
Comparative Phosphoproteomics of Neuro-2a Cells under Insulin Resistance Reveals New Molecular Signatures of Alzheimer’s Disease


Abstract
:1. Introduction
2. Results
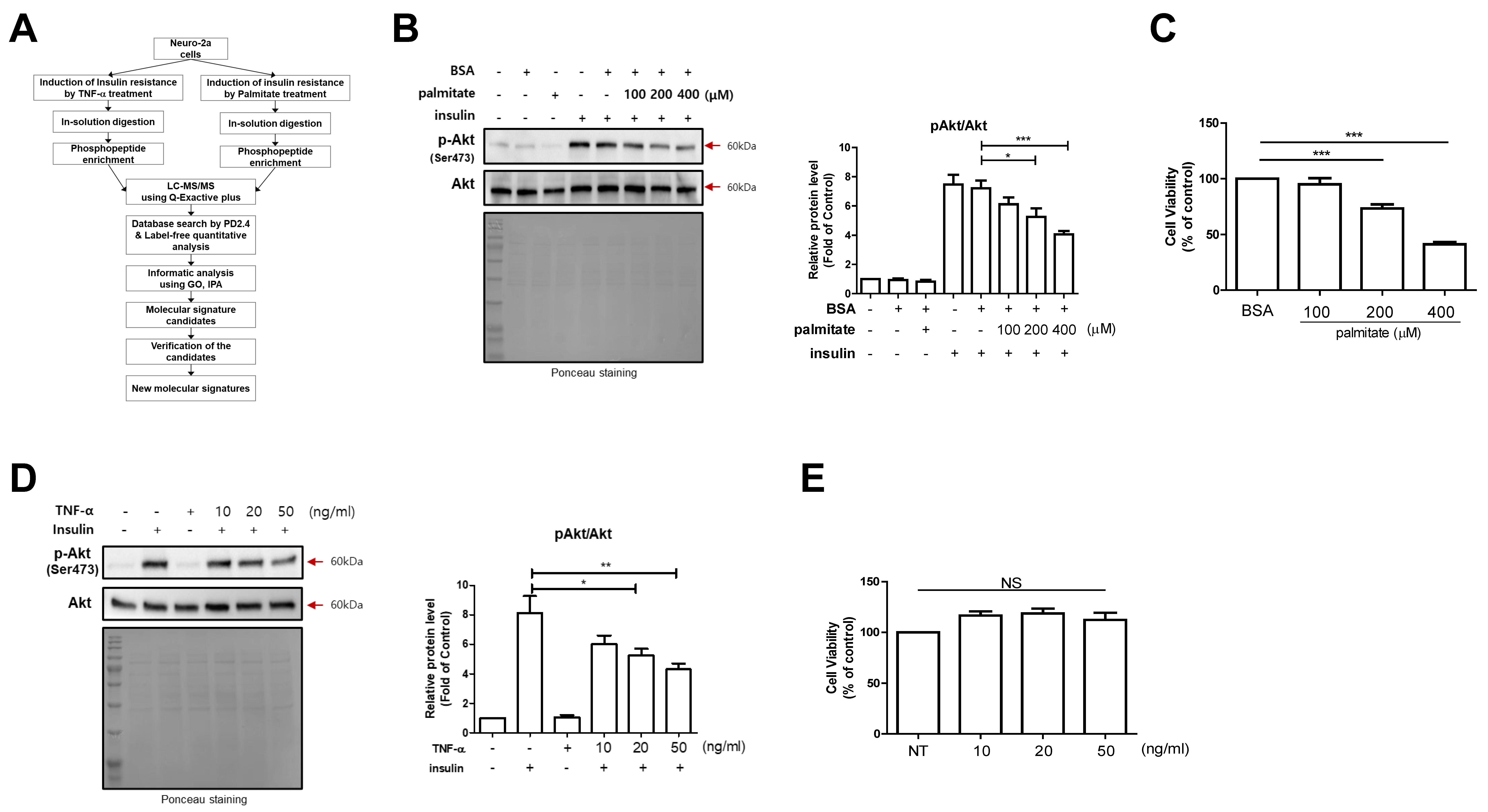
2.1. Induction of Clinically Reliable Insulin Resistance on N2a Cells
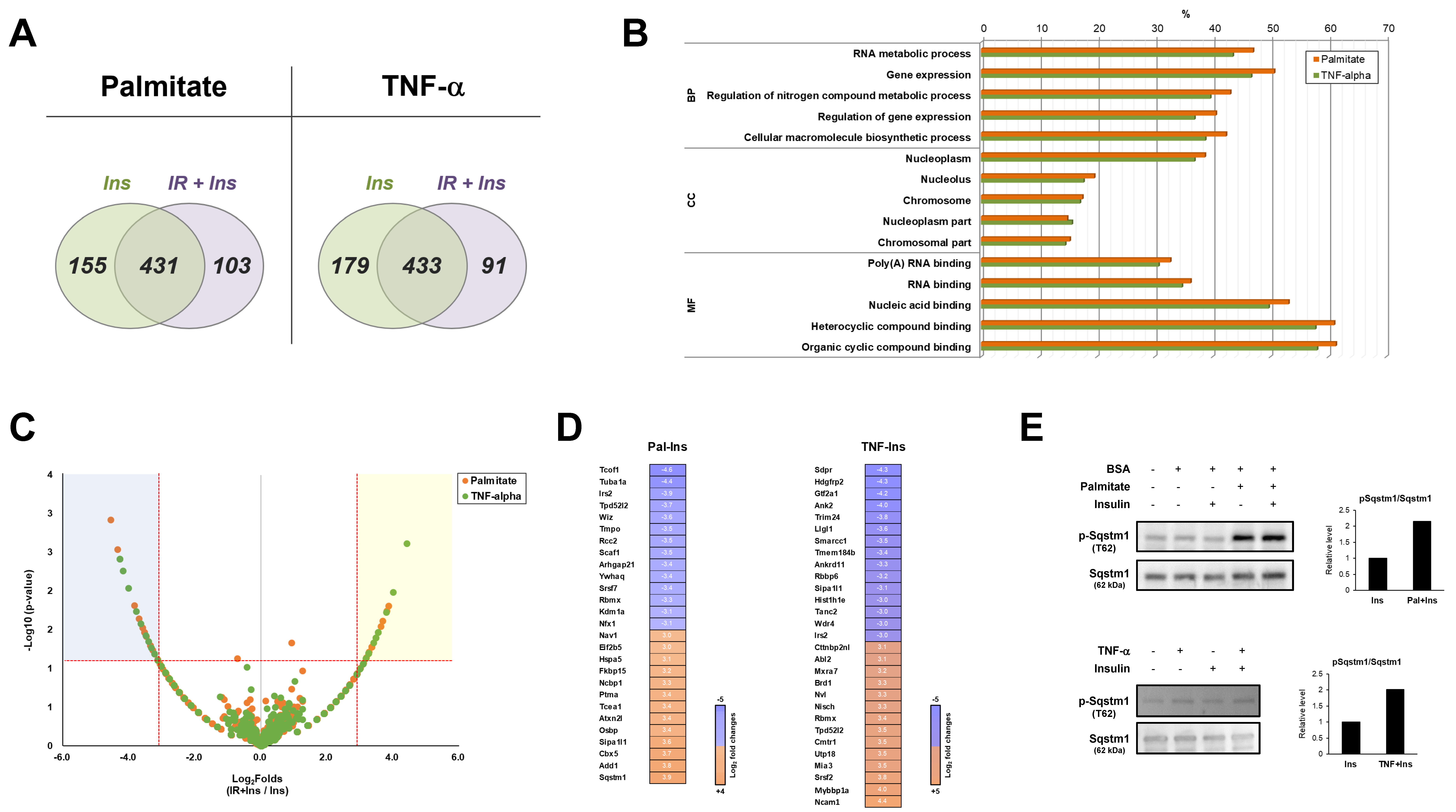
2.2. Phosphoproteomic Analysis Using Reliable Proteomics Approach
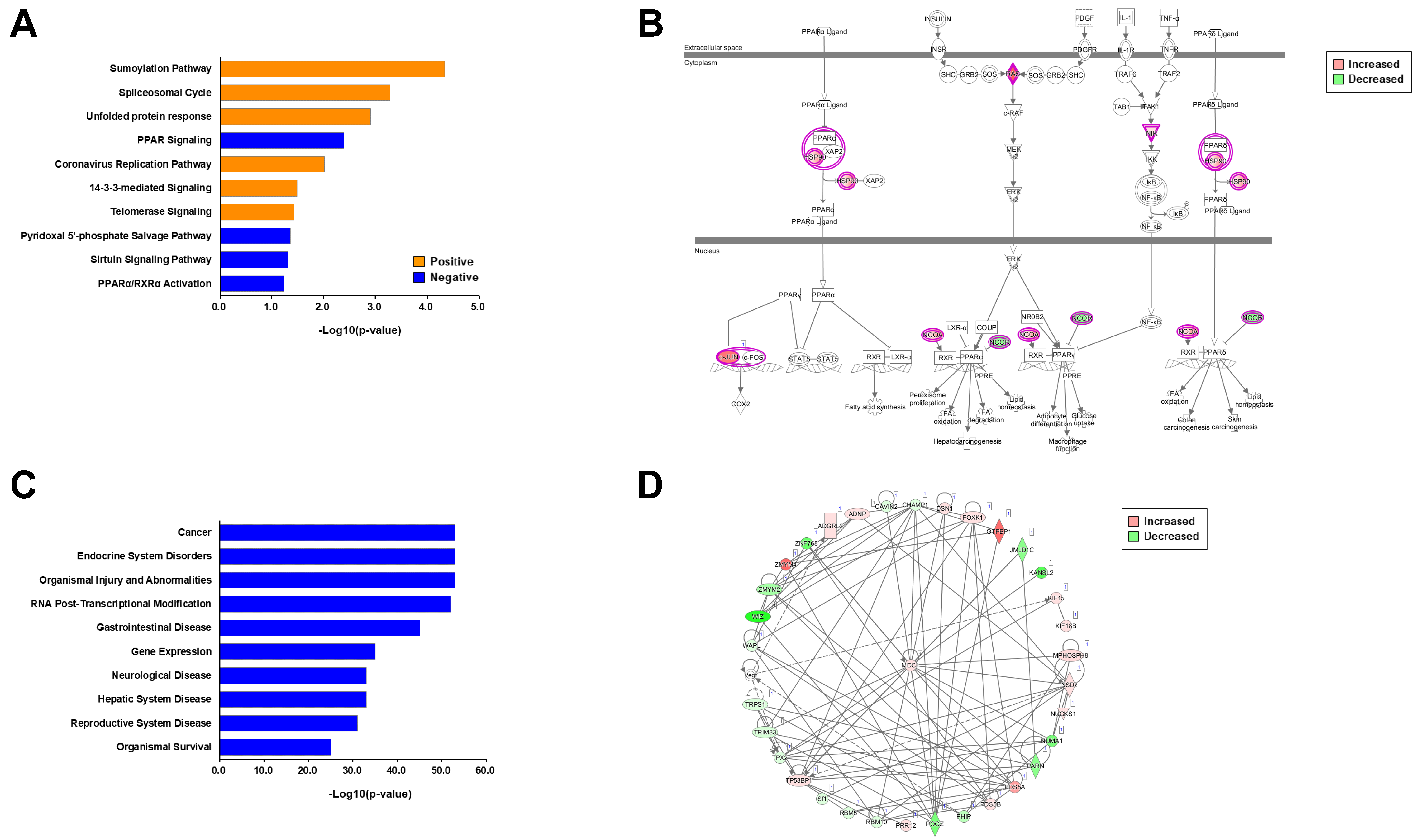
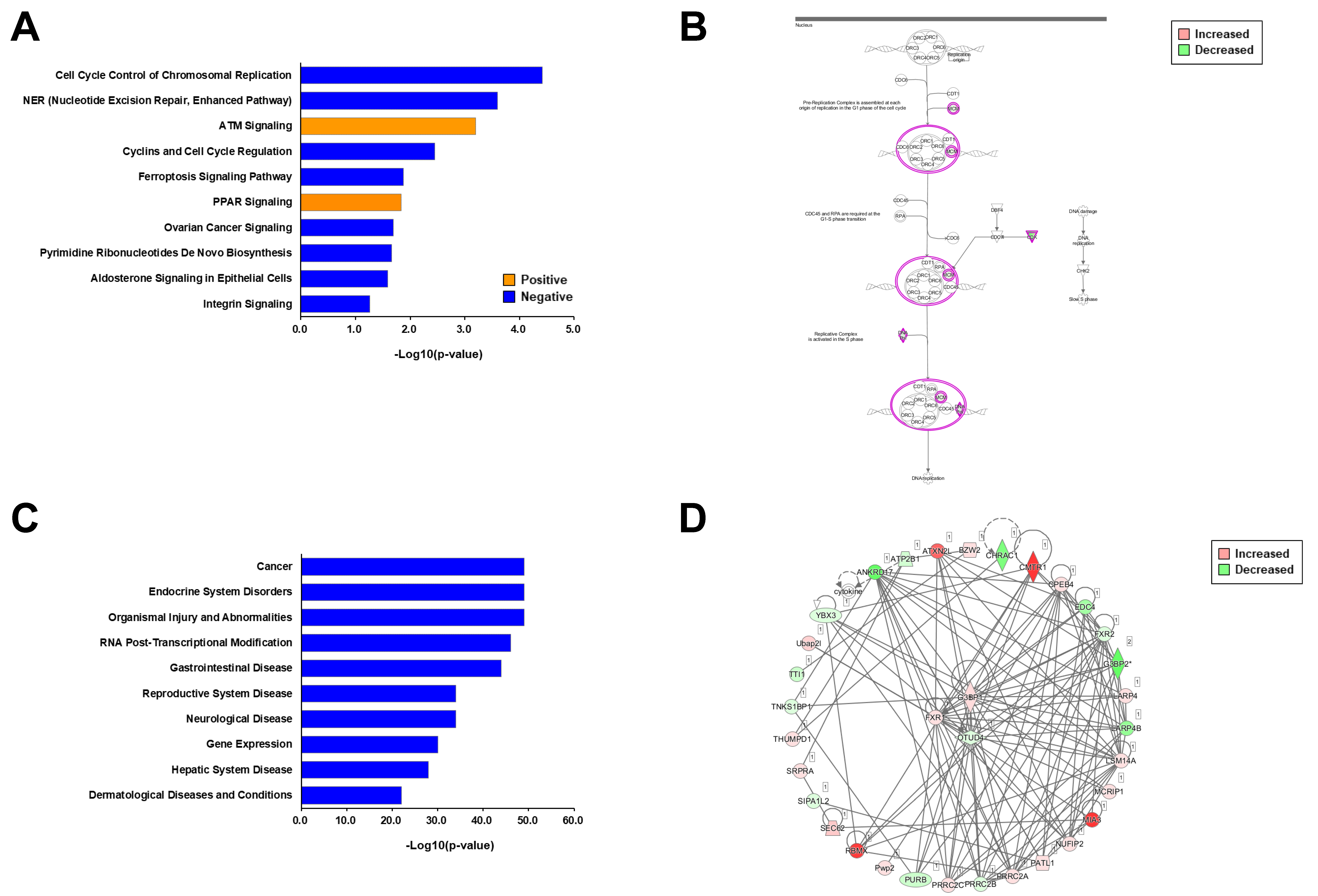
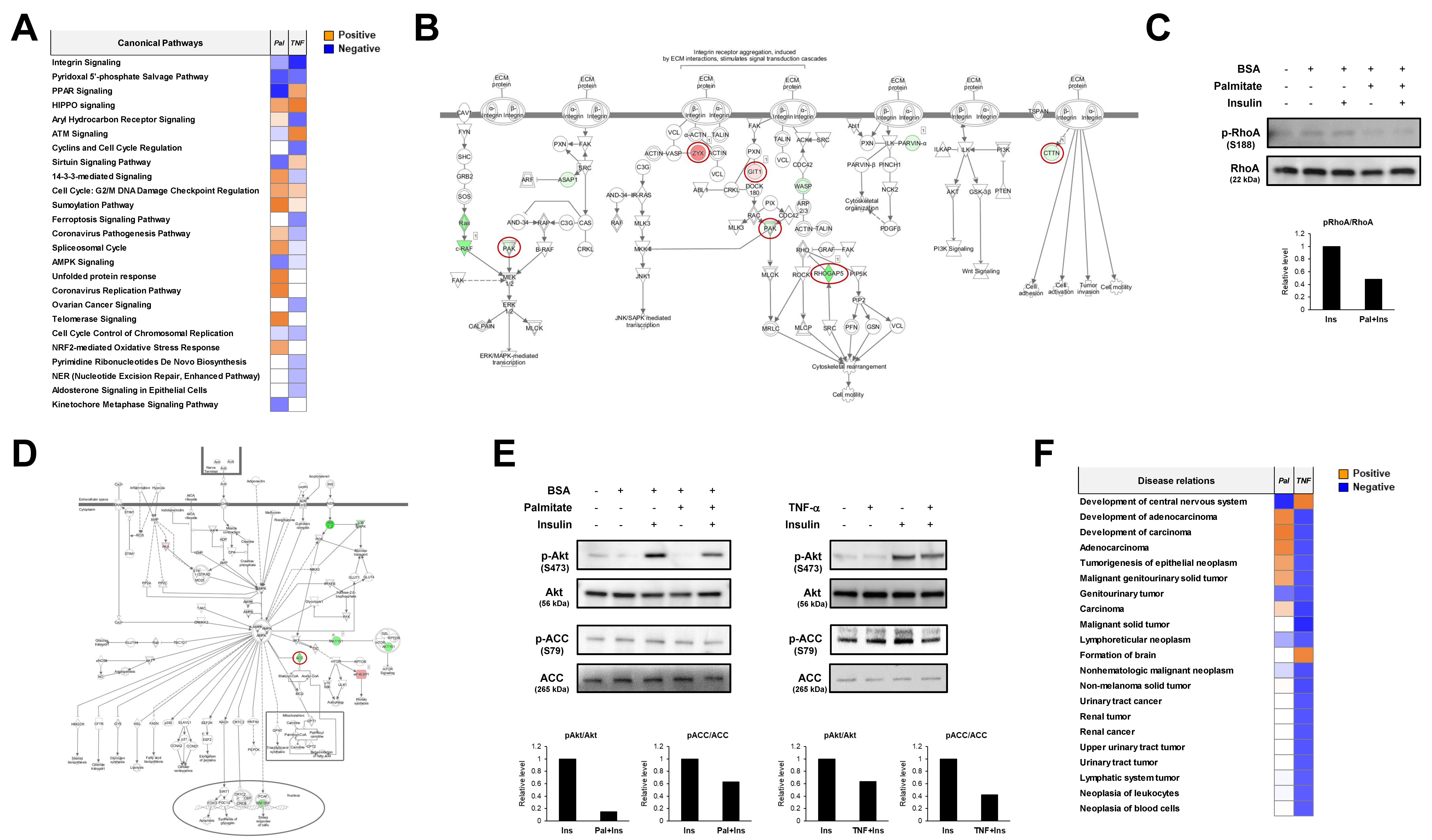
2.3. Informatics Analysis of Each Phosphoproteome
2.4. Comparative Informatics Analysis for Two Phosphoproteomes
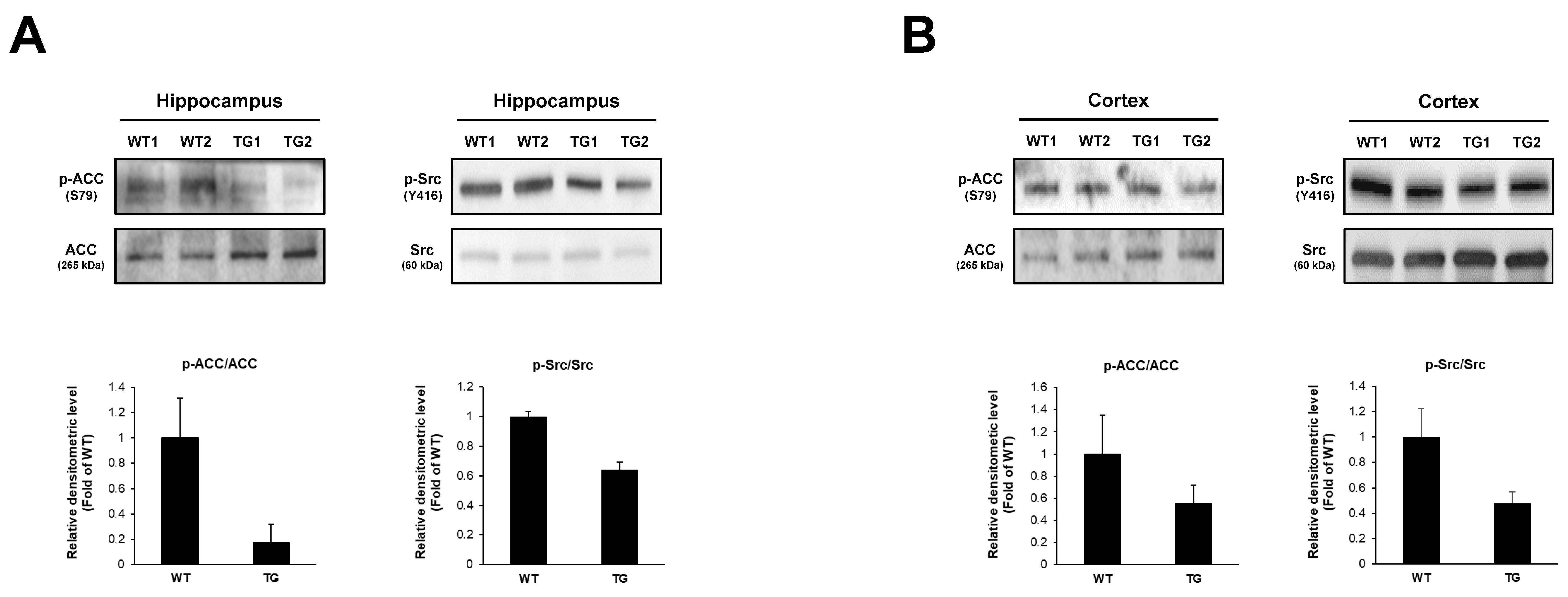
2.5. Changes of Phosphoproteins in 5xFAD Mice Brain
3. Discussion
4. Materials and Methods
4.1. Animal Management
4.2. Establishment of Two Different Insulin Resistance Conditions
4.3. Phosphopeptide Preparation
4.4. Mass Analysis and Database Searching
4.5. Bioinformatics
4.6. Western Blotting
4.7. Cell Viability Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonomi, C.G.; De Lucia, V.; Mascolo, A.P.; Assogna, M.; Motta, C.; Scaricamazza, E.; Sallustio, F.; Mercuri, N.B.; Koch, G.; Martorana, A. Brain energy metabolism and neurodegeneration: Hints from CSF lactate levels in dementias. Neurobiol. Aging 2021, 105, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Uemura, E.; Greenlee, H.W. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 2006, 198, 48–53. [Google Scholar] [CrossRef]
- Al-Zubaidi, A.; Heldmann, M.; Mertins, A.; Brabant, G.; Nolde, J.M.; Jauch-Chara, K.; Münte, T.F. Impact of Hunger, Satiety, and Oral Glucose on the Association Between Insulin and Resting-State Human Brain Activity. Front. Hum. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- Fetterly, T.L.; Oginsky, M.F.; Nieto, A.M.; Alonso-Caraballo, Y.; Santana-Rodriguez, Z.; Ferrario, C.R. Insulin Bidirectionally Alters NAc Glutamatergic Transmission: Interactions between Insulin Receptor Activation, Endogenous Opioids, and Glutamate Release. J. Neurosci. 2021, 41, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Lee, C.-C.; Hsu, K.-S. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J. Neurochem. 2004, 89, 217–231. [Google Scholar] [CrossRef]
- Van der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M.J. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Labouèbe, G.; Liu, S.; Dias, C.; Zou, H.; Wong, J.; Karunakaran, S.; Clee, S.; Phillips, A.G.; Boutrel, B.; Borgland, S.L. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci. 2013, 16, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moult, P.R.; Harvey, J. Hormonal regulation of hippocampal dendritic morphology and synaptic plasticity. Cell Adhes. Migr. 2008, 2, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forny-Germano, L.; De Felice, F.G.; Vieira, M.N.D.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2019, 12, 1027. [Google Scholar] [CrossRef] [Green Version]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.M.; Wellington, C.L.; Johnsen, R.D.; Dhaliwal, S.S.; Mamo, J.C. Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-beta. Br. J. Nutr. 2010, 103, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Barber, T.M.; Kyrou, I.; Randeva, H.S.; Weickert, M.O. Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction. Int. J. Mol. Sci. 2021, 22, 546. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef]
- Melo, H.M.; Silva, G.D.S.S.D.; Sant’Ana, M.R.; Teixeira, C.V.L.; Clarke, J.R.; Coreixas, V.S.M.; de Melo, B.C.; Fortuna, J.T.; Forny-Germano, L.; Ledo, J.H.; et al. Palmitate Is Increased in the Cerebrospinal Fluid of Humans with Obesity and Induces Memory Impairment in Mice via Pro-inflammatory TNF-α. Cell Rep. 2020, 30, 2180–2194.e8. [Google Scholar] [CrossRef] [Green Version]
- Karmi, A.; Iozzo, P.; Viljanen, A.; Hirvonen, J.; Fielding, B.A.; Virtanen, K.; Oikonen, V.; Kemppainen, J.; Viljanen, T.; Guiducci, L.; et al. Increased brain Fatty Acid Uptake in Metabolic Syndrome. Diabetes 2010, 59, 2171–2177. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Caceres, M.P.; Toledo-Valenzuela, L.; Diaz-Castro, F.; Avalos, Y.; Burgos, P.; Narro, C.; Pena-Oyarzun, D.; Espinoza-Caicedo, J.; Cifuentes-Araneda, F.; Navarro-Aguad, F.; et al. Palmitic Acid Reduces the Autophagic Flux and Insulin Sensitivity Through the Activation of the Free Fatty Acid Receptor 1 (FFAR1) in the Hypothalamic Neuronal Cell Line N43/5. Front. Endocrinol. 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.M.; Belsham, D.D. Palmitate attenuates Insulin Signaling and Induces Endoplasmic Reticulum Stress and Apoptosis in Hypothalamic Neurons: Rescue of Resistance and Apoptosis through Adenosine 5′ Monophosphate-Activated Protein Kinase Activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.K.; Salehi, A.; Clemenzi, M.N.; Belsham, D.D. Role of the saturated fatty acid palmitate in the interconnected hypothalamic control of energy homeostasis and biological rhythms. Am. J. Physiol. Metab. 2018, 315, E133–E140. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Zand, H.; Morshedzadeh, N.; Naghashian, F. Signaling pathways linking inflammation to insulin resistance. Diabetes Metab. Syndr. 2017, 11, S307–S309. [Google Scholar] [CrossRef]
- Liu, C.; Feng, X.; Li, Q.; Wang, Y.; Li, Q.; Hua, M. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: A systematic review and meta-analysis. Cytokine 2016, 86, 100–109. [Google Scholar] [CrossRef]
- Moller, D.E. Potential Role of TNF-α in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Trends Endocrinol. Metab. 2000, 11, 212–217. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2018, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Magnan, C. The central nervous system at the core of the regulation of energy homeostasis. Front. Biosci. (Sch. Ed.) 2009, 1, 448–465. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gennip, A.C.; Stehouwer, C.D.; van Boxtel, M.P.; Verhey, F.R.; Koster, A.; Kroon, A.A.; Köhler, S.; van Greevenbroek, M.M.; Wesselius, A.; Eussen, S.J.; et al. Association of Type 2 Diabetes, According to the Number of Risk Factors Within Target Range, With Structural Brain Abnormalities, Cognitive Performance, and Risk of Dementia. Diabetes Care 2021, 44, 2493–2502. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Karter, A.J.; Yaffe, K.; Quesenberry, C.P., Jr.; Selby, J.V. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 2009, 301, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef]
- Maguire, E.A.; Frackowiak, R.; Frith, C. Learning to find your way: A role for the human hippocampal formation. Proc. Biol. Sci. 1996, 263, 1745–1750. [Google Scholar] [CrossRef]
- Verret, L.; Mann, E.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, D.; Sano, M.; Marder, K.; Bell, K.; Bylsma, F.; Lafleche, G.; Albert, M.; Brandt, J.; Stern, Y. Age at onset of Alzheimer’s disease: Relation to pattern of cognitive dysfunction and rate of decline. Neurology 1994, 44, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- O’Kusky, J.R.; Ye, P.; D’Ercole, A.J. Insulin-Like Growth Factor-I Promotes Neurogenesis and Synaptogenesis in the Hippocampal Dentate Gyrus during Postnatal Development. J. Neurosci. 2000, 20, 8435–8442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Leinninger, G.M.; Russell, J.W.; Van Golen, C.M.; Berent, A.; Feldman, E.L. Insulin-like growth factor-I regulates glucose-induced mitochondrial depolarization and apoptosis in human neuroblastoma. Cell Death Differ. 2004, 11, 885–896. [Google Scholar] [CrossRef]
- Waly, M.; Olteanu, H.; Banerjee, R.; Choi, S.-W.; Mason, J.B.; Parker, B.; Sukumar, S.; Shim, S.; Sharma, A.; Benzecry, J.M.; et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: A target for neurodevelopmental toxins and thimerosal. Mol. Psychiatry 2004, 9, 358–370. [Google Scholar] [CrossRef]
- Tanno, B.; Mancini, C.; Vitali, R.; Mancuso, M.; McDowell, H.P.; Dominici, C.; Raschellà, G. Down-Regulation of Insulin-Like Growth Factor I Receptor Activity by NVP-AEW541 Has an Antitumor Effect on Neuroblastoma Cells In vitro and In vivo. Clin. Cancer Res. 2006, 12, 6772–6780. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Estévez, V.; Defterali, Ç.; Vicario-Abejón, C. IGF-I: A key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Otaegi, G.; Yusta-Boyo, M.J.; Vergaño-Vera, E.; Méndez-Gómez, H.R.; Carrera, A.; Abad, J.L.; González, M.; de la Rosa, E.J.; Vicario-Abejón, C.; de Pablo, F. Modulation of the PI 3-kinase–Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J. Cell Sci. 2006, 119, 2739–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winocur, G.; Greenwood, C.E.; Piroli, G.G.; Grillo, C.; Reznikov, L.; Reagan, L.P.; McEwen, B.S. Memory impairment in obese Zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 2005, 119, 1389–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.O.; Panda, B.P.; Parvez, S.; Kaundal, M.; Hussain, S.; Akhtar, M.; Najmi, A.K. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Abeta peptides in animal model of Alzheimer’s disease. Biomed. Pharmacother. 2019, 110, 47–58. [Google Scholar] [CrossRef]
- Akhtar, A.; Bishnoi, M.; Sah, S.P. Sodium orthovanadate improves learning and memory in intracerebroventricular-streptozotocin rat model of Alzheimer’s disease through modulation of brain insulin resistance induced tau pathology. Brain Res. Bull. 2020, 164, 83–97. [Google Scholar] [CrossRef]
- Ho, N.; Sommers, M.S.; Lucki, I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci. Biobehav. Rev. 2013, 37, 1346–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamatsu, S.; Nakamichi, Y.; Yamamura, C.; Matsushima, S.; Watanabe, T.; Ozawa, S.; Furukawa, H.; Ishida, H. Decreased expression of t-SNARE, syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: Restoration of decreased t-SNARE proteins improves impaired insulin secretion. Diabetes 1999, 48, 2367–2373. [Google Scholar] [CrossRef]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn from Diet-Induced Diabetes Models? Front. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Choi, J.; Yang, J.; Chung, W.; Kim, J.-H.; Paik, S.K.; Kim, K.; Han, S.; Won, H.; Bae, Y.-S.; et al. Enhanced NMDA Receptor-Mediated Synaptic Transmission, Enhanced Long-Term Potentiation, and Impaired Learning and Memory in Mice Lacking IRSp53. J. Neurosci. 2009, 29, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Nowak, F.V. Porf-2 = Arhgap39 = Vilse: A Pivotal Role in Neurodevelopment, Learning and Memory. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Sibson, N.R.; Dhankhar, A.; Mason, G.F.; Rothman, D.L.; Behar, K.; Shulman, R.G. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA 1998, 95, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia modulates extracellular amyloid-beta concentrations and neuronal activity in vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef]
- Rocher, A.B.; Chapon, F.; Blaizot, X.; Baron, J.-C.; Chavoix, C. Resting-state brain glucose utilization as measured by PET is directly related to regional synaptophysin levels: A study in baboons. NeuroImage 2003, 20, 1894–1898. [Google Scholar] [CrossRef]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Anchisi, D.; Borroni, B.; Franceschi, M.; Kerrouche, N.; Kalbe, E.; Beuthien-Beumann, B.; Cappa, S.; Lenz, O.; Ludecke, S.; Marcone, A.; et al. Heterogeneity of Brain Glucose Metabolism in Mild Cognitive Impairment and Clinical Progression to Alzheimer Disease. Arch. Neurol. 2005, 62, 1728–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, S.; Herance, J.R.; Gispert, J.D.; Abad, S.; Torrent, É.; Jiménez, X.; Pareto, D.; Perpiña, U.; Sarroca, S.; Rodríguez, E.; et al. In vivo evaluation of amyloid deposition and brain glucose metabolism of 5XFAD mice using positron emission tomography. Neurobiol. Aging 2013, 34, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Brewer, G.J. Amyloid-beta as a modulator of synaptic plasticity. J. Alzheimers Dis. 2010, 22, 741–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaddam, M.; Singh, A.; Jain, N.; Avanthika, C.; Jhaveri, S.; De la Hoz, I.; Sanka, S.; Goli, S.R. A Comprehensive Review of Intranasal Insulin and Its Effect on the Cognitive Function of Diabetics. Cureus 2021, 13. [Google Scholar] [CrossRef]
- Torabi, N.; Noursadeghi, E.; Shayanfar, F.; Nazari, M.; Fahanik-Babaei, J.; Saghiri, R.; Khodagholi, F.; Eliassi, A. Intranasal insulin improves the structure–function of the brain mitochondrial ATP–sensitive Ca2+ activated potassium channel and respiratory chain activities under diabetic conditions. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2021, 1867, 166075. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.; Ali, W.; Nagpal, P.; Grover, A.; Grover, S. Predicting phosphorylation sites using machine learning by integrating the sequence, structure, and functional information of proteins. J. Transl. Med. 2021, 19, 1–11. [Google Scholar] [CrossRef]
- Alexovič, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.G.; Poncet, S.; Derouiche, A.; Shi, L.; Mijakovic, I.; Noirot-Gros, M.-F. Role of Protein Phosphorylation in the Regulation of Cell Cycle and DNA-Related Processes in Bacteria. Front. Microbiol. 2016, 7, 184. [Google Scholar] [CrossRef]
- Baker, N.; Catta-Preta, C.M.C.; Neish, R.; Sadlova, J.; Powell, B.; Alves-Ferreira, E.V.C.; Geoghegan, V.; Carnielli, J.B.T.; Newling, K.; Hughes, C.; et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation or survival. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, J. The post translational modification of key regulators of ATR signaling in DNA replication. Genome Instab. Dis. 2021, 2, 92–101. [Google Scholar] [CrossRef]
- Barford, D.; Das, A.K.; Egloff, M.-P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 133–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-Dephosphorylation of Enzymes. Annu. Rev. Biochem. 1979, 48, 923–959. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Litichevskiy, L.; Peckner, R.; Abelin, J.G.; Asiedu, J.K.; Creech, A.L.; Davis, J.F.; Davison, D.; Dunning, C.M.; Egertson, J.D.; Egri, S.; et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst. 2018, 6, 424–443.e7. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wei, S.; Ji, Y.; Guo, X.; Yang, F. Quantitative proteomics using SILAC: Principles, applications, and developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kim, D.; Jang, J.-H.; Ghim, J.; Park, S.; Song, P.; Kwon, Y.; Kim, J.; Hwang, D.; Bae, Y.-S.; et al. Proteomic Analysis of the Palmitate-induced Myotube Secretome Reveals Involvement of the Annexin A1-Formyl Peptide Receptor 2 (FPR2) Pathway in Insulin Resistance. Mol. Cell. Proteom. 2015, 14, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Song, P.; Jang, J.-H.; Kim, D.-K.; Choi, S.; Kim, J.; Ghim, J.; Kim, D.; Park, S.; Lee, H.; et al. Proteomic Analysis of Tumor Necrosis Factor-Alpha (TNF-α)-Induced L6 Myotube Secretome Reveals Novel TNF-α-Dependent Myokines in Diabetic Skeletal Muscle. J. Proteome Res. 2011, 10, 5315–5325. [Google Scholar] [CrossRef]
- Cheyuo, C.; Aziz, M.; Wang, P. Neurogenesis in Neurodegenerative Diseases: Role of MFG-E8. Front. Neurosci. 2019, 13, 569. [Google Scholar] [CrossRef]
- Wennstrom, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar]
- Bozzo, C.; Graziola, F.; Chiocchetti, A.; Canonico, P.L. Estrogen and beta-amyloid toxicity: Role of integrin and PI3-K. Mol. Cell Neurosci. 2010, 45, 85–91. [Google Scholar] [CrossRef]
- Rowan, M.J.; Klyubin, I.; Wang, Q.; Hu, N.W.; Anwyl, R. Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochem. Soc. Trans. 2007, 35, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; McGeer, P.L. Brain microglia constitutively express beta-2 integrins. J. Neuroimmunol. 1990, 30, 81–93. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase—An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.-M.; Zheng, Y.-L.; Hu, B.; Zhang, Z.-F.; Shan, Q.; Zheng, Z.-H.; Liu, C.-M.; Wang, Y.-J. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J. Pathol. 2010, 222, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Seixas da Silva, G.S.; Melo, H.M.; Lourenco, M.V.; Lyra, E.S.N.M.; de Carvalho, M.B.; Alves-Leon, S.V.; de Souza, J.M.; Klein, W.L.; da-Silva, W.S.; Ferreira, S.T.; et al. Amyloid-beta oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons. J. Biol. Chem. 2017, 292, 7395–7406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, C.; Tremblay, C.; Émond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 Reduction Parallels the Accumulation of Tau in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Hu, R.; He, D.; Zhou, G.; Wu, H.; Xu, C.; He, B.; Wu, L.; Wang, Y.; Chang, Y.; et al. Bisdemethoxycurcumin inhibits oxidative stress and antagonizes Alzheimer’s disease by up-regulating SIRT1. Brain Behav. 2020, 10, e01655. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Gene Symbol | Description | Log2Fold (Pal + Ins/Ins) | Modifications |
|---|---|---|---|---|
| Q64337 | Sqstm1 | Sequestosome-1 | 3.86 | Phospho [T269(100); T271(99.5); S330(100); S334(100); S363(98.7); S367(98.7); S368(98.7)] |
| Q9QYC0 | Add1 | Alpha-adducin | 3.80 | Phospho [T610(99.1); T614(99.1); S724(100)] |
| Q61686 | Cbx5 | Chromobox protein homolog 5 | 3.69 | Phospho [S14(99.6)] |
| Q8C0T5 | Sipa1l1 | Signal-induced proliferation-associated 1-like protein 1 | 3.63 | Phospho [S1528(100); S1624(100); S1626(100)] |
| P26350 | Ptma | Prothymosin alpha | 3.35 | Met−loss + Acetyl [N-Term]; Phospho [S2(100)]; Acetyl [N-Term] |
| P10711 | Tcea1 | Transcription elongation factor A protein 1 | 3.35 | Phospho [S100(100)] |
| Q7TQH0 | Atxn2l | Ataxin-2-like protein | 3.35 | Phospho [S109(100); S304(100); S337(99.4)] |
| Q3B7Z2 | Osbp | Oxysterol-binding protein 1 | 3.35 | Phospho [S188(100); S191(100); T375(100); S377(99.5); S380(100); S383(100)] |
| Q3UYV9 | Ncbp1 | Nuclear cap-binding protein subunit 1 | 3.27 | Phospho [S22(99.6)] |
| Q6P9Q6 | Fkbp15 | FK506-binding protein 15 | 3.19 | Phospho [S1157(100); S1159(100)] |
| P20029 | Hspa5 | Endoplasmic reticulum chaperone BiP | 3.10 | Phospho [S650(98)] |
| Q8CH77 | Nav1 | Neuron navigator 1 | 3.01 | Phospho [S1247(99.2)] |
| Q8CHW4 | Eif2b5 | Translation initiation factor eIF-2B subunit epsilon | 3.01 | Phospho [S540(100)] |
| B1AY10 | Nfx1 | Transcriptional repressor NF-X1 | −3.07 | Phospho [S51(98.7); S81(100); S147(100); S149(98.6)] |
| Q6ZQ88 | Kdm1a | Lysine-specific histone demethylase 1A | −3.14 | Phospho [S132(100); S138(100); S167(100)] |
| Q9WV02 | Rbmx | RNA-binding motif protein, X chromosome | −3.29 | Phospho [S208(100)] |
| Q6DFV3 | Arhgap21 | Rho GTPase-activating protein 21 | −3.36 | Phospho [S874(100); T1621(100); S1623(100)] |
| P68254 | Ywhaq | 14-3-3 protein theta | −3.36 | Phospho [S230(96)] |
| Q8BL97 | Srsf7 | Serine/arginine-rich splicing factor 7 | −3.36 | Phospho [S208(100); S210(100)] |
| Q5U4C3 | Scaf1 | Splicing factor, arginine/serine-rich 19 | −3.48 | Phospho [S510(100); S518(99.5); S676(100); S682(100); S691(100); S695(100); S821(100)] |
| Q61029 | Tmpo | Lamina-associated polypeptide 2, isoforms beta/delta/epsilon/gamma | −3.54 | Phospho [S66(100); S67(100); T74(100); T159(98.6); S179(100); S183(100)] |
| Q8BK67 | Rcc2 | Protein RCC2 | −3.54 | Phospho [S48(95.5)] |
| O88286 | Wiz | Protein Wiz | −3.60 | Phospho [S1045(100); S1050(100)] |
| Q9CYZ2 | Tpd52l2 | Tumor protein D54 | −3.70 | Phospho [S200(99)] |
| P81122 | Irs2 | Insulin receptor substrate 2 | −3.85 | Phospho [T55(99); S66(100); T517(100); T524(100); S556(100); S573(100); S616(99.5); S1089(100)] |
| P68369 | Tuba1a | Tubulin alpha-1A chain | −4.36 | Phospho [S439(100)] |
| O08784 | Tcof1 | Treacle protein | −4.57 | Phospho [S83(97); T1114(99.2); S1191(100)] |
| Accession | Gene Symbol | Description | Log2Fold (TNF + Ins/Ins) | Modifications |
|---|---|---|---|---|
| P13595 | Ncam1 | Neural cell adhesion molecule 1 | 4.41 | Phospho [S770(100); S774(100); S1005(100)] |
| Q7TPV4 | Mybbp1a | Myb-binding protein 1A | 3.99 | Phospho [S1164(100); S1253(99.1); T1256(99.1); S1280(100)] |
| Q62093 | Srsf2 | Serine/arginine-rich splicing factor 2 | 3.79 | Phospho [T25(99.4); S206(100); S208(100); S212(100)] |
| Q5SSI6 | Utp18 | U3 small nucleolar RNA-associated protein 18 homolog | 3.55 | Phospho [S114(100); S115(100); S118(100); S206(100)] |
| Q8BI84 | Mia3 | Transport and Golgi organization protein 1 homolog | 3.55 | Phospho [S1458(100); S1765(99.4)] |
| Q9CYZ2 | Tpd52l2 | Tumor protein D54 | 3.48 | Phospho [S200(100)] |
| Q9DBC3 | Cmtr1 | Cap-specific mRNA (nucleoside-2′-O-)-methyltransferase 1 | 3.48 | Phospho [S27(99.4); S48(98.6); S50(100); S52(100); S54(100)] |
| Q9WV02 | Rbmx | RNA-binding motif protein, X chromosome | 3.41 | Phospho [S208(100)] |
| G5E8P1 | Brd1 | Bromodomain-containing protein 1 | 3.26 | Phospho [S128(100); S1052(100); S1055(100)] |
| Q9DBY8 | Nvl | Nuclear valosin-containing protein-like | 3.26 | Phospho [S190(100)] |
| Q80TM9 | Nisch | Nischarin | 3.26 | Phospho [S543(99.5); S548(99.7); S1373(100)] |
| Q9CZH7 | Mxra7 | Matrix-remodeling-associated protein 7 | 3.18 | Phospho [S79(100)] |
| Q99LJ0 | Cttnbp2nl | CTTNBP2 N-terminal-like protein | 3.09 | Phospho [S481(100); S556(100); S559(100); S562(100)] |
| Q4JIM5 | Abl2 | Tyrosine-protein kinase ABL2 | 3.09 | Phospho [S621(100); S632(99.5)] |
| P43274 | Hist1h1e | Histone H1.4 | −3.03 | Met−loss + Acetyl [N-Term]; Phospho [S2(99.2); T18(100)]; Acetyl [N-Term] |
| A2A690 | Tanc2 | Protein TANC2 | −3.03 | Phospho [S1534(100); S1538(100)] |
| Q9EP82 | Wdr4 | tRNA (guanine-N(7)-)-methyltransferase non-catalytic subunit WDR4 | −3.03 | Phospho [S397(100)] |
| P81122 | Irs2 | Insulin receptor substrate 2 | −3.03 | Phospho [S66(97.6)] |
| Q8C0T5 | Sipa1l1 | Signal-induced proliferation-associated 1-like protein 1 | −3.13 | Phospho [S1528(100); S1624(100); S1626(100); S1629(99.1)] |
| P97868 | Rbbp6 | E3 ubiquitin-protein ligase RBBP6 | −3.23 | Phospho [S1179(100); S1329(100); S1644(98.4); S1646(98.4); S1651(100)] |
| E9Q4F7 | Ankrd11 | Ankyrin repeat domain-containing protein 11 | −3.33 | Phospho [S1070(100); S1832(99); S1844(100)] |
| Q8BG09 | Tmem184b | Transmembrane protein 184B | −3.41 | Phospho [S402(100); S403(100)] |
| P97496 | Smarcc1 | SWI/SNF complex subunit SMARCC1 | −3.50 | Phospho [S327(100); S329(100)] |
| Q80Y17 | Llgl1 | Lethal(2) giant larvae protein homolog 1 | −3.65 | Phospho [S982(100); S986(100); S989(98.6)] |
| Q64127 | Trim24 | Transcription intermediary factor 1-alpha | −3.79 | Phospho [S1026(100); S1029(100)] |
| Q8C8R3 | Ank2 | Ankyrin-2 | −4.03 | Phospho [S1699(100); S1700(100); S1703(100); S2824(100); S2827(99.4); S3362(100)] |
| Q99PM3 | Gtf2a1 | Transcription initiation factor IIA subunit 1 | −4.19 | Phospho [S318(100); S323(100)] |
| Q63918 | Sdpr | Caveolae-associated protein 2 | −4.28 | Phospho [S203(100); S204(100); S218(100); S293(100); S359(100); S363(100); T368(100)] |
| Q3UMU9-2 | Hdgfrp2 | Isoform 2 of Hepatoma-derived growth factor-related protein 2 | −4.28 | Phospho [S365(100); S366(100); S627(100); S628(100); S638(100)] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Jo, Y.S.; Jo, H.-S.; Bae, S.; Kwon, Y.W.; Oh, Y.-S.; Yoon, J.H. Comparative Phosphoproteomics of Neuro-2a Cells under Insulin Resistance Reveals New Molecular Signatures of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1006. https://doi.org/10.3390/ijms23021006
Kim D, Jo YS, Jo H-S, Bae S, Kwon YW, Oh Y-S, Yoon JH. Comparative Phosphoproteomics of Neuro-2a Cells under Insulin Resistance Reveals New Molecular Signatures of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(2):1006. https://doi.org/10.3390/ijms23021006
Chicago/Turabian StyleKim, Dayea, Yeon Suk Jo, Han-Seul Jo, Sungwon Bae, Yang Woo Kwon, Yong-Seok Oh, and Jong Hyuk Yoon. 2022. "Comparative Phosphoproteomics of Neuro-2a Cells under Insulin Resistance Reveals New Molecular Signatures of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 2: 1006. https://doi.org/10.3390/ijms23021006