
Conformational Preferences of Pyridone Adenine Dinucleotides from Molecular Dynamics Simulations


Abstract
:1. Introduction
2. Results
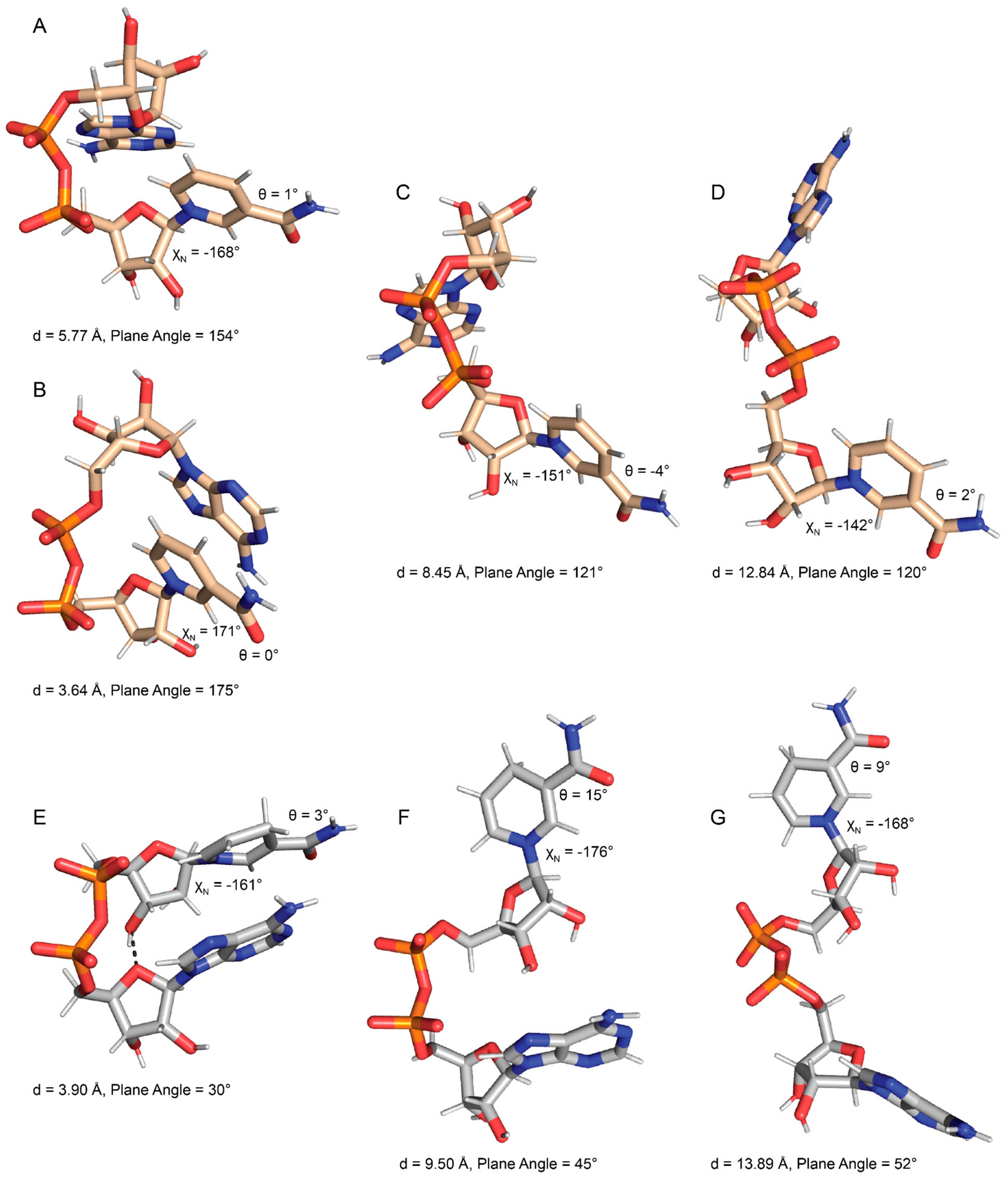
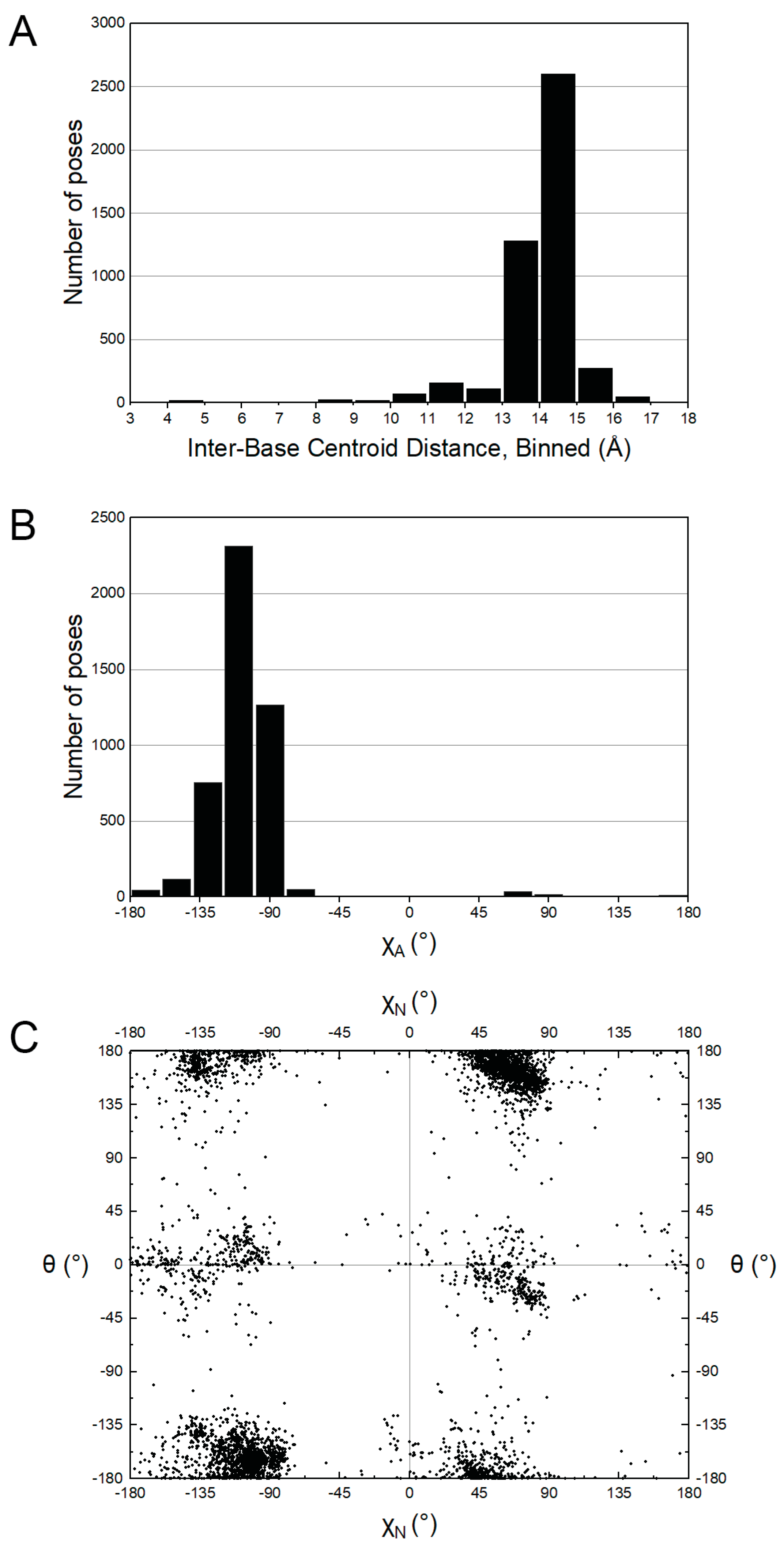
2.1. Conformational Preferences of NAD+ and NADH
2.2. Foldedness of ox-NADs
2.3. Pyridone Ribose Conformation of ox-NADs
2.4. Puckering of the Pyridone Ribose
2.5. Conformations of Protein-Bound NAD(H)
3. Discussion
4. Computational Methods
4.1. Molecular Dynamics (MD) Simulations of NAD+, NADH, and ox-NADs
4.2. Analysis of Conformations
4.3. PDB Data Mining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| ox-NAD | pyridone adenine dinucleotide |
References
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, H.B.S.; Williams, C.; King, S.J.; Allison, S.J. Nicotinamide adenine dinucleotide (NAD+): Essential redox metabolite, co-substrate and an anti-cancer and anti-ageing therapeutic target. Biochem. Soc. Trans. 2020, 48, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.A.G.; Li, H.; Johnson, M.; Sobrado, P. New frontiers in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2021, 699, 108765. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A.; Schramm, V.L. SIR2: The biochemical mechanism of NAD(+)-dependent protein deacetylation and ADP-ribosyl enzyme intermediates. Curr. Med. Chem. 2004, 11, 807–826. [Google Scholar] [CrossRef]
- Liu, C.; Yu, X. ADP-ribosyltransferases and poly ADP-ribosylation. Curr. Protein Pept. Sci. 2015, 16, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Oppenheimer, N.J.; Arnold, L.J.; Kaplan, N.O. A structure of pyridine nucleotides in solution. Proc. Nat. Acad. Sci. USA 1971, 68, 3200–3205. [Google Scholar] [CrossRef] [Green Version]
- Jardetzky, O.; Wade-Jardetzky, N.G. The conformation of pyridine dinucleotides in solution. J. Biol. Chem. 1966, 241, 85–91. [Google Scholar] [CrossRef]
- Meyer, W.L.; Mahler, H.R.; Baker, R.H. Nuclear magnetic resonance spectra and conformation of 1,4-dihydropyridines. Biochim. Et Biophys. Acta 1962, 64, 353–358. [Google Scholar] [CrossRef]
- Couprie, M.E.; Mérola, F.; Tauc, P.; Garzella, D.; Delboulbé, A.; Hara, T.; Billardon, M. First use of the UV Super-ACO free-electron laser: Fluorescence decays and rotational dynamics of the NADH coenzyme. Rev. Sci. Instrum. 1994, 65, 1485–1495. [Google Scholar] [CrossRef]
- Cadena-Caicedo, A.; Gonzalez-Cano, B.; Lopez-Arteaga, R.; Esturau-Escofet, N.; Peon, J. Ultrafast Fluorescence Signals from beta-Dihydronicotinamide Adenine Dinucleotide: Resonant Energy Transfer in the Folded and Unfolded Forms. J. Phys. Chem. B 2020, 124, 519–530. [Google Scholar] [CrossRef]
- Zheng, W.; Li, D.; Qu, J. Monitoring changes of cellular metabolism and microviscosity in vitro based on time-resolved endogenous fluorescence and its anisotropy decay dynamics. J. Biomed. Opt. 2010, 15, 037013. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.; Brown, B.; Hollis, D.; Walter, C. Some effects of environment on the folding of nicotinamide-adenine dinucleotides in aqueous solutions. Biochemistry 1972, 11, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Saenger, W.; Mühlegger, K.; Weimann, G. Crystal and molecular structure of the lithium salt of nicotinamide adenine dinucleotide dihydrate (NAD+, DPN+, cozymase, codehydraseI). J. Am. Chem. Soc. 1981, 103, 907–914. [Google Scholar] [CrossRef]
- Saenger, W.; Reddy, B.S.; Muhlegger, K.; Weimann, G. X-ray study of the lithium complex of NAD. Nature 1977, 267, 225–229. [Google Scholar] [CrossRef]
- Hayat, F.; Sonavane, M.; Makarov, M.V.; Trammell, S.A.J.; McPherson, P.; Gassman, N.R.; Migaud, M.E. The Biochemical Pathways of Nicotinamide-Derived Pyridones. Int. J. Mol. Sci. 2021, 22, 1145. [Google Scholar] [CrossRef]
- Foster, K.A.; Margraf, R.R.; Turner, D.A. NADH hyperoxidation correlates with enhanced susceptibility of aged rats to hypoxia. Neurobiol. Aging 2008, 29, 598–613. [Google Scholar] [CrossRef] [Green Version]
- Shetty, P.K.; Galeffi, F.; Turner, D.A. Nicotinamide pre-treatment ameliorates NAD(H) hyperoxidation and improves neuronal function after severe hypoxia. Neurobiol. Dis 2014, 62, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierzejewska, P.; Kunc, M.; Zabielska-Kaczorowska, M.A.; Kutryb-Zajac, B.; Pelikant-Malecka, I.; Braczko, A.; Jablonska, P.; Romaszko, P.; Koszalka, P.; Szade, J.; et al. An unusual nicotinamide derivative, 4-pyridone-3-carboxamide ribonucleoside (4PYR), is a novel endothelial toxin and oncometabolite. Exp. Mol. Med. 2021, 53, 1402–1412. [Google Scholar] [CrossRef]
- Koszalka, P.; Kutryb-Zajac, B.; Mierzejewska, P.; Tomczyk, M.; Wietrzyk, J.; Serafin, P.K.; Smolenski, R.T.; Slominska, E.M. 4-Pyridone-3-carboxamide-1-beta-D-ribonucleoside (4PYR)-A Novel Oncometabolite Modulating Cancer-Endothelial Interactions in Breast Cancer Metastasis. Int. J. Mol. Sci 2022, 23. [Google Scholar]
- Gelbin, A.; Schneider, B.; Clowney, L.; Hsieh, S.-H.; Olson, W.K.; Berman, H.M. Geometric Parameters in Nucleic Acids: Sugar and Phosphate Constituents. J. Am. Chem. Soc. 1996, 118, 519–529. [Google Scholar] [CrossRef]
- Il’icheva, I.A.; Polyakov, K.M.; Mikhailov, S.N. Strained Conformations of Nucleosides in Active Sites of Nucleoside Phosphorylases. Biomolecules 2020, 10, 552. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Mizutani, R. Structural biology of DNA (6-4) photoproducts formed by ultraviolet radiation and interactions with their binding proteins. Int. J. Mol. Sci. 2014, 15, 20321–20338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, C.S.; Lim, C. Efficient Binding of Flexible and Redox-Active Coenzymes by Oxidoreductases. ACS Catal. 2016, 6, 3469–3472. [Google Scholar] [CrossRef] [Green Version]
- Kuppuraj, G.; Sargsyan, K.; Hua, Y.H.; Merrill, A.R.; Lim, C. Linking distinct conformations of nicotinamide adenine dinucleotide with protein fold/function. J. Phys. Chem. B 2011, 115, 7932–7939. [Google Scholar] [CrossRef]
- Luo, M.; Gamage, T.T.; Arentson, B.W.; Schlasner, K.N.; Becker, D.F.; Tanner, J.J. Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. J. Biol. Chem. 2016, 291, 24065–24075. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.J. Structural Biology of Proline Catabolic Enzymes. Antioxid. Redox. Signal. 2019, 30, 650–673. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.E.; Tanner, J.J. Molecular dynamics simulations of NAD+ in solution. J. Amer. Chem. Soc. 1999, 121, 8637–8644. [Google Scholar] [CrossRef]
- Pavelites, J.J.; Gao, J.; Bash, P.A.; Mackerell, A.D., Jr. A Molecular Mechanics Force Field for NAD+, NADH, and the Pyrophosphate Groups of Nucleotides. J. Comput. Chem. 1997, 18, 221–239. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2018, 1, 5068. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rossum, G. Python reference manual; Centrum Wiskunde & Informatica (CWI): Amsterdam, The Netherlands, 1995. [Google Scholar]
- Hagberg, A.; Swart, P.; S. Chult, D. Exploring network structure, dynamics, and function using networkx. In Proceedings of the SCIPY 2008, Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference (SciPy 2016), Austin, TX, USA, 11–17 July 2016. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Brunger, A.T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007, 2, 2728–2733. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
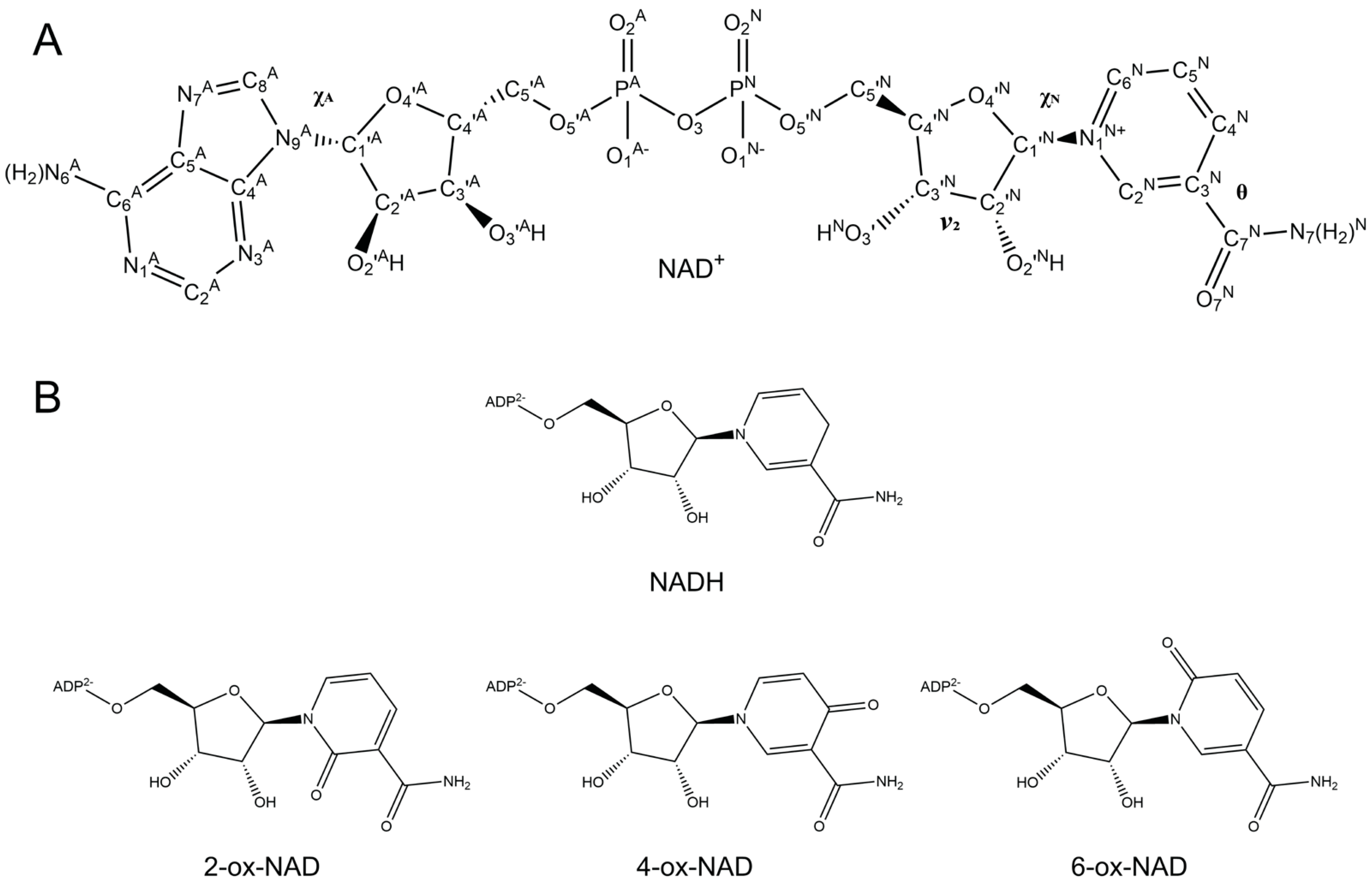
| Dihedral Angle | Adenine (A) or Nicotinamide (N) | Atoms Associated |
|---|---|---|
| θ | N | C4N-C3N-C7N-N7N |
| ν2 | N | C1′N-C2′N-C3′N-C4′N |
| χN | N | O4′N-C1′N-N1N-C2N |
| χA | A | C4A-N9A-C1′A-O4′A |
| NAD+ | NADH | 2-ox-NAD | 4-ox-NAD | 6-ox-NAD | |
|---|---|---|---|---|---|
| Simulation time per simulation (ns) | 100 | 100 | 100 | 100 | 100 |
| Number of simulations | 10 | 10 | 10 | 10 | 10 |
| Time step (fs) | 2 | 2 | 2 | 2 | 2 |
| Number of dinucleotide atoms | 70 | 71 | 70 | 70 | 70 |
| Number of water molecules | 1221 | 1115 | 1227 | 1234 | 1220 |
| Number of Na+ ions | 1 | 2 | 2 | 2 | 2 |
| Total number of atoms in system | 3734 | 3418 | 3753 | 3774 | 3732 |
| Ensemble | NPT | NPT | NPT | NPT | NPT |
| Pressure (bar) | 1 | 1 | 1 | 1 | 1 |
| Temperature (K) | 300 | 300 | 300 | 300 | 300 |
| Box size (nm3) | 40.64 | 35.48 | 40.93 | 41.69 | 40.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckley, D.P.; Migaud, M.E.; Tanner, J.J. Conformational Preferences of Pyridone Adenine Dinucleotides from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 11866. https://doi.org/10.3390/ijms231911866
Buckley DP, Migaud ME, Tanner JJ. Conformational Preferences of Pyridone Adenine Dinucleotides from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2022; 23(19):11866. https://doi.org/10.3390/ijms231911866
Chicago/Turabian StyleBuckley, David P., Marie E. Migaud, and John J. Tanner. 2022. "Conformational Preferences of Pyridone Adenine Dinucleotides from Molecular Dynamics Simulations" International Journal of Molecular Sciences 23, no. 19: 11866. https://doi.org/10.3390/ijms231911866