
Neuroinflammation: A Potential Risk for Dementia

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Diagnosis of Cognitive Decline
2.1. Cognitive Decline
2.2. Aetiology of Neurodegenerative Disorders
2.3. Cognitive Downturn
2.4. Chronic Decrease in Functionality
2.5. Mild Cognitive Impairment
3. Types of Dementias
3.1. Primary Dementias
3.2. Rapidly Progressive Dementia
4. Risk Factors for Dementia
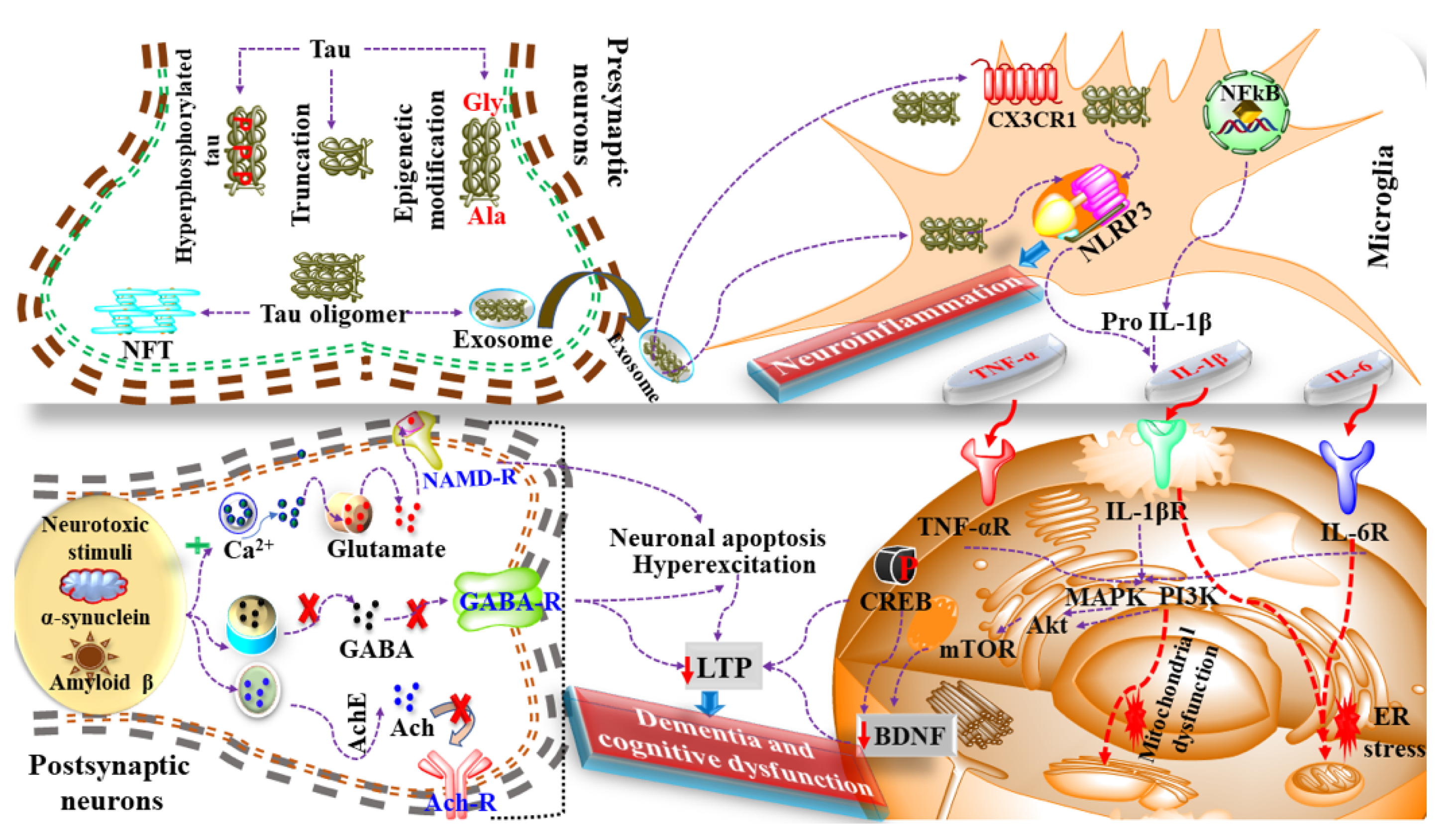
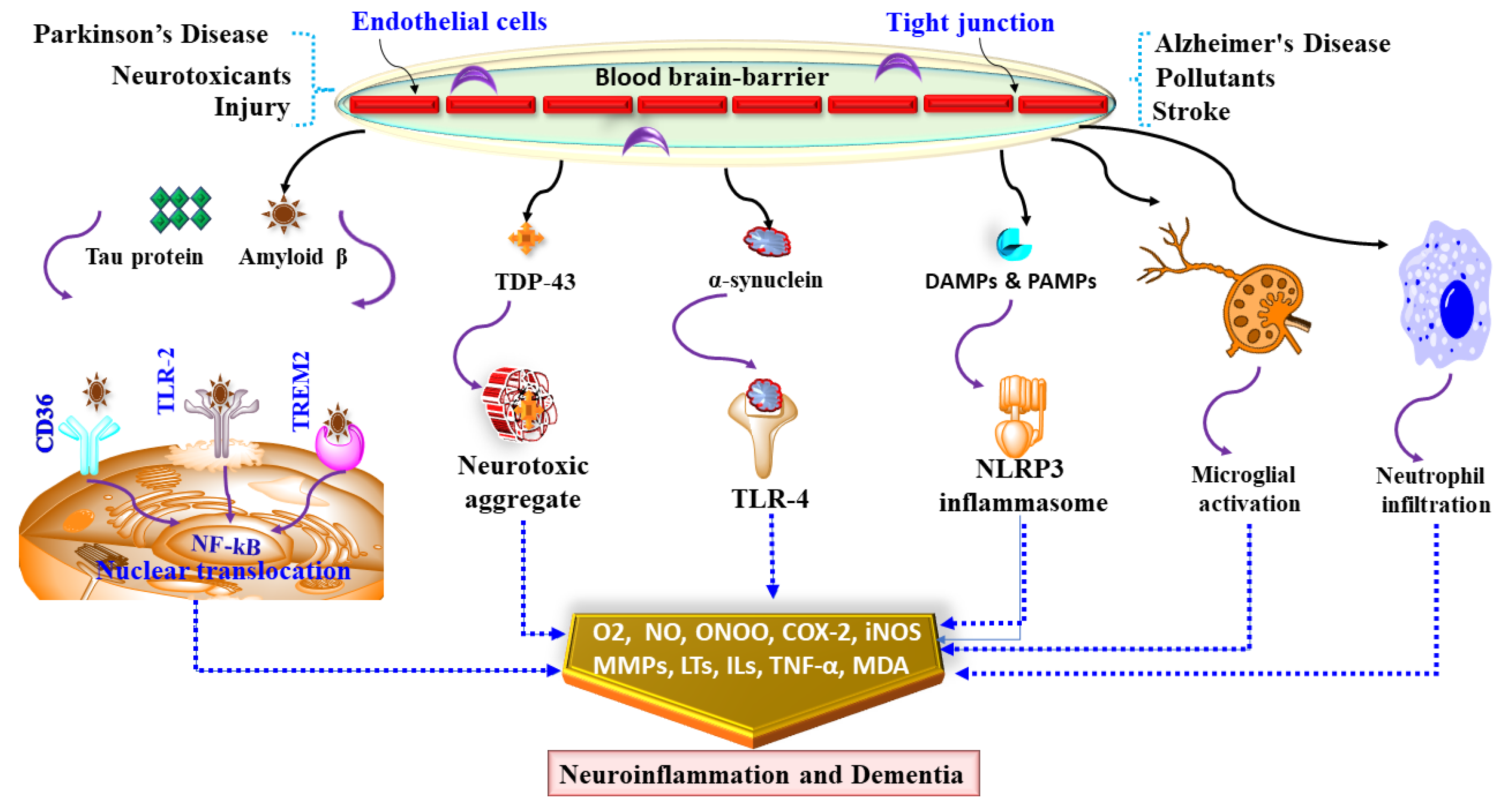
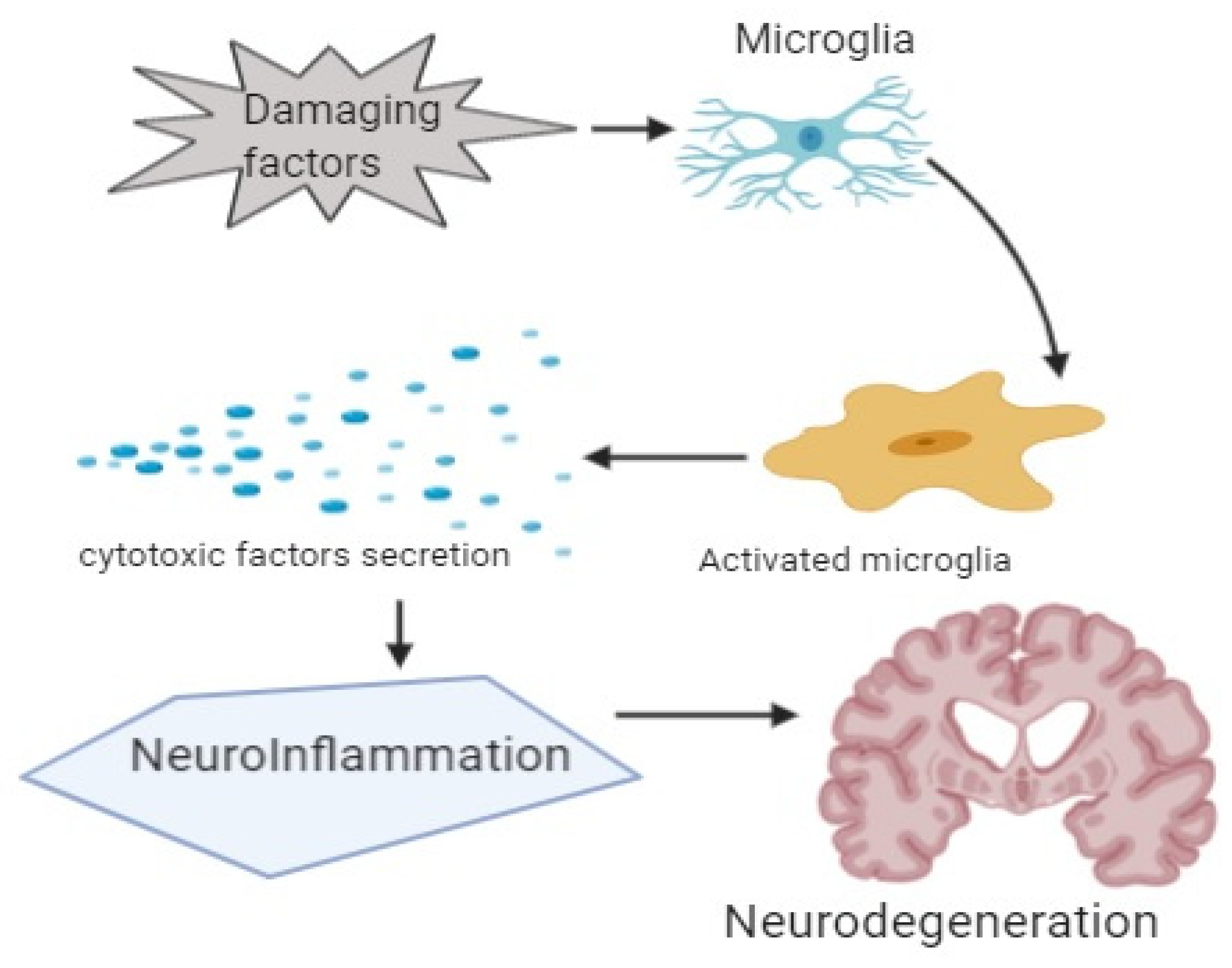
5. Neuroinflammation in Dementia
6. Mediators and Neuroinflammatory Modulators
7. Future Directions
8. Need for Guidelines
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- United Nations. World Population Ageing 2019; Economic and Social Affairs, Population Division: New York, NY, USA, 2019; pp. 1–111. [Google Scholar]
- Rahman, M.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh Thi, T.; Akter, R.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Redox Effects of Molecular Hydrogen and Its Therapeutic Efficacy in the Treatment of Neurodegenerative Diseases. Processes 2021, 9, 308. [Google Scholar] [CrossRef]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C. Living too long: The current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Rep. 2015, 16, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Australia, D.; Baker, S.; Banerjee, S. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Salardini, A. (Ed.) An Overview of Primary Dementias as Clinicopathological Entities. Seminars in Neurology; Thieme Medical Publishers: New York, NY, USA, 2019. [Google Scholar]
- Kumar, B.; Pandey, M.; Mishra, A.K.; Sharma, A.; Fayaz, F.; Pottoo, F.H. Autophagic Dysfunction in Dementia: Scope for the Development of Potential Remedies. CNS Neurol. Disord.-Drug Targets (Formerly Curr. Drug Targets-CNS Neurol. Disord.) 2021, 20, 704–722. [Google Scholar] [CrossRef]
- Sartori, A.C.; Vance, D.E.; Slater, L.Z.; Crowe, M. The impact of inflammation on cognitive function in older adults: Implications for health care practice and research. J. Neurosci. Nurs. 2012, 44, 206. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Al Mamun, A.; Iqbal, M.A.; Islam, A.; Hossain, M.F.; Khanum, S.; Rashid, M. Analyzing nootropic effect of Phyllanthus reticulatus Poir. on cognitive functions, brain antioxidant enzymes and acetylcholinesterase activity against aluminium-induced Alzheimer’s model in rats: Applicable for controlling the risk factors of Alzheimer’s disease. Adv. Alzheimer’s Dis. 2016, 5, 87. [Google Scholar]
- Geschwind, M.D.; Shu, H.; Haman, A.; Sejvar, J.J.; Miller, B.L. Rapidly progressive dementia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, G.; Giovannetti, T.; Price, C.; Tanner, J.; Mitchell, S.; Eppig, J.; Libon, D. Neuroimaging correlates of everyday action in dementia. J. Clin. Exp. Neuropsychol. 2013, 35, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Chiba, Y. A case of treatable dementia with Lewy bodies remarkably improved by immunotherapy. J. Neuroimmunol. 2019, 330, 35–37. [Google Scholar] [CrossRef]
- Garre-Olmo, J.; Ponjoan, A.; Inoriza, J.M.; Blanch, J.; Sánchez-Pérez, I.; Cubí, R.; de Eugenio, R.; Turró-Garriga, O.; Vilalta-Franch, J. Survival, effect measures, and impact numbers after dementia diagnosis: A matched cohort study. Clin. Epidemiol. 2019, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L., Jr.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T., Jr.; et al. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension 2003, 42, 1206–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, A.; Kunik, M.E.; Schulz, P.; Williams, S.P.; Singh, H. Missed and delayed diagnosis of dementia in primary care: Prevalence and contributing factors. Alzheimer Dis. Assoc. Disord. 2009, 23, 306. [Google Scholar] [CrossRef]
- Das, S.; Akbar, S.; Ahmed, B.; Dewangan, R.P.; Iqubal, A.; Pottoo, F.H.; Joseph, A. Structural Activity Relationship-based Medicinal perspectives of Pyrimidine derivatives as Anti-Alzheimer’s Agent: A Comprehensive Review. CNS Neurol. Disord. Drug Targets 2021. [Google Scholar] [CrossRef]
- Collins, R.; Silarova, B.; Clare, L. Dementia primary prevention policies and strategies and their local implementation: A scoping review using England as a case study. J. Alzheimer’s Dis. 2019, 70 (Suppl. 1), S303–S318. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Morales, I.; Farías, G.A.; Cortes, N.; Maccioni, R.B. Neuroinflammation and Neurodegeneration. In Update on Dementia; IntechOpen: London, UK, 2016; p. 13. [Google Scholar]
- Uddin, M.S.; Mamun, A.A.; Labu, Z.K.; Hidalgo-Lanussa, O.; Barreto, G.E.; Ashraf, G.M. Autophagic dysfunction in Alzheimer’s disease: Cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J. Cell. Physiol. 2019, 234, 8094–8112. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Abdel-Daim, M.M.; Barreto, G.E.; Ashraf, G.M. APOE and Alzheimer’s disease: Evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol. Neurobiol. 2019, 56, 2450–2465. [Google Scholar] [CrossRef]
- Lannfelt, L.; Relkin, N.; Siemers, E. Amyloid-ß-directed immunotherapy for Alzheimer’s disease. J. Intern. Med. 2014, 275, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Mohi-Ud-Din, R.; Mir, R.H.; Wani, T.U.; Shah, A.J.; Banday, N.; Pottoo, F.H. Berberine in the Treatment of Neurodegenerative Diseases and Nanotechnology Enabled Targeted Delivery. Comb. Chem. High Throughput Screen. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mohi-Ud-Din, R.; Mir, R.H.; Wani, T.U.; Shah, A.J.; Mohi-Ud-Din, I.; Dar, M.A.; Pottoo, F.H. Novel Drug Delivery System for Curcumin: Implementation to Improve Therapeutic Efficacy against Neurological Disorders. Comb. Chem. High Throughput Screen. 2021. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqubal, A.; Rahman, S.O.; Ahmed, M.; Bansal, P.; Haider, M.R.; Iqubal, M.K.; Najmi, A.K.; Pottoo, F.H.; Haque, S.E. Current Quest in Natural Bioactive Compounds for Alzheimer’s Disease: Multi-Targeted-Designed-Ligand Based Approach with Preclinical and Clinical Based Evidence. Curr. Drug Targets 2021, 22, 685–720. [Google Scholar] [CrossRef]
- Iqubal, A.; Iqubal, M.K.; Fazal, S.A.; Pottoo, F.H.; Haque, S.E. Nutraceuticals and their Derived Nano-formulations for the Prevention and Treatment of Alzheimer’s disease. Curr. Mol. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Action Plan on the Public Health Response to Dementia 2017–2025; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Pottoo, F.H.; Sharma, S.; Javed, M.N.; Barkat, M.A.; Harshita, A.M.S.; Naim, M.J.; Alam, O.; Ansari, M.A.; Barreto, G.E.; Ashraf, G.M. Lipid-based nanoformulations in the treatment of neurological disorders. Drug Metab. Rev. 2020, 52, 185–204. [Google Scholar] [CrossRef]
- Mir, R.H.; Shah, A.J.; Mohi-Ud-Din, R.; Potoo, F.H.; Dar, M.; Jachak, S.M.; Masoodi, M.H. Natural Anti-inflammatory compounds as Drug candidates in Alzheimer’s disease. Curr. Med. Chem. 2021, 28, 4799–4825. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Sharma, S.; Sharma, K.; Bhavsar, A.; Hussain, I.; Iqubal, M.K.; Kumar, R. Intranasally administered pitavastatin ameliorates pentylenetetrazol-induced neuroinflammation, oxidative stress and cognitive dysfunction. Life Sci. 2018, 211, 172–181. [Google Scholar] [CrossRef]
- Iqubal, A.; Syed, M.A.; Haque, M.M.; Najmi, A.K.; Ali, J.; Haque, S.E. Effect of nerolidol on cyclophosphamide-induced bone marrow and hematologic toxicity in Swiss albino mice. Exp. Hematol. 2020, 82, 24–32. [Google Scholar] [CrossRef]
- Iqubal, A.; Haque, S.E.; Sharma, S.; Ansari, M.A.; Khan, V.; Iqubal, M.K. Clinical updates on drug-induced cardiotoxicity. Int. J. Pharm. Sci. Res. 2018, 9, 16–26. [Google Scholar]
- Iqubal, A.; Iqubal, M.K.; Khan, A.; Ali, J.; Baboota, S.; Haque, S.E. Gene therapy, a novel therapeutic tool for neurological disorders: Current progress, challenges and future prospective. Curr. Gene Ther. 2020, 20, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Proteolytic activation of proapoptotic kinase protein kinase Cδ by tumor necrosis factor α death receptor signaling in dopaminergic neurons during neuroinflammation. J. Neuroinflamm. 2012, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Pan, S.; Crapser, J.; Hammond, M.; Jellison, E.; McCullough, L.D. Age-and location-related changes in microglial function. Neurobiol. Aging 2015, 36, 2153–2163. [Google Scholar] [CrossRef]
- Gorlovoy, P.; Larionov, S.; Pham, T.T.H.; Neumann, H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J. 2009, 23, 2502–2513. [Google Scholar] [CrossRef]
- Rabbitt, P.; Lowe, C. Patterns of cognitive ageing. Psychol. Res. 2000, 63, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Rehman, S.; Nabi, B.; Iqubal, A.; Nehal, N.; Fahmy, U.A.; Kotta, S.; Baboota, S.; Md, S.; Ali, J. Boosting the brain delivery of Atazanavir through nanostructured lipid carrier-based approach for mitigating neuroaids. Pharmaceutics 2020, 12, 1059. [Google Scholar] [CrossRef]
- Tangestani Fard, M.; Stough, C. A review and hypothesized model of the mechanisms that underpin the relationship between inflammation and cognition in the elderly. Front. Aging Neurosci. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, C.N.; Love, M.C.N.; Triebel, K.L. Normal cognitive aging. Clin. Geriatr. Med. 2013, 29, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Series Introduction: Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh, T.T.; Akter, R.; Jeong, Y.J.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Therapeutic Potential of Natural Products in Treating Neurodegenerative Disorders and Their Future Prospects and Challenges. Molecules 2021, 26, 5327. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Akter, R.; Bhattacharya, T.; Abdel-Daim, M.M.; Alkahtani, S.; Arafah, M.W.; Al-Johani, N.S.; Alhoshani, N.M.; Alkeraishan, N.; Alhenaky, A.; et al. Resveratrol and Neuroprotection: Impact and its Therapeutic Potential in Alzheimer’s disease. Front. Pharmacol. 2020, 11, 2272. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef]
- Dreher, M.L. Dietary Patterns, Foods and Beverages in Age-Related Cognitive Performance and Dementia. In Dietary Patterns and Whole Plant Foods in Aging and Disease; Springer: Cham, Switzerland, 2018; pp. 471–518. [Google Scholar]
- Wesnes, K.A.; Harrison, J.E. The evaluation of cognitive function in the dementias: Methodological and regulatory considerations. Dialogues Clin. Neurosci. 2003, 5, 77. [Google Scholar] [PubMed]
- Desai, A.K.; Grossberg, G.T.; Sheth, D.N. Activities of daily living in patients with dementia. CNS Drugs 2004, 18, 853–875. [Google Scholar] [CrossRef]
- Tucker, A.M.; Stern, Y. Cognitive reserve in aging. Curr. Alzheimer Res. 2011, 8, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Iqubal, A.; Wasim, M.; Ashraf, M.; Najmi, A.K.; Syed, M.A.; Ali, J.; Haque, S.E. Natural Bioactive as a Potential Therapeutic Approach for the Management of Cyclophosphamide-induced Cardiotoxicity. Curr. Top. Med. Chem. 2021, 21, 2647–2670. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Iqubal, A.; Baboota, S.; Ali, J. Natural Bioactives as Potential Therapeutic Modalities Against NeuroAIDS. Curr. Top. Med. Chem. 2021, 21, 1052–1066. [Google Scholar] [CrossRef]
- Uddin, M.; Asaduzzaman, M.; Mamun, A.; Iqbal, M.; Wahid, F.; Rony, R. Neuroprotective activity of Asparagus racemosus Linn. against ethanol-induced cognitive impairment and oxidative stress in rats brain: Auspicious for controlling the risk of Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 2016, 6, 1000245. [Google Scholar] [CrossRef]
- Iqubal, M.K.; Saleem, S.; Iqubal, A.; Chaudhuri, A.; Pottoo, F.H.; Ali, J.; Baboota, S. Natural, synthetic and their combinatorial nanocarriers based drug delivery system in the treatment paradigm for wound healing via dermal targeting. Curr. Pharm. Des. 2020, 26, 4551–4568. [Google Scholar] [CrossRef]
- Pottoo, F.; Bhowmik, M.; Vohora, D. Raloxifene protects against seizures and neurodegeneration in a mouse model mimicking epilepsy in postmenopausal woman. Eur. J. Pharm. Sci. 2014, 65, 167–173. [Google Scholar] [CrossRef]
- Ahmad, M.A.; Pottoo, F.H.; Akbar, M. Gene Therapy Repairs for the Epileptic Brain: Potential for Treatment and Future Directions. Curr. Gene Ther. 2019, 19, 367–375. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román, G.C. Vascular dementia may be the most common form of dementia in the elderly. J. Neurol. Sci. 2002, 203, 7–10. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Kim, S.-J.; Robinson, J.L.; Sumsuzzman, D.M.; Islam, M.S.; Barreto, G.E.; Mathew, B.; Ashraf, G.M. Detection of Alzheimer’s disease (AD) specific tau pathology with conformation-selective anti-tau monoclonal antibody in co-morbid frontotemporal lobar degeneration-tau (FTLD-tau). Acta Neuropathol. Commun. 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.F.; Uddin, M.S.; Uddin, G.S.; Sumsuzzman, D.M.; Islam, M.S.; Barreto, G.E.; Mathew, B.; Ashraf, G.M. Melatonin in Alzheimer’s disease: A latent endogenous regulator of neurogenesis to mitigate Alzheimer’s neuropathology. Mol. Neurobiol. 2019, 56, 8255–8276. [Google Scholar] [CrossRef]
- Paterson, R.W.; Takada, L.T.; Geschwind, M.D. Diagnosis and treatment of rapidly progressive dementias. Neurol. Clin. Pract. 2012, 2, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Mathew, B.; Parambi, D.G.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019, 352, 1900177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, A.; Fayaz, F.; Wakode, S.; Pottoo, F.H. Biological signatures of Alzheimer’s disease. Curr. Top. Med. Chem. 2020, 20, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Singh, T.G.; Garg, N.; Dhiman, S.; Gupta, S.; Rahman, M.; Najda, A.; Walasek-Janusz, M.; Kamel, M.; Albadrani, G.M.; et al. Dysbiosis and Alzheimer’s Disease: A Role for Chronic Stress? Biomolecules 2021, 11, 678. [Google Scholar] [CrossRef]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.D.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef] [Green Version]
- Snyder, H.M.; Asthana, S.; Bain, L.; Brinton, R.; Craft, S.; Dubal, D.B.; Espeland, M.A.; Gatz, M.; Mielke, M.M.; Raber, J.; et al. Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the Women’s Alzheimer’s Research Initiative. Alzheimer’s Dement. 2016, 12, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lü, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665. [Google Scholar] [CrossRef] [Green Version]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.; Haque, A.; Mamun, A.; Iqbal, M.; Kabir, M.; Rony, R.; Begum, M. Searching the linkage between high fat diet and Alzheimer’s disease: A debatable proof stand for ketogenic diet to alleviate symptoms of Alzheimer’s patient with APOE ε4 allele. J. Neurol. Neurophysiol. 2016, 7, 1000397. [Google Scholar] [CrossRef]
- Uddin, M.S.; Hossain, M.F.; Al Mamun, A.; Shah, M.A.; Hasana, S.; Bulbul, I.J.; Sarwar, M.S.; Mansouri, R.A.; Ashraf, G.M.; Rauf, A.; et al. Exploring the multimodal role of phytochemicals in the modulation of cellular signaling pathways to combat age-related neurodegeneration. Sci. Total Environ. 2020, 725, 138313. [Google Scholar] [CrossRef] [PubMed]
- Crotty, M.; Killington, M.; Liu, E.; Cameron, I.D.; Kurrle, S.; Kaambwa, B.; Davies, O.; Miller, M.; Chehade, M.; Ratcliffe, J. Should we provide outreach rehabilitation to very old people living in Nursing Care Facilities after a hip fracture? A randomised controlled trial. Age Ageing 2019, 48, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, A.M.; Sharrett, A.R.; Albert, M.S.; Coresh, J.; Windham, B.G.; Power, M.C.; Knopman, D.S.; Walker, K.; Burgard, S.; Mosley, T.H.; et al. The association of late-life diabetes status and hyperglycemia with incident mild cognitive impairment and dementia: The ARIC study. Diabetes Care 2019, 42, 1248–1254. [Google Scholar] [CrossRef]
- Fukushima, A.; Nakamura, M.; Suzuki, H.; Saito, K.; Yamazaki, M. High-throughput sequencing and de novo assembly of red and green forms of the Perilla frutescens var. crispa transcriptome. PLoS ONE 2015, 10, e0129154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-H.; Lin, K.-P.; Chen, Y.-C. Risk factors for dementia. J. Formos. Med Assoc. 2009, 108, 754–764. [Google Scholar] [CrossRef] [Green Version]
- Campbell, N.L.; Unverzagt, F.; LaMantia, M.A.; Khan, B.A.; Boustani, M.A. Risk factors for the progression of mild cognitive impairment to dementia. Clin. Geriatr. Med. 2013, 29, 873–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabia, S.; Fayosse, A.; Dumurgier, J.; Dugravot, A.; Akbaraly, T.; Britton, A.; Kivimäki, M.; Singh-Manoux, A. Alcohol consumption and risk of dementia: 23 year follow-up of Whitehall II cohort study. BMJ 2018, 362, k2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crichton, G.E.; Elias, M.F.; Buckley, J.; Murphy, K.J.; Bryan, J.; Frisardi, V. Metabolic syndrome cognitive performance and dementia. J. Alzheimers Dis. 2012, 30, 77–87. [Google Scholar] [CrossRef]
- Duff, C. Dementia: Assessment, Management and Support for People Living with Dementia and Their Carers; National Institute for Health and Care Excellence: London, UK, 2018. [Google Scholar]
- Kane, R.L.; Butler, M.; Fink, H.A.; Brasure, M.; Davila, H.; Desai, P.; Jutkowitz, E.; McCreedy, E.; Nelson, V.A.; McCarten, J.R.; et al. Interventions to Prevent Age-Related Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer’s-Type Dementia. In Comparative Effectiveness Review; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2017. [Google Scholar]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Tang, Z.; Ma, L. Related factors of cognitive impairment in community-dwelling older adults in Beijing Longitudinal Study of Aging. Aging Clin. Exp. Res. 2019, 31, 95–100. [Google Scholar] [CrossRef]
- Shah, A.J.; Mir, R.H.; Mohi-Ud-Din, R.; Pottoo, F.H.; Masoodi, M.H.; Bhat, Z.A. Depression: An insight into Heterocyclic and Cyclic Hydrocarbon Compounds inspired from Natural Sources. Curr. Neuropharmacol. 2021, 19, 2020–2037. [Google Scholar] [CrossRef]
- Iqubal, A.; Ahmed, M.; Iqubal, M.K.; Pottoo, F.H.; Haque, S.E. Polyphenols as Potential Therapeutics for Pain and Inflammation in Spinal Cord Injury. Curr. Mol. Pharmacol. 2020, 14, 714–730. [Google Scholar] [CrossRef]
- Godbout, J.P.; Johnson, R.W. Age and neuroinflammation: A lifetime of psychoneuroimmune consequences. Neurol. Clin. 2006, 24, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Kareem, O.; Bader, G.N.; Pottoo, F.H.; Amir, M.; Barkat, M.A.; Pandey, M. Beclin 1 Complex and Neurodegenerative Disorders. Quality Control of Cellular Protein in Neurodegenerative Disorders; IGI Global: Hershey, PA, USA, 2020; pp. 236–260. [Google Scholar]
- Ahmad, M.; Najmi, A.; Mujeeb, M.; Akhtar, M. Protective effect of guggulipid in high fat diet and middle cerebral artery occlusion (MCAO) induced ischemic cerebral injury in rats. Drug Res. 2016, 66, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ptacek, S.; Farahmand, B.; Kåreholt, I.; Religa, D.; Cuadrado, M.L.; Eriksdotter, M. Mortality risk after dementia diagnosis by dementia type and underlying factors: A cohort of 15,209 patients based on the Swedish Dementia Registry. J. Alzheimer’s Dis. 2014, 41, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Nigar, S.; Pottoo, F.H.; Tabassum, N.; Verma, S.K.; Javed, M.N. Molecular insights into the role of inflammation and oxidative stress in epilepsy. J. Adv. Med Pharm. Sci. 2016, 10, 1132. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs. 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Jakaria, M.; Mathew, B.; Barreto, G.E.; Ashraf, G.M. Nootropic and anti-Alzheimer’s actions of medicinal plants: Molecular insight into therapeutic potential to alleviate Alzheimer’s neuropathology. Mol. Neurobiol. 2019, 56, 4925–4944. [Google Scholar] [CrossRef]
- Zaplatic, E.; Bule, M.; Shah, S.Z.A.; Uddin, M.S.; Niaz, K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer’s disease. Life Sci. 2019, 224, 109–119. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Versijpt, J.J.; Dumont, F.; Van Laere, K.J.; Decoo, D.; Santens, P.; Audenaert, K.; Achten, E.; Slegers, G.; Dierckx, R.A.; Korf, J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. Eur. Neurol. 2003, 50, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R) PK11195-PET and [11C] PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef]
- Yasuno, F.; Ota, M.; Kosaka, J.; Ito, H.; Higuchi, M.; Doronbekov, T.K.; Nozaki, S.; Fujimura, Y.; Koeda, M.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C] DAA1106. Biol. Psychiatry 2008, 64, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqubal, A.; Ahmed, M.; Ahmad, S.; Sahoo, C.R.; Iqubal, M.K.; Haque, S.E. Environmental neurotoxic pollutants: Review. Environ. Sci. Pollut. Research Int. 2020, 27, 41175–41198. [Google Scholar] [CrossRef] [PubMed]
- Giacobbo, B.L.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.; Bromberg, E.; de Vries, E.F. Brain-derived neurotrophic factor in brain disorders: Focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, S.; Singh, A.; Ibrahim, A.M.; Alhajri, N.; Abu Izneid, T.; Pottoo, F.H. Role of triggering receptor expressed on myeloid cells 2 (TREM2) in neurodegenerative dementias. Eur. J. Neurosci. 2021, 53, 3294–3310. [Google Scholar] [CrossRef] [PubMed]
- Haukedal, H.; Freude, K. Implications of microglia in amyotrophic lateral sclerosis and frontotemporal dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef]
- Uddin, M.; Kabir, M.; Niaz, K.; Jeandet, P.; Clément, C.; Mathew, B.; Rauf, A.; Rengasamy, K.R.; Sobarzo-Sánchez, E.; Ashraf, G.M.; et al. Molecular insight into the therapeutic promise of flavonoids against Alzheimer’s disease. Molecules 2020, 25, 1267. [Google Scholar] [CrossRef] [Green Version]
- Akter, R.; Rahman, H.; Behl, T.; Chowdhury, M.; Rahman, A.; Manirujjaman, M.; Bulbul, I.J.; Elshenaw, S.E.; Tit, D.M.; Bungau, S. Prospective role of polyphenolic compounds in the treatment of neurodegenerative diseases. CNS Neurol. Disord.-Drug Targets (Formerly Curr. Drug Targets-CNS Neurol. Disord.) 2021, 20, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Machulda, M.M.; Lundt, E.S.; Albertson, S.M.; Spychalla, A.J.; Schwarz, C.G.; Mielke, M.M.; Jack, C.R., Jr.; Kremers, W.K.; Vemuri, P.; Knopman, D.S.; et al. Cortical atrophy patterns of incident MCI subtypes in the Mayo Clinic Study of Aging. Alzheimer’s Dement. 2020, 16, 1013–1022. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s disease: Implications for microglial activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Rawat, C.; Kukal, S.; Dahiya, U.R.; Kukreti, R. Cyclooxygenase-2 (COX-2) inhibitors: Future therapeutic strategies for epilepsy management. J. Neuroinflamm. 2019, 16, 197. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, W.J.; Grossberg, G.T. Targeting neuroinflammation in Alzheimer’s disease: Evidence for NSAIDs and novel therapeutics. Expert Rev. Neurother. 2017, 17, 17–32. [Google Scholar] [CrossRef]
- Pleen, J.; Townley, R. Alzheimer’s disease clinical trial update 2019–2021. J. Neurol. 2021, 1–14. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Apte, R.N.; Fleisher-Berkovich, S. Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Hou, J.; Zhang, C.; Xu, C.; Wang, L.; Zou, X.; Yu, H.; Shi, Y.; Yin, Z.; Chen, G. Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci. Rep. 2015, 5, 10535. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Uddin, M.; Ashraf, G.M. Quality Control of Cellular Protein in Neurodegenerative Disorders; IGI Global: Hershey, PA, USA, 2020. [Google Scholar]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarin-Vuaillat, C.; Ransohoff, R.M. Chemokines and chemokine receptors in neurological disease: Raise, retain, or reduce? Neurotherapeutics 2007, 4, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Sharma, S.; Najmi, A.K.; Syed, M.A.; Ali, J.; Alam, M.M.; Haque, S.E. Nerolidol ameliorates cyclophosphamide-induced oxidative stress, neuroinflammation and cognitive dysfunction: Plausible role of Nrf2 and NF-κB. Life Sci. 2019, 236, 116867. [Google Scholar] [CrossRef]
- Iqubal, M.K.; Iqubal, A.; Imtiyaz, K.; Rizvi, M.M.A.; Gupta, M.M.; Ali, J.; Baboota, S. Combinatorial lipid-nanosystem for dermal delivery of 5-fluorouracil and resveratrol against skin cancer: Delineation of improved dermatokinetics and epidermal drug deposition enhancement analysis. Eur. J. Pharm. Biopharm. 2021, 163, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Wanrooy, B.J.; Bruce, D.G. Neuroinflammation, Type 2 Diabetes, and Dementia. In Type 2 Diabetes and Dementia; Elsevier: Amsterdam, The Netherlands, 2018; pp. 195–209. [Google Scholar]
- Iqubal, A.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Syed, M.A.; Ali, J.; Alam, M.M.; Ahmad, S.; Haque, S.E. Nerolidol attenuates cyclophosphamide-induced cardiac inflammation, apoptosis and fibrosis in Swiss Albino mice. Eur. J. Pharmacol. 2019, 863, 172666. [Google Scholar] [CrossRef]
- Xia, M.; Qin, S.; Wu, L.; Mackay, C.R.; Hyman, B.T. Immunohistochemical study of the β-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 1998, 153, 31–37. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Iqubal, A.; Syed, M.A.; Ali, J.; Najmi, A.K.; Haque, M.M.; Haque, S.E. Nerolidol protects the liver against cyclophosphamide-induced hepatic inflammation, apoptosis, and fibrosis via modulation of Nrf2, NF-κB p65, and caspase-3 signaling molecules in Swiss albino mice. BioFactors 2020, 46, 963–973. [Google Scholar] [CrossRef]
- Nehal, N.; Nabi, B.; Rehman, S.; Pathak, A.; Iqubal, A.; Khan, S.A.; Yar, M.S.; Parvez, S.; Baboota, S.; Ali, J. Chitosan coated synergistically engineered nanoemulsion of Ropinirole and nigella oil in the management of Parkinson’s disease: Formulation perspective and In vitro and In vivo assessment. Int. J. Biol. Macromol. 2021, 167, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Husain, I.; Zameer, S.; Madaan, T.; Minhaj, A.; Ahmad, W.; Iqubaal, A.; Ali, A.; Najmi, A.K. Exploring the multifaceted neuroprotective actions of Emblica officinalis (Amla): A review. Metab. Brain Dis. 2019, 34, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Syed, M.A.; Najmi, A.K.; Azam, F.; Barreto, G.E.; Iqubal, M.K.; Ali, J.; Haque, S.E. Nano-engineered nerolidol loaded lipid carrier delivery system attenuates cyclophosphamide neurotoxicity–Probable role of NLRP3 inflammasome and caspase-1. Exp. Neurol. 2020, 334, 113464. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Syed, M.A.; Najmi, A.K.; Ali, J.; Haque, S.E. Ameliorative effect of nerolidol on cyclophosphamide-induced gonadal toxicity in Swiss Albino mice: Biochemical-, histological- and immunohistochemical-based evidences. Andrologia 2020, 52, e13535. [Google Scholar] [CrossRef]
- Verma, R.; Hoda, F.; Arshad, M.; Iqubal, A.; Siddiqui, A.N.; Khan, M.A.; Haque, S.E.; Akhtar, M.; Najmi, A.K. Cannabis, a Miracle Drug with Polyvalent Therapeutic Utility: Preclinical and Clinical-Based Evidence. Med. Cannabis Cannabinoids 2021, 4, 43–60. [Google Scholar] [CrossRef]
- Md, S.; Alhakamy, N.A.; Alfaleh, M.A.; Afzal, O.; Altamimi, A.S.; Iqubal, A.; Shaik, R.A. Mechanisms Involved in Microglial-Interceded Alzheimer’s Disease and Nanocarrier-Based Treatment Approaches. J. Pers. Med. 2021, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Nitrini, R. Frontotemporal dementia and amyotrophic lateral sclerosis: Revisiting one of the first case reports with neuropathological examination. Dement. Neuropsychol. 2014, 8, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Rahman, M.R.; Zaman, T.; Uddin, M.; Islam, R.; Abdel-Daim, M.M.; Rhim, H. Emerging potential of naturally occurring autophagy modulators against neurodegeneration. Curr. Pharm. Des. 2020, 26, 772–779. [Google Scholar] [CrossRef]
- Kabir, M.; Uddin, M.; Mamun, A.A.; Jeandet, P.; Aleya, L.; Mansouri, R.A.; Ashraf, G.M.; Mathew, B.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Combination drug therapy for the management of Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3272. [Google Scholar] [CrossRef]
- Uddin, M.; Kabir, M. Emerging signal regulating potential of genistein against Alzheimer’s disease: A promising molecule of interest. Front. Cell Dev. Biol. 2019, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Asaduzzaman, M. Innovation and validation of neuropsychopharmacological testing methods for the assessment of memory, attention and cognition in human participants. Neurosci. Med. 2016, 7, 83. [Google Scholar] [CrossRef] [Green Version]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Wang, H.-F.; Tan, C.-C.; Meng, X.-F.; Wang, C.; Tang, S.-W.; Yu, J.-T. Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimer’s Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef]
- Al Mamun, A.; Uddin, M.S.; Mathew, B.; Ashraf, G.M. Toxic tau: Structural origins of tau aggregation in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 1417. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Risk Indicators | Cumulative Lesion | Neurodegeneration | Decline in Cognition |
|---|---|---|---|
| Non-modifiable | Extracellular amyloid proteins | Loss of synaptic | |
| Age Genetics | Tau (AD and FTD) intracellular protein | Neuroinflammation death in neurons | A decline in one or more memory visuospatial functions Language executive social cognition functions Complex attention |
| Modifiable | |||
| Vascular risk, factor head injury, low education, poor hearing, depreciation, social isolation | Synuclein (LPD, PDD, MSA) PrPSC (prion disease) TDP-43(FTD) FUS(FTD) | Glial reaction |
| S.NO | Neuroanatomical Areas with Substantially Greater Inflammation Compared to Controls in the Dementia Group | Other Findings | References |
|---|---|---|---|
| 1 | Inferior and middle temporal gyri, fusiform gyri, putamen, left amygdala, left posterior cingulate, left parahippocampal gyrus, inferior parietal lobes, and right pallidum in AD relative to controls. Inferior temporal gyri, fusiform gyri, and left parahippocampal in MCI compared with controls | Enhanced neuroinflammation in the left inferior temporal lobe differentiated AD relative to locomotion Regions with elevated inflammation exhibited the highest atrophy rate in AD for 12–24 months. | [97] |
| 2 | Lateral frontal cortices, prefrontal cortices, right mesial temporal cortex, left orbitofrontal cortex | Specific regional inflammation was linked with particular cognitive impairments. The task of image recognition and right superior frontal cortex, the role of orientation and left frontal lateral, left parietal cortex, and right superior frontal. | [98] |
| 3 | Temporal, frontal, parietal, cingulate cortices, and occipital cortices | Inflammation of cingulate gyrus posterior, parietal frontal cortices are associated with MMSE scores. | [99] |
| 4 | Striatum and cerebellum, anterior cingulate, dorsal parietal, lateral, temporal occipital cortices and medial prefrontal cortices. | No MMSE and inflammation association in any amount of interest. | [100] |
| 5 | Basal ganglia, substantia nigra, frontal lateral cortices. The lateral parietal, lateral temporal, cingulate, anterior and posterior cortices, medial occipital and lateral cortices in the occipital, temporal DLB poles, and precuneus compared to the controls | None reported | [101] |
| 6 | Sensorimotor cortices, prefrontal cortices, superior parietal, parietal cortices, middle, temporal, occipital cortices and posterior cingulate cortices in AD relative to controls. Inferior parietal lobules, middle and inferior temporal cortices, and precuneus had the greatest variations, but no difference was seen in thalamus, striatum, or cerebellum, white matter. | Age-related inflammation in the parietal cortex and striatum. Appearance in MCI and AD. | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, M.A.; Kareem, O.; Khushtar, M.; Akbar, M.; Haque, M.R.; Iqubal, A.; Haider, M.F.; Pottoo, F.H.; Abdulla, F.S.; Al-Haidar, M.B.; et al. Neuroinflammation: A Potential Risk for Dementia. Int. J. Mol. Sci. 2022, 23, 616. https://doi.org/10.3390/ijms23020616
Ahmad MA, Kareem O, Khushtar M, Akbar M, Haque MR, Iqubal A, Haider MF, Pottoo FH, Abdulla FS, Al-Haidar MB, et al. Neuroinflammation: A Potential Risk for Dementia. International Journal of Molecular Sciences. 2022; 23(2):616. https://doi.org/10.3390/ijms23020616
Chicago/Turabian StyleAhmad, Md Afroz, Ozaifa Kareem, Mohammad Khushtar, Md Akbar, Md Rafiul Haque, Ashif Iqubal, Md Faheem Haider, Faheem Hyder Pottoo, Fatima S. Abdulla, Mahia B. Al-Haidar, and et al. 2022. "Neuroinflammation: A Potential Risk for Dementia" International Journal of Molecular Sciences 23, no. 2: 616. https://doi.org/10.3390/ijms23020616