

Analysis of Host–Bacteria Protein Interactions Reveals Conserved Domains and Motifs That Mediate Fundamental Infection Pathways

Abstract
:1. Introduction
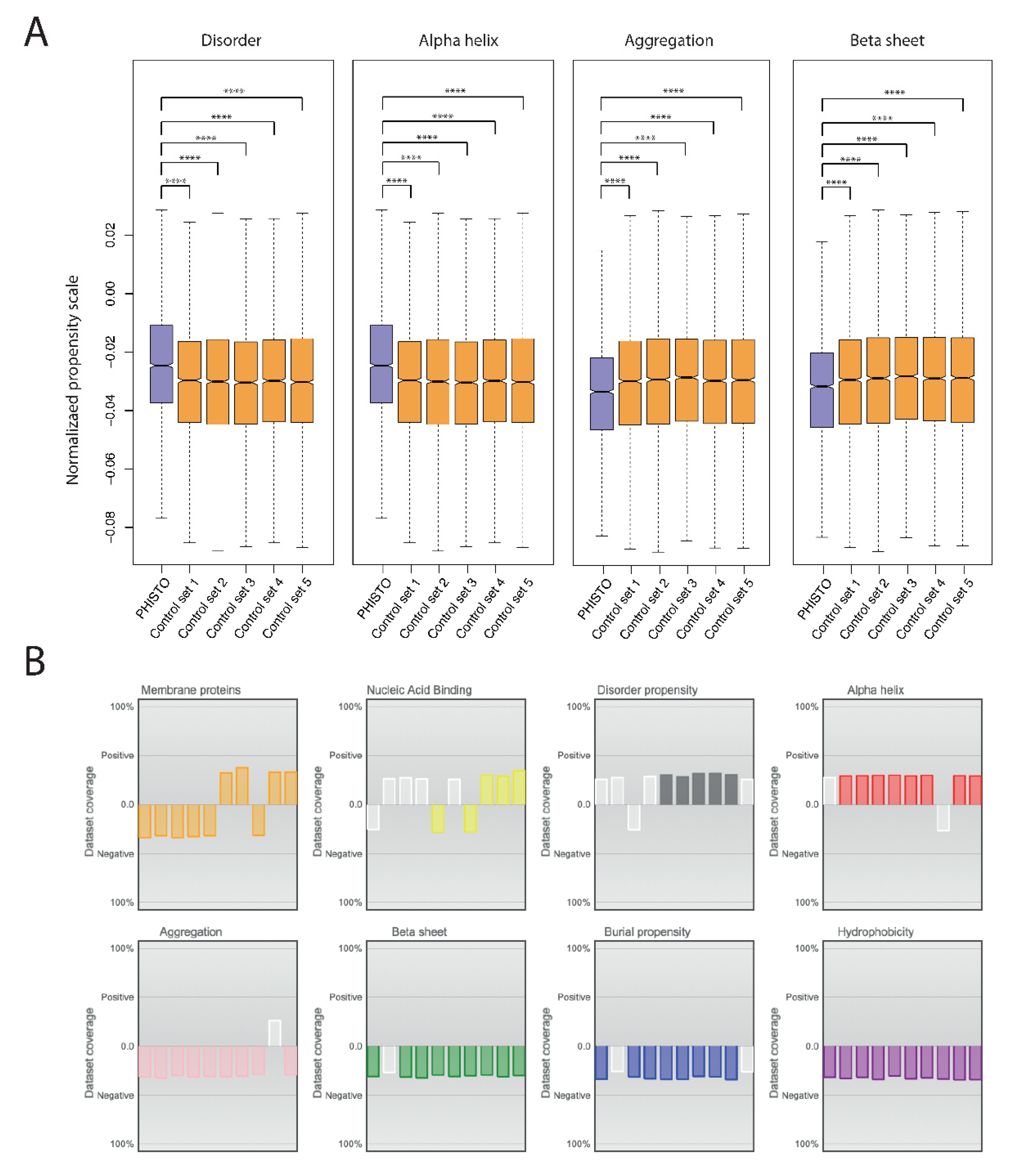
2. Results and Discussion
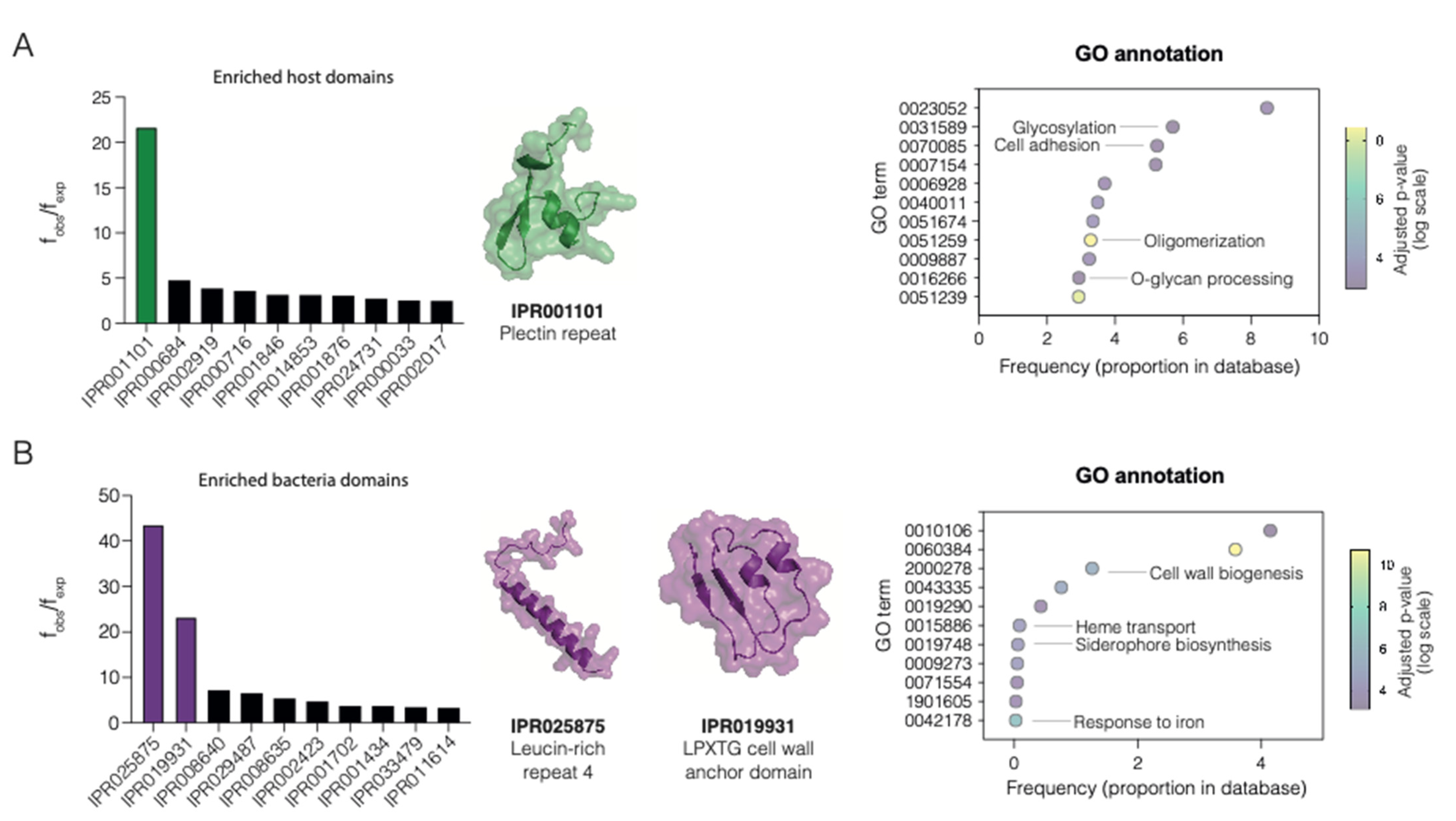
2.1. Enriched Domains in Host–Pathogen PPIs
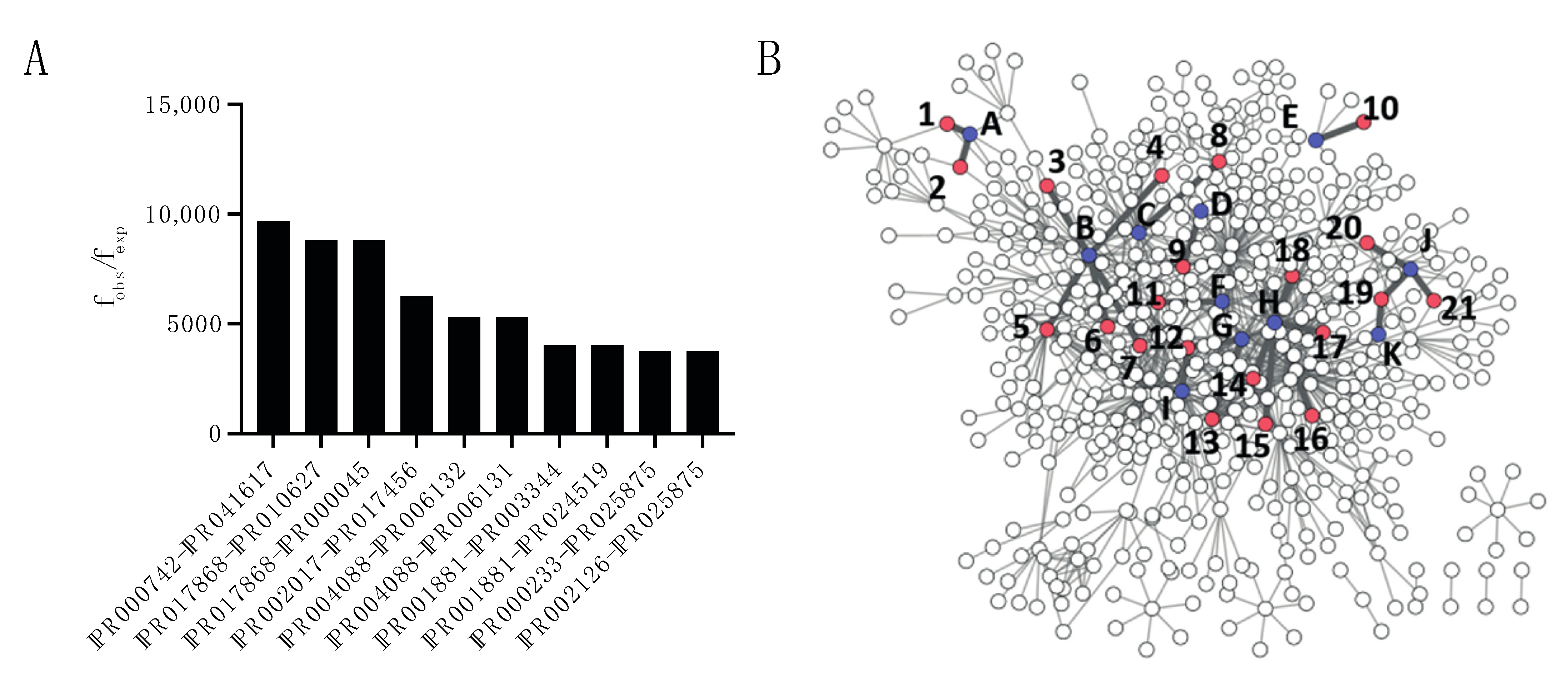
2.2. Domain–Domain Associations in Host–Pathogen PPIs
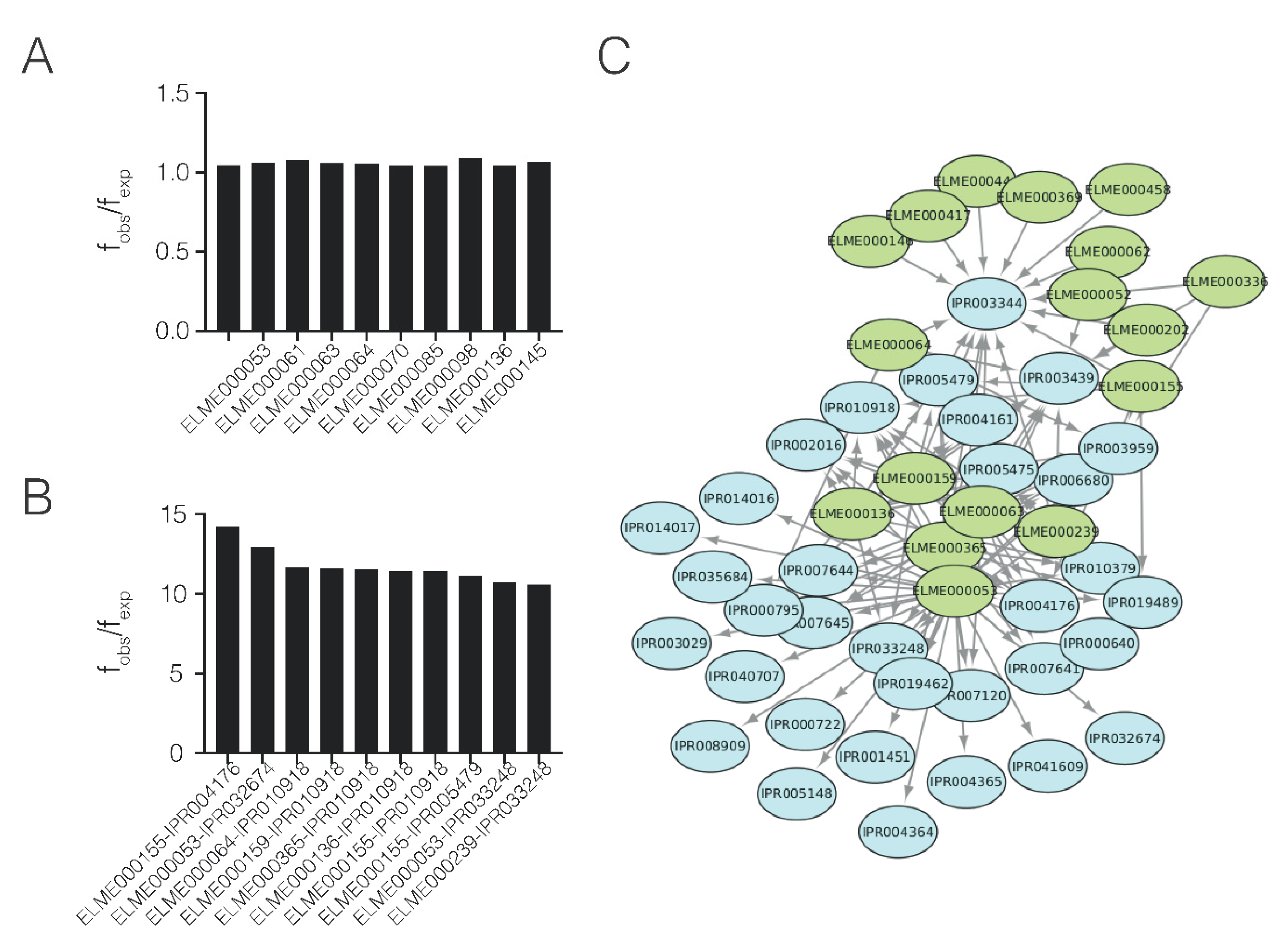
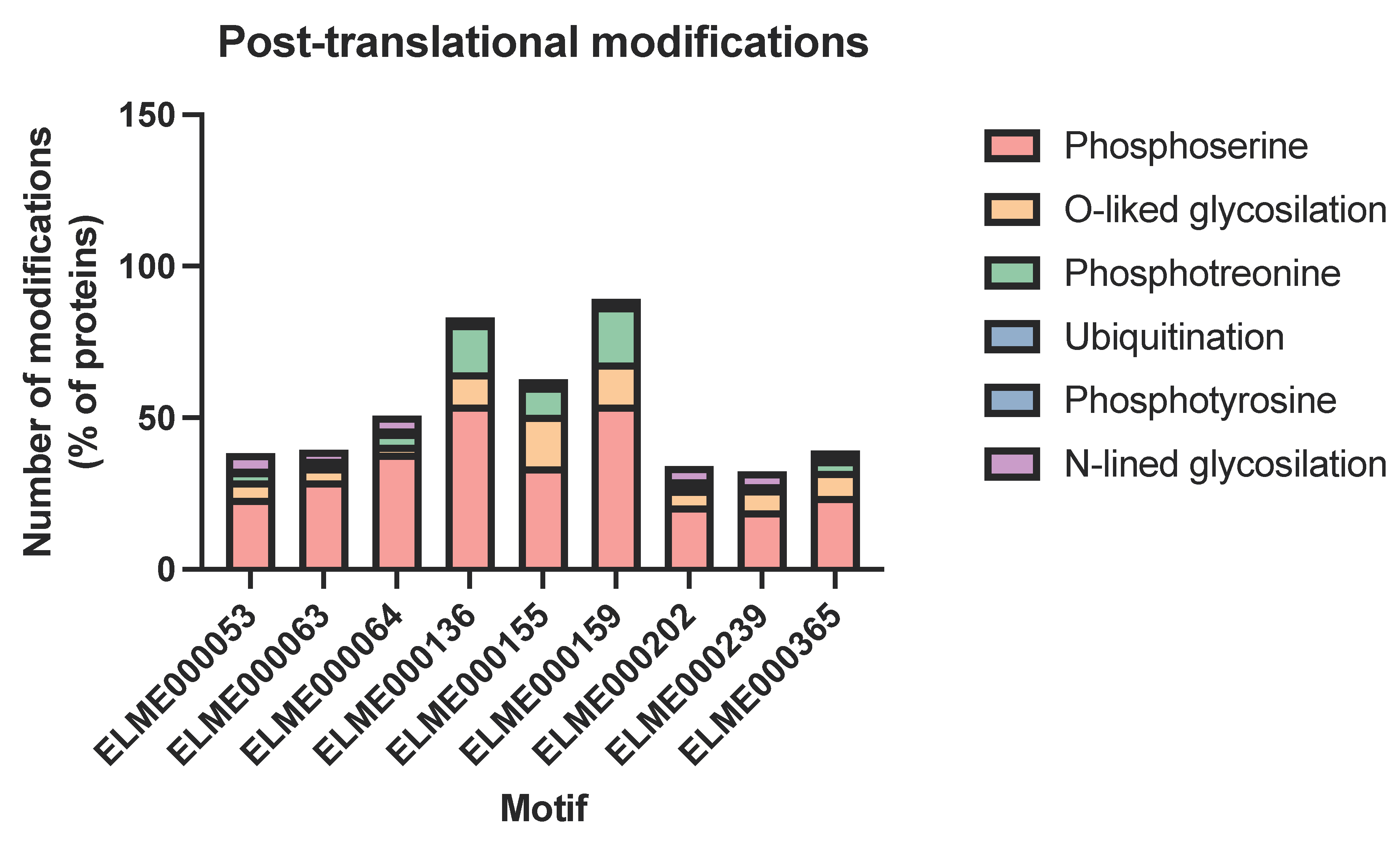
2.3. Domain–Motif Associations in Host–Pathogen PPIs
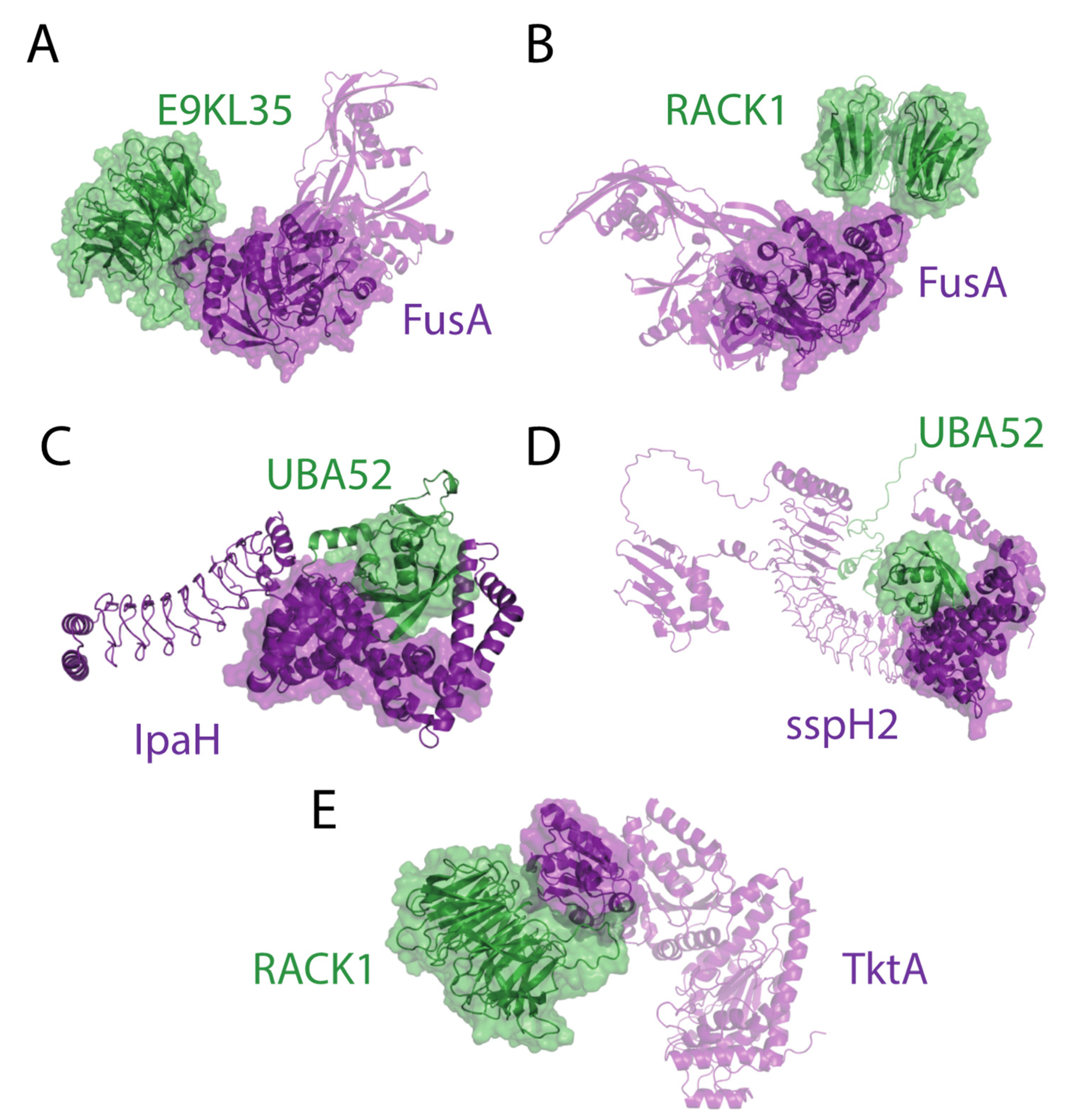
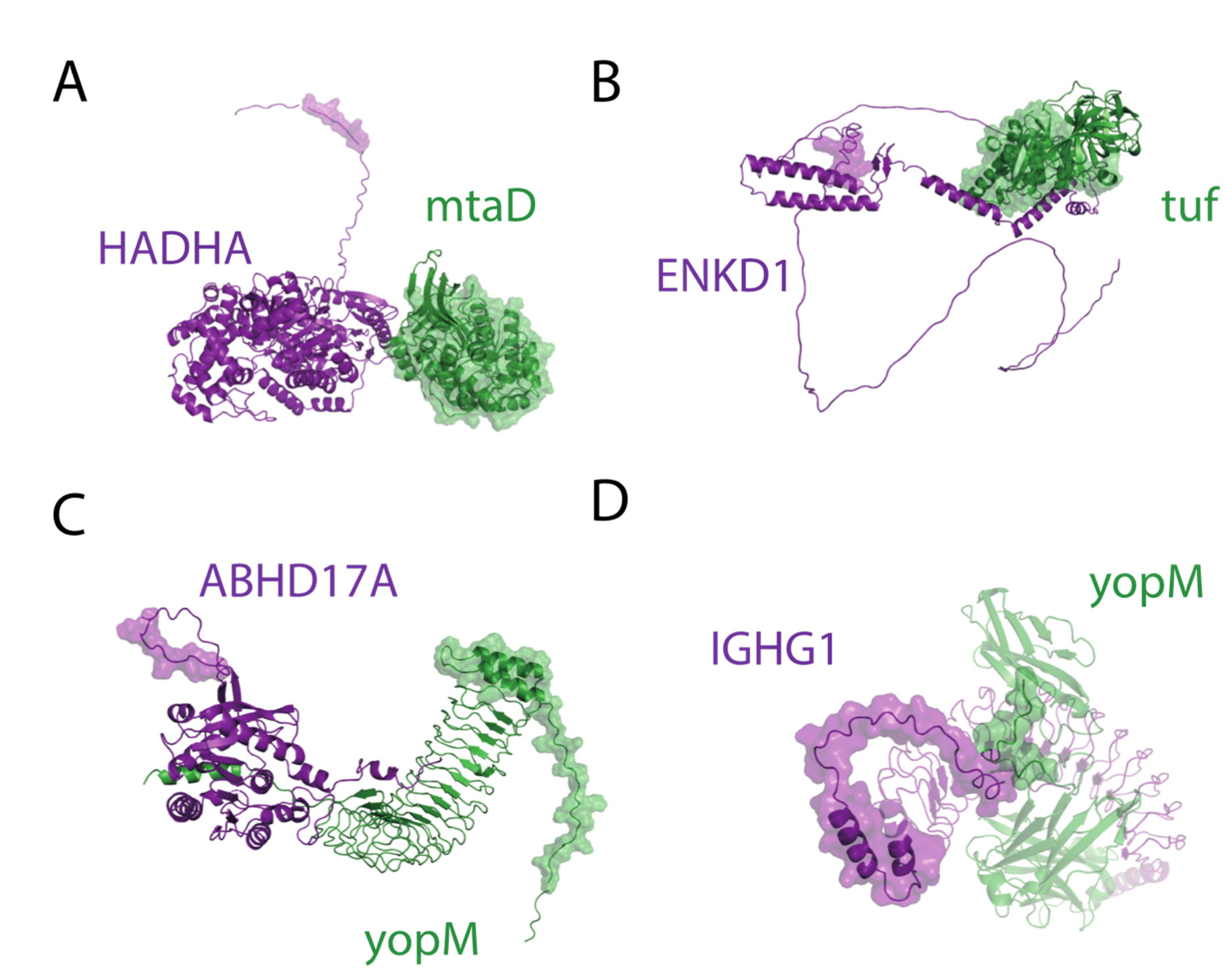
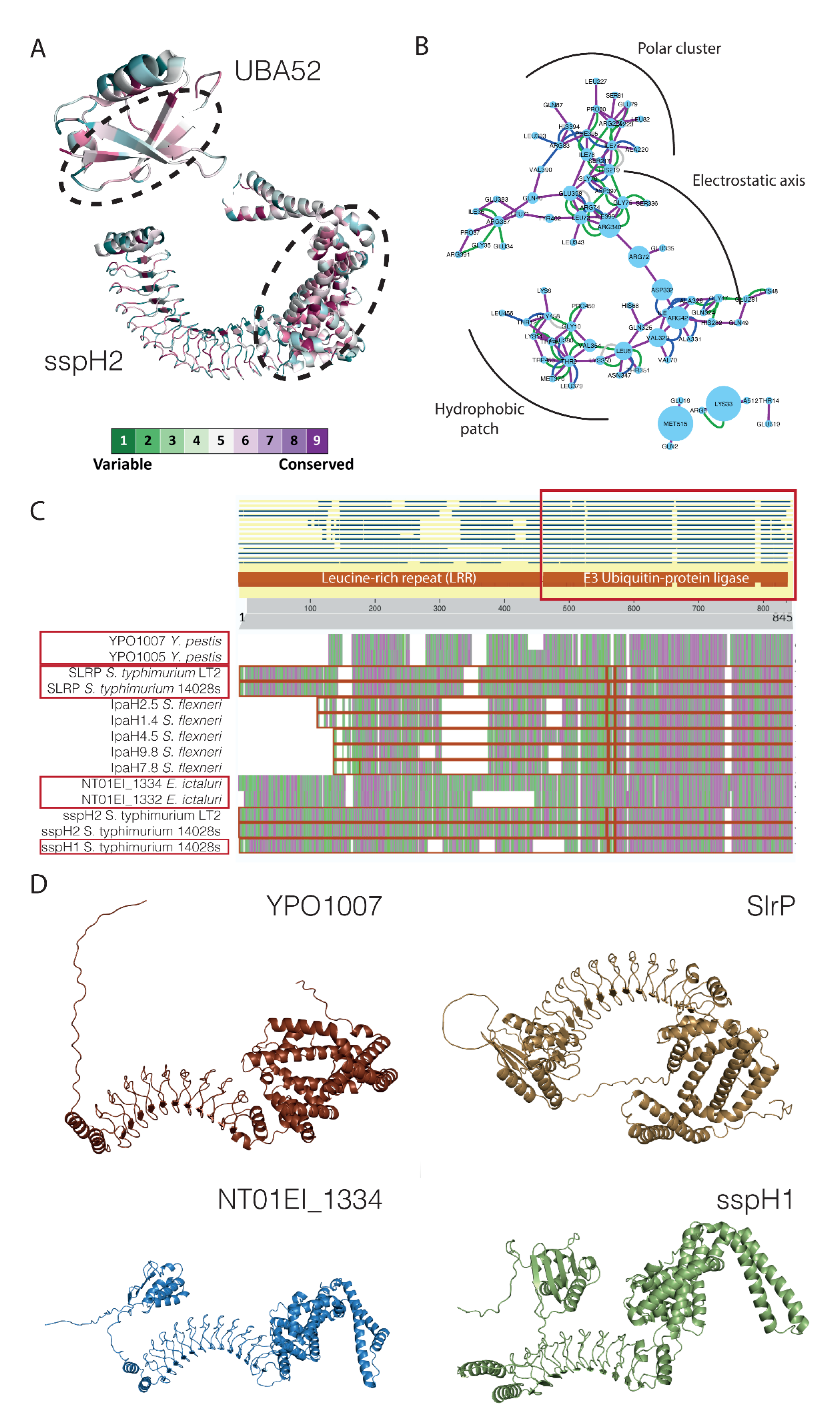
2.4. Structural Analysis of DD and DM Associations
2.5. Virulence Factors with Ubiquitin–Protein Ligase Activity as a Case Example
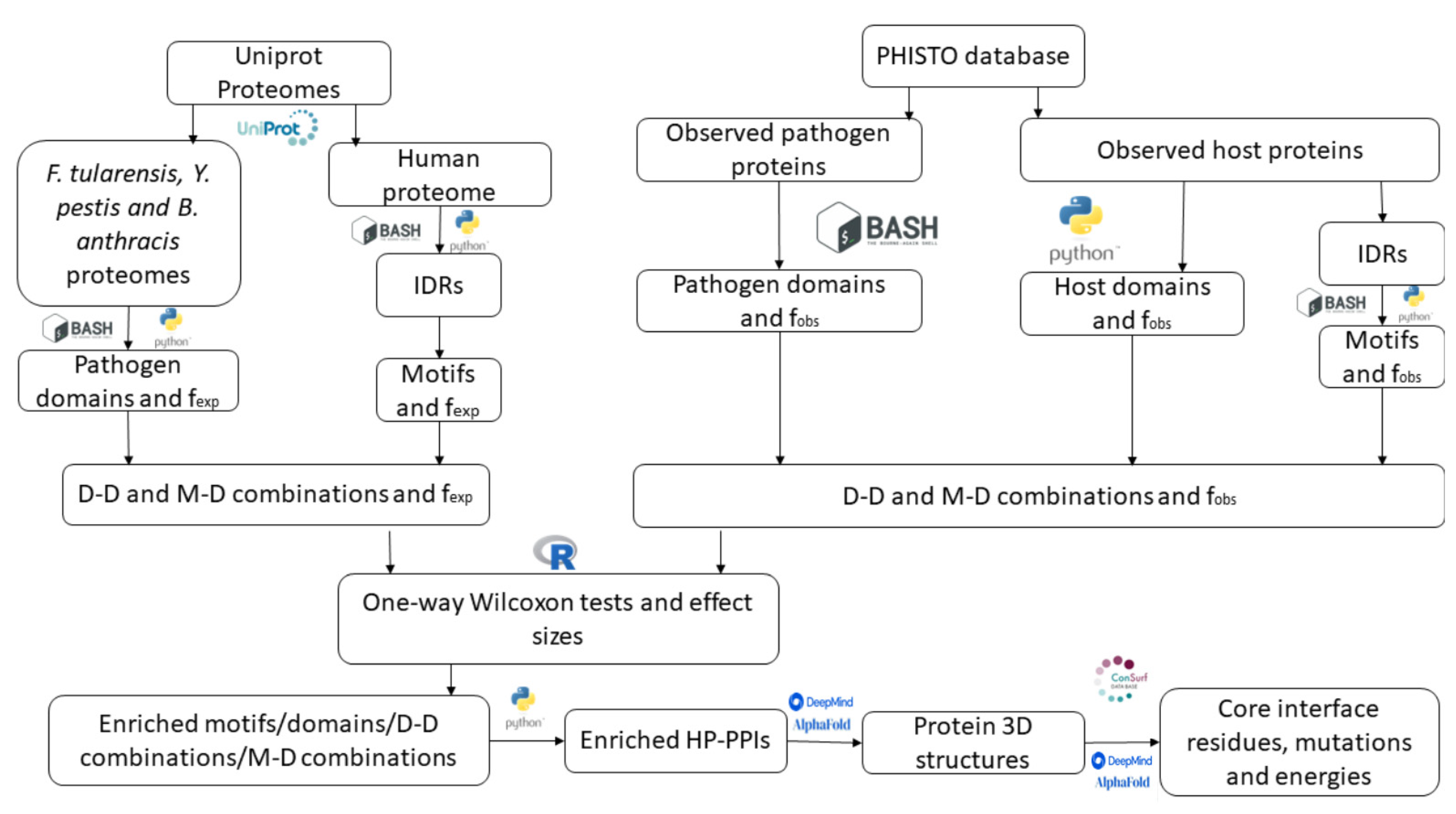
3. Materials and Methods
3.1. Databases
3.2. Domain and Motif Scanning
3.3. Domain–Domain (DD) and Domain–Motif (DM) Interactions
3.4. Domain and Motif Enrichment Analysis
3.5. Gene Ontology Analysis
3.6. Structural and Conservation Analysis of Selected PPIs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Crua Asensio, N.; Macho Rendon, J.; Torrent Burgas, M. Time-Resolved Transcriptional Profiling of Epithelial Cells Infected by Intracellular Acinetobacter baumannii. Microorganisms 2021, 9, 354. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Torrent Burgas, M. A Coordinated Response at The Transcriptome and Interactome Level is Required to Ensure Uropathogenic Escherichia coli Survival during Bacteremia. Microorganisms 2019, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Crua Asensio, N.; Munoz Giner, E.; de Groot, N.S.; Torrent Burgas, M. Centrality in the host-pathogen interactome is associated with pathogen fitness during infection. Nat. Commun. 2017, 8, 14092. [Google Scholar] [CrossRef] [PubMed]
- Akhter, Y.; Hussain, R. Protein-protein complexes as targets for drug discovery against infectious diseases. Adv. Protein. Chem. Struct. Biol. 2020, 121, 237–251. [Google Scholar]
- Carro, L. Protein-protein interactions in bacteria: A promising and challenging avenue towards the discovery of new antibiotics. Beilstein. J. Org. Chem. 2018, 14, 2881–2896. [Google Scholar] [CrossRef]
- Kahan, R.; Worm, D.J.; de Castro, G.V.; Ng, S.; Barnard, A. Modulators of protein-protein interactions as antimicrobial agents. RSC Chem. Biol. 2021, 2, 387–409. [Google Scholar] [CrossRef]
- Cossar, P.J.; Lewis, P.J.; McCluskey, A. Protein-protein interactions as antibiotic targets: A medicinal chemistry perspective. Med. Res. Rev. 2020, 40, 469–494. [Google Scholar] [CrossRef]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host-pathogen interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef]
- Durmus Tekir, S.; Cakir, T.; Ardic, E.; Sayilirbas, A.S.; Konuk, G.; Konuk, M.; Sariyer, H.; Ugurlu, A.; Karadeniz, I.; Ozgur, A.; et al. PHISTO: Pathogen-host interaction search tool. Bioinformatics 2013, 29, 1357–1358. [Google Scholar] [CrossRef]
- Cook, H.V.; Doncheva, N.T.; Szklarczyk, D.; von Mering, C.; Jensen, L.J. Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses 2018, 10, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583-7. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bhatia, V.; Singh, S.; Bhatnagar, S. MorCVD: A Unified Database for Host-Pathogen Protein-Protein Interactions of Cardiovascular Diseases Related to Microbes. Sci. Rep. 2019, 9, 4039. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.K.; Light, S.; Hedin, L.; Elofsson, A. Quantitative assessment of the structural bias in protein-protein interaction assays. Proteomics 2008, 8, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bader, J.S. Precision and recall estimates for two-hybrid screens. Bioinformatics 2009, 25, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jedynak, B.M.; Bader, J.S. Where have all the interactions gone? Estimating the coverage of two-hybrid protein interaction maps. PLoS Comput. Biol. 2007, 3, e214. [Google Scholar] [CrossRef]
- Finn, R.D.; Miller, B.L.; Clements, J.; Bateman, A. iPfam: A database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014, 42, D364-73. [Google Scholar] [CrossRef]
- Mosca, R.; Ceol, A.; Stein, A.; Olivella, R.; Aloy, P. 3did: A catalog of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2014, 42, D374-9. [Google Scholar] [CrossRef]
- Yellaboina, S.; Tasneem, A.; Zaykin, D.V.; Raghavachari, B.; Jothi, R. DOMINE: A comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res. 2011, 39, D730-5. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pawson, T.; Raina, M.; Nash, P. Interaction domains: From simple binding events to complex cellular behavior. FEBS Lett. 2002, 513, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A million peptide motifs for the molecular biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Meszaros, B.; Samano-Sanchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Ord, M.; Nagpal, A.; et al. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 2022, 50, D497–D508. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Wiche, G.; Winter, L. Plectin isoforms as organizers of intermediate filament cytoarchitecture. Bioarchitecture 2011, 1, 14–20. [Google Scholar] [CrossRef]
- Schweppe, D.K.; Harding, C.; Chavez, J.D.; Wu, X.; Ramage, E.; Singh, P.K.; Manoil, C.; Bruce, J.E. Host-Microbe Protein Interactions during Bacterial Infection. Chem. Biol. 2015, 22, 1521–1530. [Google Scholar] [CrossRef]
- Ireton, K.; Mortuza, R.; Gyanwali, G.C.; Gianfelice, A.; Hussain, M. Role of internalin proteins in the pathogenesis of Listeria monocytogenes. Mol. Microbiol. 2021, 116, 1407–1419. [Google Scholar] [CrossRef]
- Siegel, S.D.; Reardon, M.E.; Ton-That, H. Anchoring of LPXTG-Like Proteins to the Gram-Positive Cell Wall Envelope. Curr. Top Microbiol. Immunol. 2017, 404, 159–175. [Google Scholar]
- Fang, H.; Gough, J. DcGO: Database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 2013, 41, D536-44. [Google Scholar] [CrossRef] [PubMed]
- Klus, P.; Bolognesi, B.; Agostini, F.; Marchese, D.; Zanzoni, A.; Tartaglia, G.G. The cleverSuite approach for protein characterization: Predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics 2014, 30, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska-Mieszkowska, S.; Zolkiewski, M. Hsp100 Molecular Chaperone ClpB and Its Role in Virulence of Bacterial Pathogens. Int. J. Mol. Sci. 2021, 22, 5319. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.R.; Breitkreutz, A.; Chenal, A.; Sansonetti, P.J.; Parsot, C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 2007, 1, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Alegado, R.A.; Campbell, M.C.; Chen, W.C.; Slutz, S.S.; Tan, M.W. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell Microbiol. 2003, 5, 435–444. [Google Scholar] [CrossRef]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res 2020, 48, W140–W146. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein. Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Guharoy, M.; Chakrabarti, P. Conservation and relative importance of residues across protein-protein interfaces. Proc. Natl. Acad. Sci. USA 2005, 102, 15447–15452. [Google Scholar] [CrossRef]
- de Groot, N.S.; Torrent Burgas, M. Bacteria use structural imperfect mimicry to hijack the host interactome. PLoS Comput. Biol. 2020, 16, e1008395. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Identifier (Host) | InterPro Identifier (Host) | Description | Network Identifier (Pathogen) | InterPro Identifier (Pathogen) | Description |
|---|---|---|---|---|---|
| A | IPR001715 | Calponin homology domain | 1 | IPR014016 | UvrD-like helicase, ATP-binding domain |
| 2 | IPR014017 | UvrD-like DNA helicase, C-terminal | |||
| B | IPR000504 | RNA recognition motif domain | 3 | IPR003343 | Bacterial Ig-like domain, group 2 |
| 4 | IPR032781 | ABC-transporter extension domain | |||
| 5 | IPR003344 | Big-1 domain | |||
| 6 | IPR002314 | Aminoacyl-tRNA synthetase, class II (G/P/S/T) | |||
| 7 | IPR018392 | LysM domain | |||
| C | IPR003961 | Fibronectin type III | 8 | IPR019931 | LPXTG cell wall anchor domain |
| D | IPR001245 | S-T/Y-protein kinase | 9 | IPR010918 | PurM-like, C-terminal domain |
| E | IPR000626 | Ubiquitin-like domain | 10 | IPR029487 | Novel E3 ligase domain |
| F | IPR001781 | Zinc finger, LIM-type | 11 | IPR006680 | Amidohydrolase-related |
| G | IPR001007 | VWFC domain | 12 | IPR001036 | Acriflavin resistance protein |
| 13 | IPR007642 | RNA polymerase Rpb2, domain 2 | |||
| H | IPR001680 | WD40 repeat | 14 | IPR004161 | Translation elongation factor EFTu-like, domain 2 |
| 15 | IPR005475 | Transketolase-like, pyrimidine-binding domain | |||
| 16 | IPR033248 | Transketolase, C-terminal | |||
| 17 | IPR005474 | Transketolase, N-terminal | |||
| 18 | IPR000795 | Translational (tr)-type GTP-binding domain | |||
| I | IPR001881 | EGF-like calcium-binding domain | 12 | IPR001036 | Acriflavin resistance protein |
| J | IPR000157 | Toll/interleukin-1 receptor homology (TIR) domain | 19 | IPR001029 | Flagellin, N-terminal domain |
| 20 | IPR002423 | Chaperonin Cpn60/GroEL/TCP-1 family | |||
| 21 | IPR001702 | Porin, Gram-negative type | |||
| K | IPR000488 | Death domain | 19 | IPR001029 | Flagellin, N-terminal domain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez Borrego, J.; Torrent Burgas, M. Analysis of Host–Bacteria Protein Interactions Reveals Conserved Domains and Motifs That Mediate Fundamental Infection Pathways. Int. J. Mol. Sci. 2022, 23, 11489. https://doi.org/10.3390/ijms231911489
Gómez Borrego J, Torrent Burgas M. Analysis of Host–Bacteria Protein Interactions Reveals Conserved Domains and Motifs That Mediate Fundamental Infection Pathways. International Journal of Molecular Sciences. 2022; 23(19):11489. https://doi.org/10.3390/ijms231911489
Chicago/Turabian StyleGómez Borrego, Jordi, and Marc Torrent Burgas. 2022. "Analysis of Host–Bacteria Protein Interactions Reveals Conserved Domains and Motifs That Mediate Fundamental Infection Pathways" International Journal of Molecular Sciences 23, no. 19: 11489. https://doi.org/10.3390/ijms231911489