, D. Inflammation in new-onset atrial fibrillation after cardiac surgery: A systematic review. Eur. J. Clin. Investig. 2014, 44, 402–428. [Google Scholar] [CrossRef] [Green Version]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-Derived Serum Lipopolysaccharide is Associated with Enhanced Risk of Major Adverse Cardiovascular Events in Atrial Fibrillation: Effect of Adherence to Mediterranean Diet. J. Am. Heart Assoc. 2017, 6, e005784. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Yamashita, T.; Hosomi, K.; Park, J.; Hayashi, T.; Yoshida, N.; Saito, Y.; Fukuzawa, K.; Konishi, K.; Murakami, H.; et al. Gut microbial composition in patients with atrial fibrillation: Effects of diet and drugs. Heart Vessel. 2021, 36, 105–114. [Google Scholar] [CrossRef]
- Ghosh, S.; Lertwattanarak, R.; Garduño, J.D.J.; Galeana, J.J.; Li, J.; Zamarripa, F.; Lancaster, J.L.; Mohan, S.; Hussey, S.; Musi, N. Elevated Muscle TLR4 Expression and Metabolic Endotoxemia in Human Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Li, B.; Luo, Y.; Gong, Y.; Jin, X.; Zhang, J.; Zhou, Y.; Zhuo, X.; Wang, Z.; et al. Gut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc. Res. 2021, cvab114. [Google Scholar] [CrossRef]
- Chen, P.B.; Black, A.S.; Sobel, A.L.; Zhao, Y.; Mukherjee, P.; Molparia, B.; Moore, N.E.; Aleman Muench, G.R.; Wu, J.; Chen, W.; et al. Directed remodeling of the mouse gut microbiome inhibits the development of atherosclerosis. Nat. Biotechnol. 2020, 38, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The effects of vegetarian and vegan diets on gut microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Franco-de-Moraes, A.C.; de Almeida-Pititto, B.; da Rocha Fernandes, G.; Gomes, E.P.; da Costa Pereira, A.; Ferreira, S.R.G. Worse inflammatory profile in omnivores than in vegetarians associates with the gut microbiota composition. Diabetol. Metab. Syndr. 2017, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.-Q.; Kong, X.-S.; Shen, X.-B.; Huang, M.-Z.; Zheng, J.-P.; Sun, J.; Xu, S.-H. Identification of Differentially Expressed Genes and Signaling Pathways in Acute Myocardial Infarction Based on Integrated Bioinformatics Analysis. Cardiovasc. Ther. 2019, 2019, 8490707. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Huang, Z.; Liu, X.; Zhang, J.; Ye, P.; Wu, C.; Lu, S.; Jia, S.; Zhang, X.; Chen, X.; et al. Biodata Mining of Differentially Expressed Genes between Acute Myocardial Infarction and Unstable Angina Based on Integrated Bioinformatics. BioMed Res. Int. 2021, 2021, 5584681. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.S.; Hu, X.F.; Chen, T.; Shen, G.L.; Cheng, D. Integrated network analysis to explore the key mRNAs and lncRNAs in acute myocardial infarction. Math. Biosci. Eng. 2019, 16, 6426–6437. [Google Scholar] [CrossRef]
- Wang, S.; Wang, E.; Chen, Q.; Yang, Y.; Xu, L.; Zhang, X.; Wu, R.; Hu, X.; Wu, Z. Uncovering Potential lncRNAs and mRNAs in the Progression From Acute Myocardial Infarction to Myocardial Fibrosis to Heart Failure. Front. Cardiovasc. Med. 2021, 8, 664044. [Google Scholar] [CrossRef]
- McCoubrey, L.E.; Elbadawi, M.; Orlu, M.; Gaisford, S.; Basit, A.W. Harnessing machine learning for development of microbiome therapeutics. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lehto, M.; Groop, P.-H. The Gut-Kidney Axis: Putative Interconnections between Gastrointestinal and Renal Disorders. Front. Endocrinol. 2018, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, L.; Guo, H.; Xu, Y.; Xu, Y. The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metabolism 2017, 68, 20–30. [Google Scholar] [CrossRef]
- Lekawanvijit, S. Role of gut-derived protein-bound uremic toxins in cardiorenal syndrome and potential treatment modalities. Circ. J. 2015, 79, 2088–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanto-Hara, F.; Kanemitsu, Y.; Fukuda, S.; Kikuchi, K.; Asaji, K.; Saigusa, D.; Iwasaki, T.; Ho, H.-J.; Mishima, E.; Suzuki, T.; et al. The guanylate cyclase C agonist linaclotide ameliorates the gut–cardio–renal axis in an adenine-induced mouse model of chronic kidney disease. Nephrol. Dial. Transpl. 2020, 35, 250–264. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Trial/Author, Year | Cohort | Dosage, Timing | Outcomes | Ref |
|---|---|---|---|---|
| Colchicine | ||||
| COLCOT, 2019 | 4745 subjects with recent MI | 0.5 mg QD | Colchicine reduced further ischaemic events at 22.6 months. | [39] |
| LoDoCo2, 2020 | 5522 subjects with chronic CAD | 0.5 mg QD | Colchicine remarkably attenuated MACEs at 28.6 months. | [40] |
| Deftereos, 2015 | 151 subjects with STEMI | 2 mg loading after PCI, then 0.5 mg BID for 5 days | Colchicine decreased accumulative level of CK-MB and infarction size after primary PCI against STEMI. | [42] |
| LoDoCo-MI, 2019 | 237 patients admitted for acute MI | 0.5 mg QD | Colchicine reduced neither the decrease nor the absolute level of CRP at 30 days. | [43] |
| COLIN, 2017 | 44 patients underwent PCI against STEMI | 1 mg QD following acute MI, lasting 1 month | Colchicine failed to reduce the peak level of CRP. | [44] |
| COLCHICINE-PCI, 2020 | 714 patients referred for possible PCI | 1.8 mg acute pre-procedural | Colchicine reduced the serum level of IL-6 and CRP but failed to decrease PCI-related MI. | [45] |
| CLEAR SYNERGY, ongoing | Patients referred for PCI against STEMI | 0.5 mg BID | To validate the efficacy of colchicine against MACEs. | [46] |
| Interleukin-1 antagonist | ||||
| CANTOS, 2017 | 10,061 patients with prior MI and CRP ≧ 2 mg/L | Canakinumab (50, 150, 300 mg) per 3 months subcutaneously | Canakinumab effectively reduced the recurrent cardiovascular events. | [47] |
| CIRT, 2019 | 4786 patients with prior CAD and DM or MetS | Methotrexate 15–20 mg per week | Methotrexate did not improve composite cardiovascular outcomes. | [48] |
| Abbate, 2013 | 30 patients with STEMI | Anakinra 100 mg loading acutely after PCI, then maintained for 14 days | Anakinra reduced CRP level, mortality rate, and new-onset heart failure. | [49] |
| MRC-ILA Heart Study, 2015 | 182 patients with NSTEMI | Anakinra 100 mg within 2 days of symptom onset, lasting 14 days | Anakinra reduced CRP and IL-6 levels, both of which rebounded after drug discontinuation. | [50] |
| Interleukin-6 antagonist | ||||
| Kleveland, 2016 | 117 patients with NSTEMI | Tocilizumab 280 mg, single dose | Tocilizumab curtailed inflammation and PCI-related troponin rise. | [57] |
| RESCUE, 2021 | 264 patients with CKD and elevated CRP | Ziltivekimab 7.5, 15, or 30 mg per 4 weeks, up to 24 weeks | Ziltivekimab attenuated the expression of inflammatory markers and thromboembolism. | [58] |
| ZEUS, ongoing | Patients with atherosclerosis, renal insufficiency, and elevated CRP | Ziltivekimab 15 mg for up to 4 years | Aimed to demonstrate that ziltivekimab would reduce cardiovascular events. | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, T.-W.; Huang, C.-C. Inflammatory Burden and Immunomodulative Therapeutics of Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 804. https://doi.org/10.3390/ijms23020804
Kao T-W, Huang C-C. Inflammatory Burden and Immunomodulative Therapeutics of Cardiovascular Diseases. International Journal of Molecular Sciences. 2022; 23(2):804. https://doi.org/10.3390/ijms23020804
Chicago/Turabian StyleKao, Ting-Wei, and Chin-Chou Huang. 2022. "Inflammatory Burden and Immunomodulative Therapeutics of Cardiovascular Diseases" International Journal of Molecular Sciences 23, no. 2: 804. https://doi.org/10.3390/ijms23020804