
Selective Anti-Leishmanial Strathclyde Minor Groove Binders Using an N-Oxide Tail-Group Modification
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
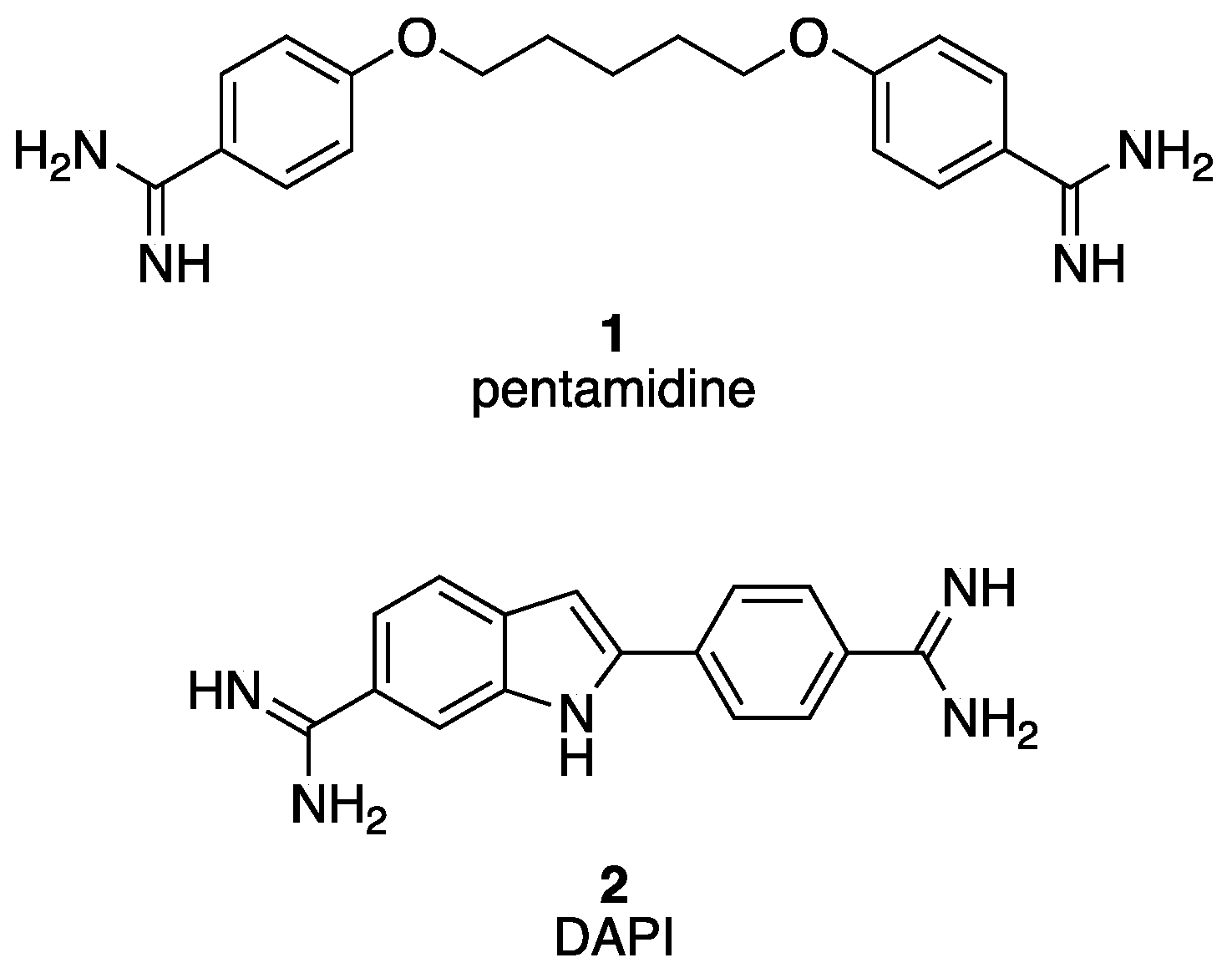
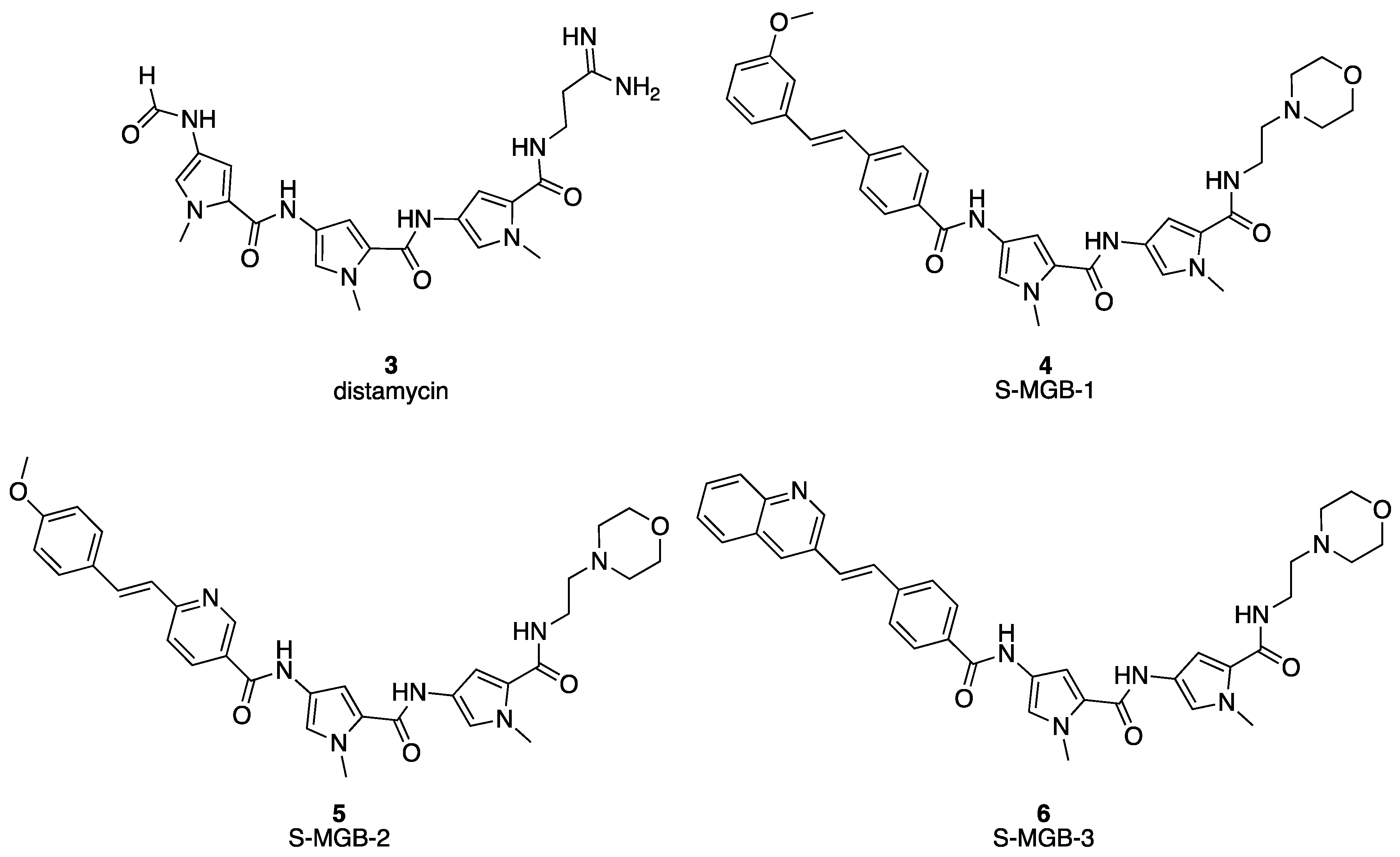
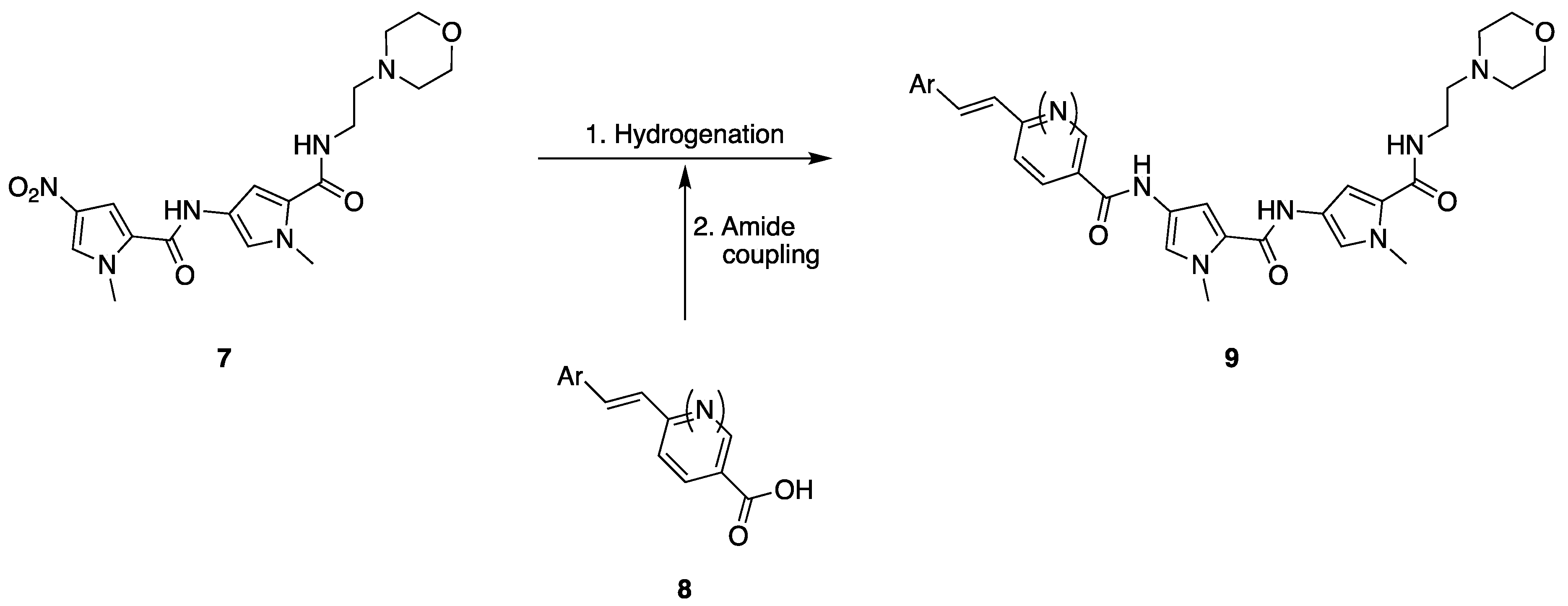
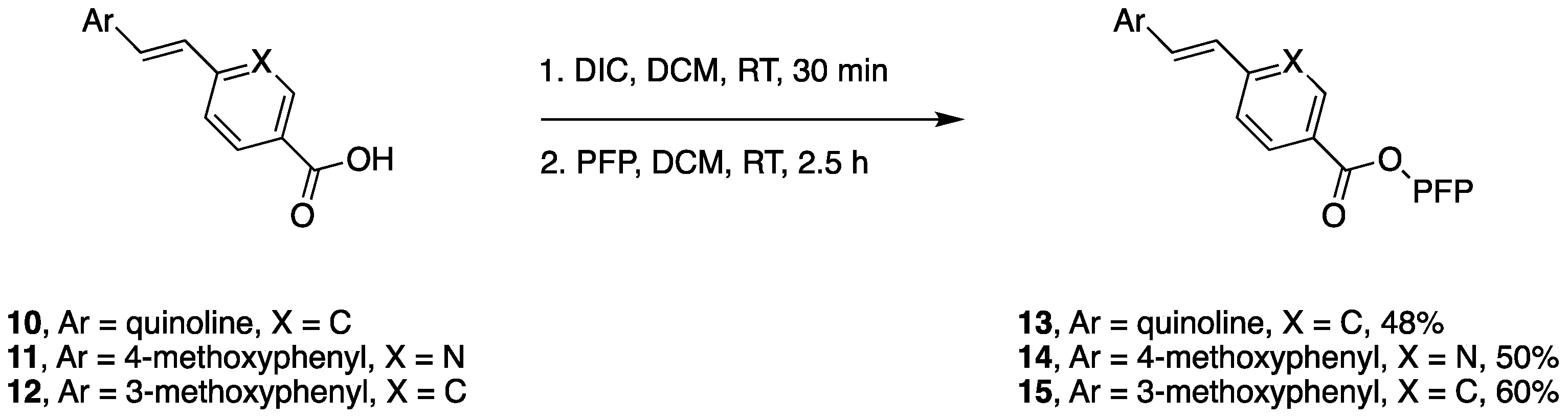
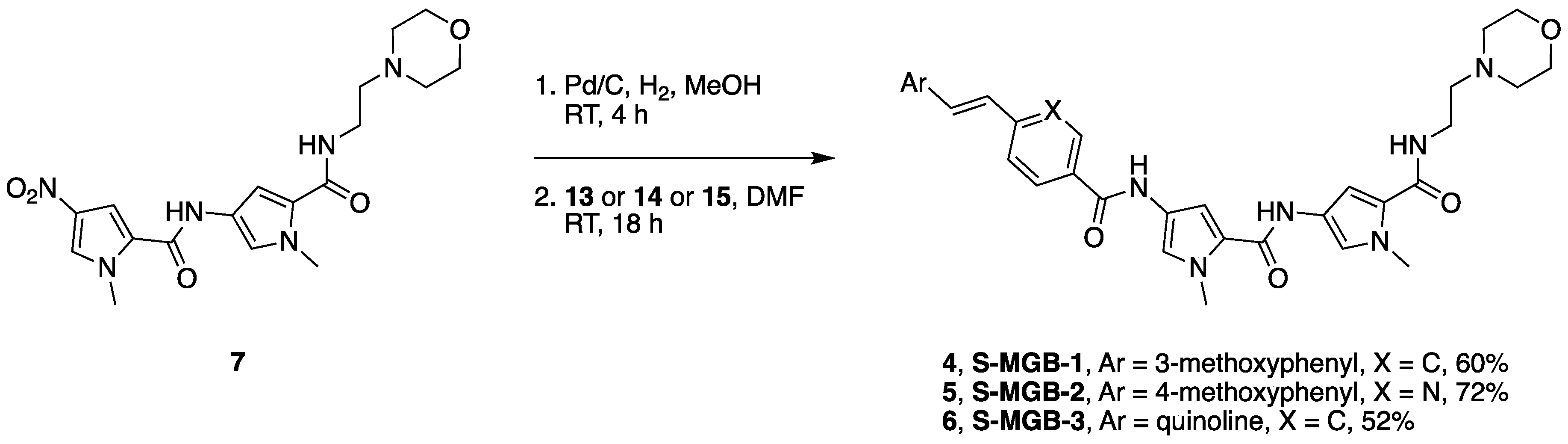
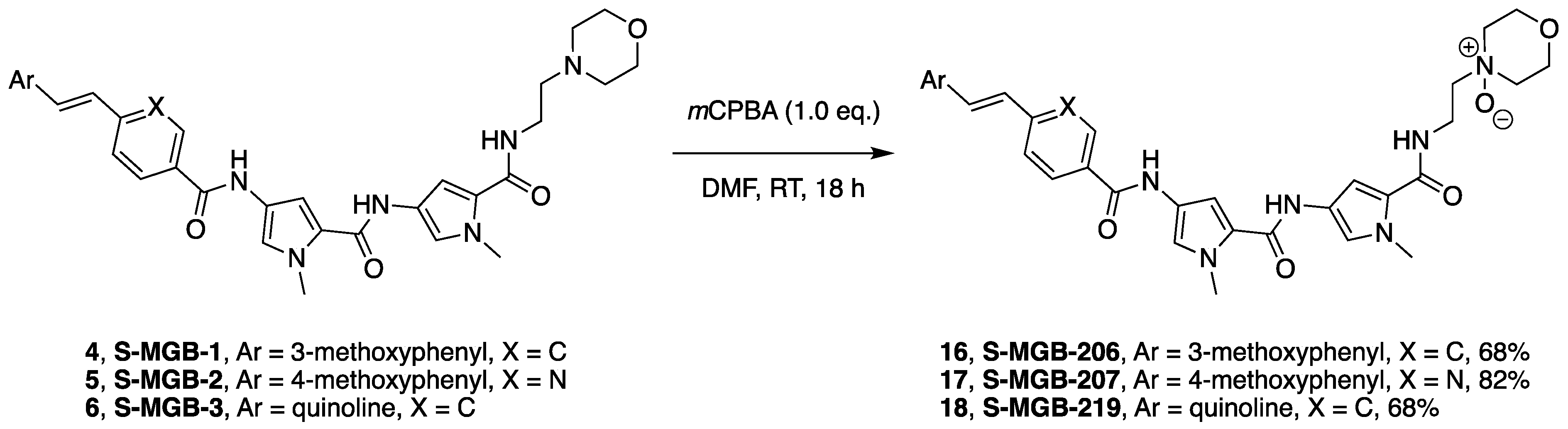
2.1. Chemistry
2.2. Biology
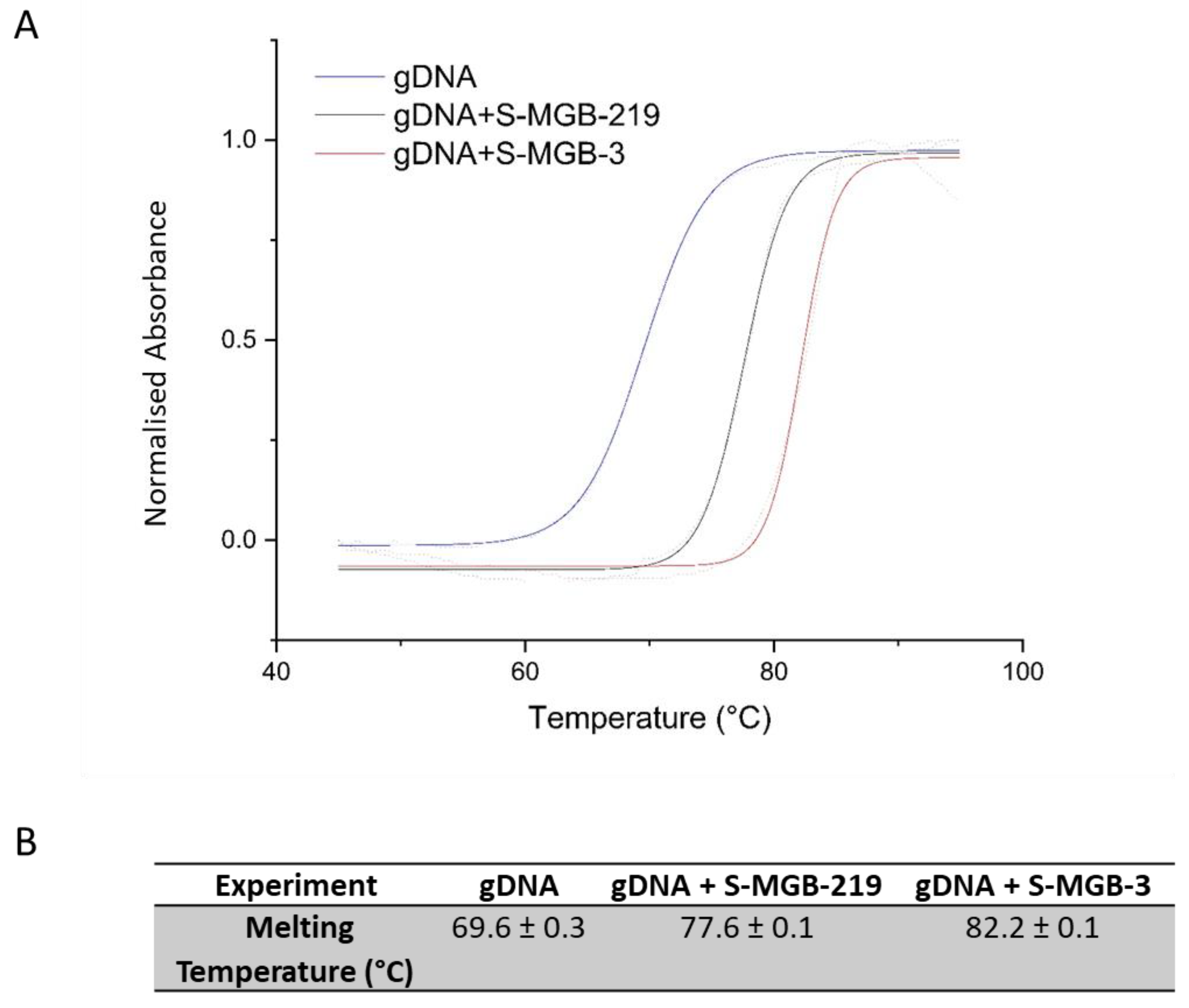
2.3. Thermal Melting Study

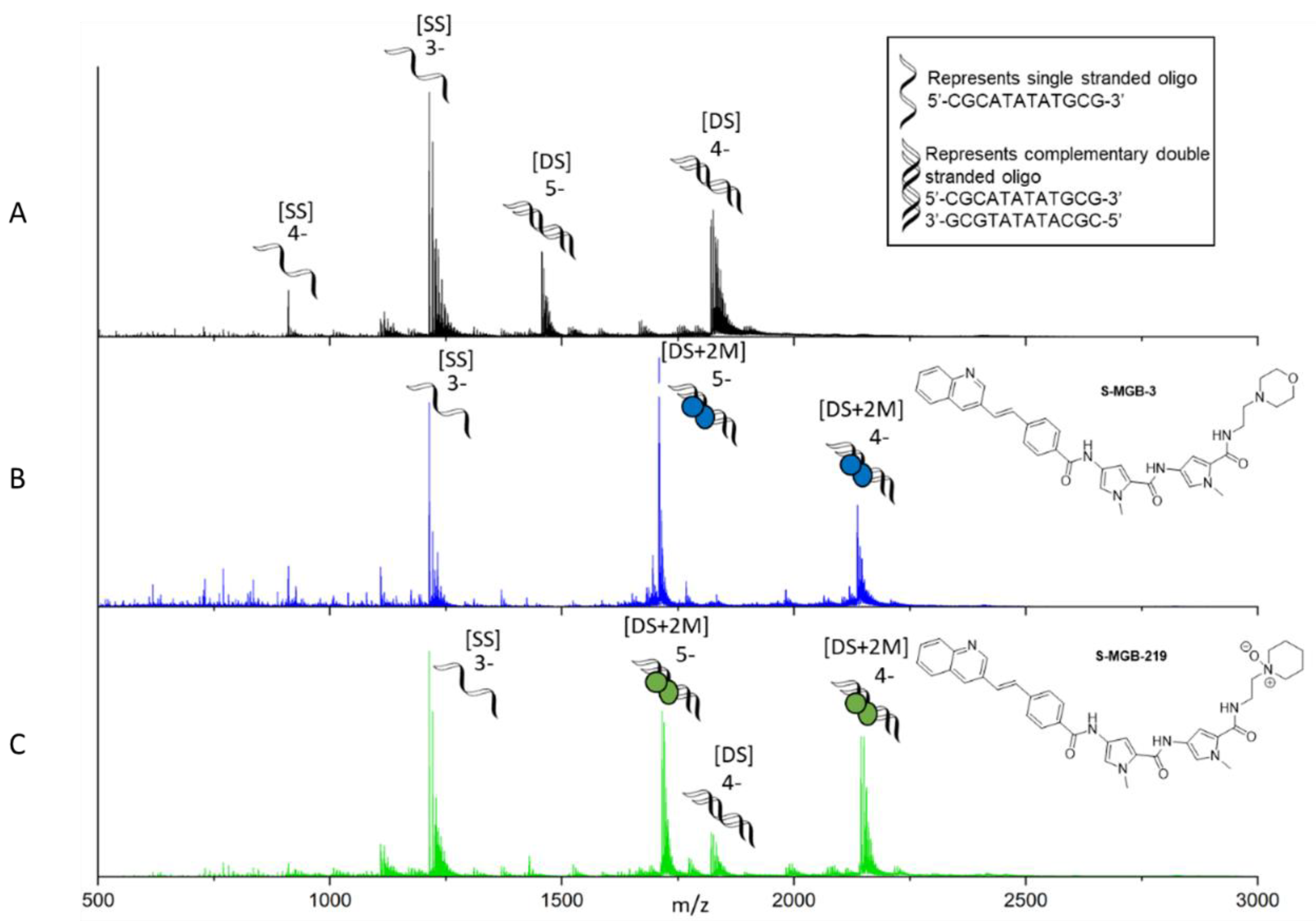
2.4. Native Mass Spectrometry
3. Conclusions
4. Experimental Procedure
4.1. Biological Evaluation
4.1.1. Antibacterial Assay
4.1.2. Anti-Trypanosoma Assay
4.1.3. Anti-Leishmania Assay
4.2. hERG Evaluation
4.2.1. Preparation of Cells
4.2.2. hERG Cell-Binding Assay
4.3. UV-Vis DNA Thermal Melting Experiments
4.3.1. Short DNA Oligo
4.3.2. Genomic DNA
4.4. Native Mass Spectrometry Experiments
4.5. Chemistry
4.5.1. General Experimental Methods
4.5.2. General Pentafluorophenol Ester Synthesis
4.5.3. General MGB Synthesis
4.5.4. General MGB Tail-Group Modification
[(E)-2-(4-methoxyphenyl)ethenyl]-5-({[1-methyl-5-({[1-methyl-5-({[2-(4-oxido-4-morpholinyl)ethyl]amino}carbonyl)-1H-pyrrol-3-yl]amino}carbonyl)-1H-pyrrol-3-yl]amino}carbonyl)pyridinium trifluoroacetate S-MGB-207 (17)
{(E)-2-[4-({[1-methyl-5-({[1-methyl-5-({[2-(4-oxido-4-morpholinyl)ethyl]amino}carbonyl)-1H-pyrrol-3-yl]amino}carbonyl)-1H-pyrrol-3-yl]amino}carbonyl)phenyl]ethenyl}quinolinium trifluoroacetate S-MGB-219 (16)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Leishmaniasis. 2020. Available online: www.cdc.gov/parasites/leishmaniasis/gen_info/faqs.html (accessed on 2 August 2022).
- Bhattacharya, S.K.; Sur, D.; Sinha, P.K.; Karbwang, J. Elimination of leishmaniasis (kala-azar) from the Indian subcontinent is technically feasible & operationally achievable. Indian J. Med Res. 2006, 123, 195. [Google Scholar] [PubMed]
- Toor, J.; Adams, E.R.; Aliee, M.; Amoah, B.; Anderson, R.M.; Ayabina, D.; Bailey, R.; Basáñez, M.-G.; Blok, D.J.; Blumberg, S.; et al. Predicted Impact of COVID-19 on Neglected Tropical Disease Programs and the Opportunity for Innovation. Clin. Infect. Dis. 2021, 72, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Bern, C.; Ghosh, D.; Rashid, M.; Molina, R.; Chowdhury, R.; Nath, R.; Ghosh, P.; Chapman, L.; Alim, A.; et al. Quantifying the Infectiousness of Post-Kala-Azar Dermal Leishmaniasis Toward Sand Flies. Clin. Infect. Dis. 2019, 69, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, N.K.; Datta, A.; Podder, I.; Das, A.; Sil, A. Therapeutic modalities in post kala-azar dermal leishmaniasis: A systematic review of the effectiveness and safety of the treatment options. Indian J. Dermatol. 2021, 66, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Erber, A.C.; Arana, B.; Ben Salah, A.; Bennis, I.; Boukthir, A.; Noriega, M.D.M.C.; Cissé, M.; Cota, G.F.; Handjani, F.; López-Carvajal, L.; et al. Patients’ preferences of cutaneous leishmaniasis treatment outcomes: Findings from an international qualitative study. PLoS Negl. Trop. Dis. 2020, 14, e0007996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olias-Molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Antileishmanial Drug Discovery and Development: Time to Reset the Model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLoS Negl. Trop. Dis. 2018, 12, e0006980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basselin, M.; Denise, H.; Coombs, G.H.; Barrett, M.P. Resistance to Pentamidine in Leishmania mexicana Involves Exclusion of the Drug from the Mitochondrion. Antimicrob. Agents Chemother. 2002, 46, 3731–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlaka, L.; Rosslee, M.-J.; Ozturk, M.; Kumar, S.; Parihar, S.P.; Brombacher, F.; Khalaf, A.I.; Carter, K.C.; Scott, F.J.; Suckling, C.J.; et al. Evaluation of minor groove binders (MGBs) as novel anti-mycobacterial agents and the effect of using non-ionic surfactant vesicles as a delivery system to improve their efficacy. J. Antimicrob. Chemother. 2017, 72, 3334–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieswetter, N.S.; Ozturk, M.; Hlaka, L.; Chia, J.E.; Nichol, R.J.O.; Cross, J.M.; McGee, L.; Tyson-Hirst, I.; Beveridge, R.; Brombacher, F.; et al. Intranasally administered S-MGB-364 displays antitubercular activity and modulates the host immune response to Mycobacterium tuberculosis infection. J. Antimicrob. Chemother. 2022, 77, 1061–1071. [Google Scholar] [CrossRef]
- Scott, F.J.; Khalaf, A.I.; Duffy, S.; Avery, V.M.; Suckling, C.J. Selective anti-malarial minor groove binders. Bioorganic Med. Chem. Lett. 2016, 26, 3326–3329. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.J.; Khalaf, A.; Giordani, F.; Wong, P.E.; Duffy, S.; Barrett, M.; Avery, V.; Suckling, C. An evaluation of Minor Groove Binders as anti-Trypanosoma brucei brucei therapeutics. Eur. J. Med. Chem. 2016, 116, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.J.; Nichol, R.J.; Khalaf, A.I.; Giordani, F.; Gillingwater, K.; Ramu, S.; Elliott, A.; Zuegg, J.; Duffy, P.; Rosslee, M.-J.; et al. An evaluation of Minor Groove Binders as anti-fungal and anti-mycobacterial therapeutics. Eur. J. Med. Chem. 2017, 136, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suckling, J.C.; Hunter, I.S.; Scott, F.J. Multitargeted anti-infective drugs: Resilience to resistance in the antimicrobial resistance era. Future Drug Discov. 2022, 4, FDD73. [Google Scholar] [CrossRef] [PubMed]
- Giordani, F.; Khalaf, A.I.; Gillingwater, K.; Munday, J.C.; de Koning, H.P.; Suckling, C.J.; Barrett, M.P.; Scott, F.J. Novel Minor Groove Binders Cure Animal African Trypanosomiasis in an In Vivo Mouse Model. J. Med. Chem. 2019, 62, 3021–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, L.; Browning, D.F.; Lemonidis, K.; Salih, T.; Hunter, I.S.; Suckling, C.J.; Tucker, N.P. Novel antibiotic mode of action by repression of promoter isomerisation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Anthony, N.G.; Breen, D.; Clarke, J.; Donoghue, G.; Drummond, A.J.; Ellis, E.M.; Gemmell, C.G.; Helesbeux, J.-J.; Hunter, I.S.; Khalaf, A.I.; et al. Antimicrobial Lexitropsins Containing Amide, Amidine, and Alkene Linking Groups. J. Med. Chem. 2007, 50, 6116–6125. [Google Scholar] [CrossRef] [Green Version]
- Brooke, D.P.; McGee, L.M.C.; Giordani, F.; Cross, J.M.; Khalaf, A.I.; Irving, C.; Gillingwater, K.; Shaw, C.D.; Carter, K.C.; Barrett, M.P.; et al. Truncated S-MGBs: Towards a parasite-specific and low aggregation chemotype. RSC Med. Chem. 2021, 12, 1391–1401. [Google Scholar] [CrossRef]
- Khalaf, A.I.; Bourdin, C.; Breen, D.; Donoghue, G.; Scott, F.J.; Suckling, C.J.; MacMillan, D.; Clements, C.; Fox, K.; Sekibo, D.A. Design, synthesis and antibacterial activity of minor groove binders: The role of non-cationic tail groups. Eur. J. Med. Chem. 2012, 56, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, A.I.; Anthony, N.; Breen, D.; Donoghue, G.; Mackay, S.P.; Scott, F.J.; Suckling, C.J. Amide isosteres in structure-activity studies of antibacterial minor groove binders. Eur. J. Med. Chem. 2011, 46, 5343–5355. [Google Scholar] [CrossRef]
- Anthony, N.G.; Johnston, B.F.; Khalaf, A.I.; MacKay, S.P.; Parkinson, J.A.; Suckling, C.J.; Waigh, R.D. Short Lexitropsin that Recognizes the DNA Minor Groove at 5′-ACTAGT-3′: Understanding the Role of Isopropyl-Thiazole. J. Am. Chem. Soc. 2004, 126, 11338–11349. [Google Scholar] [CrossRef]
- Alsaadi, M.; Italia, J.; Mullen, A.; Kumar, M.R.; Candlish, A.; Williams, R.; Shaw, C.; Al Gawhari, F.; Coombs, G.; Wiese, M.; et al. The efficacy of aerosol treatment with non-ionic surfactant vesicles containing amphotericin B in rodent models of leishmaniasis and pulmonary aspergillosis infection. J. Control. Release 2012, 160, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Carter, K.; Williams, R.A.M. Structure and Antiparasitic Activity Relationship of Alkylphosphocholine Analogues against Leishmania donovani. Microorganisms 2020, 8, 1117. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.D.; Lonchamp, J.; Downing, T.; Imamura, H.; Freeman, T.M.; Cotton, J.A.; Sanders, M.; Blackburn, G.; Dujardin, J.C.; Rijal, S.; et al. In vitro selection of miltefosine resistance in promastigotes of Leishmania donovani from Nepal: Genomic and metabolomic characterization. Mol. Microbiol. 2016, 99, 1134–1148. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S-MGB | L. donovani IC50 (μM) | T. b. brucei MIC (μM) | S. aureus MIC (μM) | hERG % |
|---|---|---|---|---|
| 1 | 3.5 ± 0.1 | 0.78 * | 50 | 65 ** |
| 206 | 2.4 ± 0.3 | 6.25 | >100 | 44 |
| 2 | 5.7 ± 0.4 | 1.56 * | 3.12 | 58 ** |
| 207 | 3.1 ± 1.0 | 12.5 | >100 | 45 |
| 3 | 5.1 ± 0.7 | <0.19 * | 0.78 | 64 ** |
| 219 | 1.0 ± 0.7 | 3.12 | >100 | 48 |
| S-MGB-ID | ΔTm (°C) |
|---|---|
| 1 | 11 |
| 206 | 1 |
| 2 | 15 |
| 207 | 1 |
| 3 | 17 |
| 219 | 12 |
| Time (min) | % Water (with 0.1% TFA) | % MeCN (with 0.1% TFA) |
|---|---|---|
| 0 | 70 | 30 |
| 25 | 50 | 50 |
| 30 | 70 | 30 |
| 35 | 70 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perieteanu, M.C.; McGee, L.M.C.; Shaw, C.D.; MacMillan, D.S.; Khalaf, A.I.; Gillingwater, K.; Beveridge, R.; Carter, K.C.; Suckling, C.J.; Scott, F.J. Selective Anti-Leishmanial Strathclyde Minor Groove Binders Using an N-Oxide Tail-Group Modification. Int. J. Mol. Sci. 2022, 23, 11912. https://doi.org/10.3390/ijms231911912
Perieteanu MC, McGee LMC, Shaw CD, MacMillan DS, Khalaf AI, Gillingwater K, Beveridge R, Carter KC, Suckling CJ, Scott FJ. Selective Anti-Leishmanial Strathclyde Minor Groove Binders Using an N-Oxide Tail-Group Modification. International Journal of Molecular Sciences. 2022; 23(19):11912. https://doi.org/10.3390/ijms231911912
Chicago/Turabian StylePerieteanu, Marina C., Leah M. C. McGee, Craig D. Shaw, Donna S. MacMillan, Abedawn I. Khalaf, Kirsten Gillingwater, Rebecca Beveridge, Katharine C. Carter, Colin J. Suckling, and Fraser J. Scott. 2022. "Selective Anti-Leishmanial Strathclyde Minor Groove Binders Using an N-Oxide Tail-Group Modification" International Journal of Molecular Sciences 23, no. 19: 11912. https://doi.org/10.3390/ijms231911912