

Cellular Biogenetic Law and Its Distortion by Protein Interactions: A Possible Unified Framework for Cancer Biology and Regenerative Medicine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
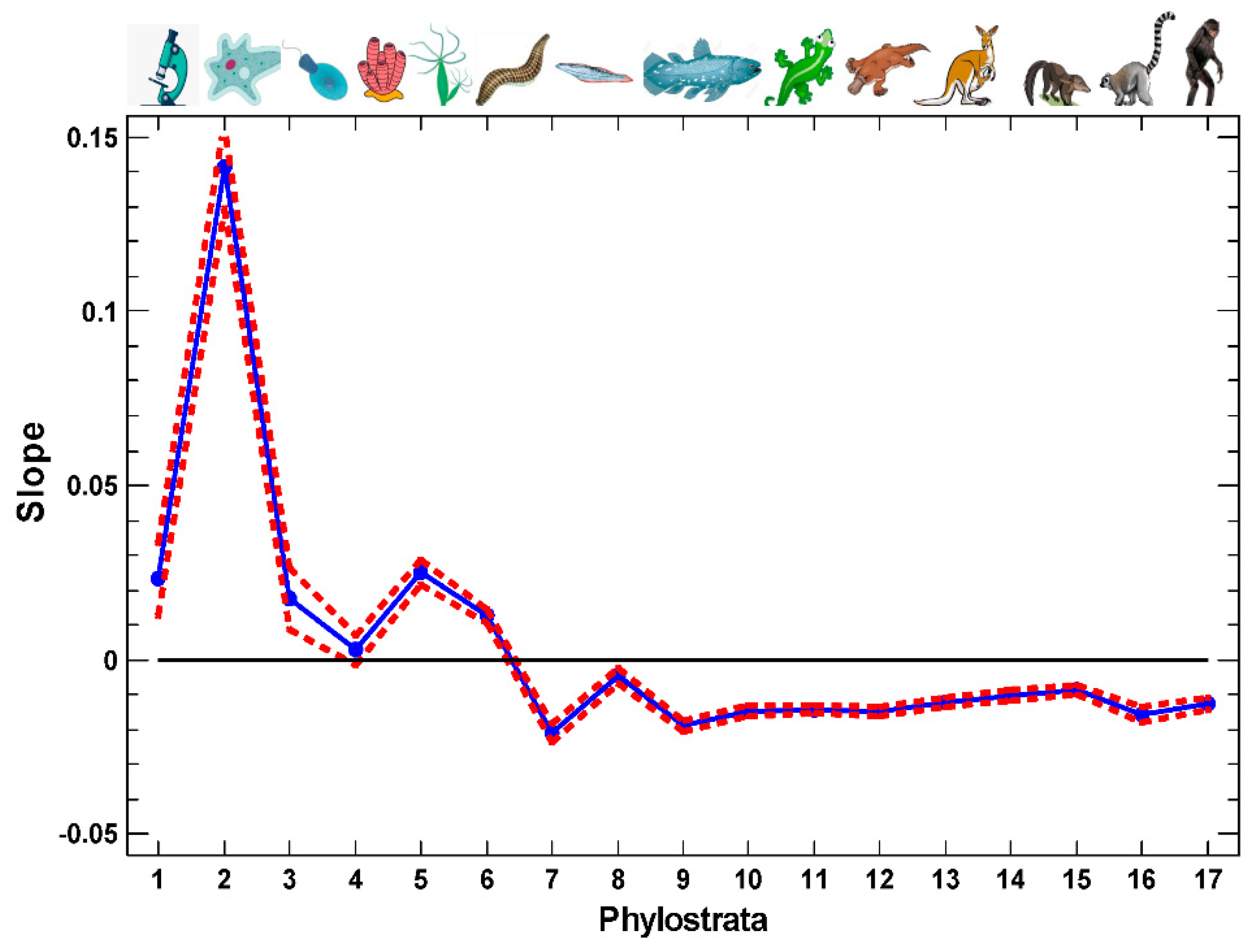
2.1. The Proof of Concept
2.2. Artificial Ontogenetic Reversal
2.3. Ab Ovo
2.4. Regulatory Gene Groups
2.5. The Strength of Old and New Ties
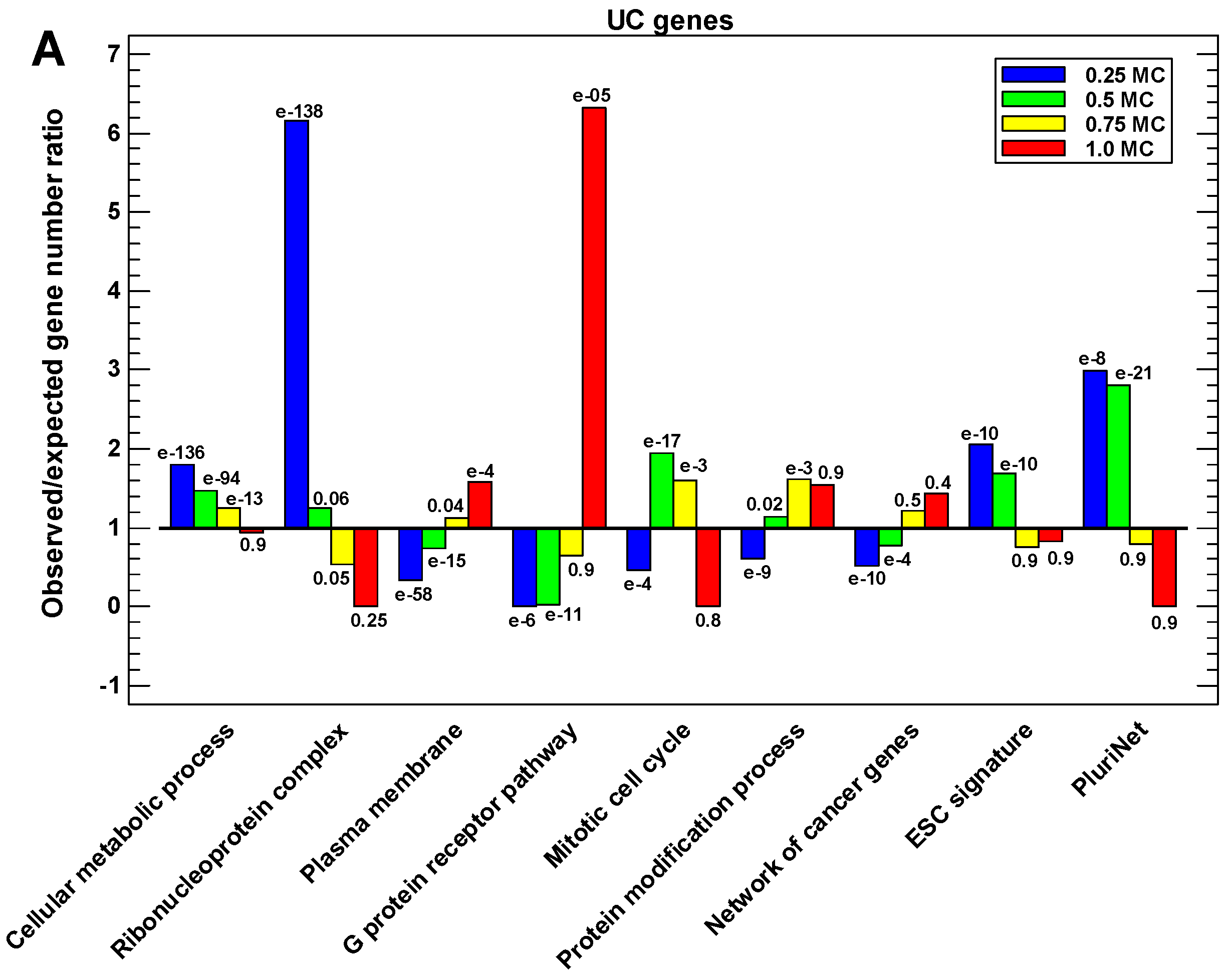
2.6. Functional Analysis of the Proteins in Different One-Step Interactome Neighborhoods
3. Discussion
3.1. Cellular Biogenetic Law
3.2. Modification of Development
3.3. A Unified Framework for Cancer Biology and Regenerative Medicine
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abzhanov, A. Von Baer’s Law for the Ages: Lost and Found Principles of Developmental Evolution. Trends Genet. TIG 2013, 29, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Dobreva, M.P.; Camacho, J.; Abzhanov, A. Time to Synchronize Our Clocks: Connecting Developmental Mechanisms and Evolutionary Consequences of Heterochrony. J. Exp. Zoolog. B Mol. Dev. Evol. 2022, 338, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Uesaka, M.; Irie, N. Beyond Recapitulation: Past, Present, and Future. J. Exp. Zoolog. B Mol. Dev. Evol. 2022, 338, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.S.; Miller, W.B. Terminal Addition in a Cellular World. Prog. Biophys. Mol. Biol. 2018, 135, 1–10. [Google Scholar] [CrossRef]
- Uesaka, M.; Kuratani, S.; Takeda, H.; Irie, N. Recapitulation-like Developmental Transitions of Chromatin Accessibility in Vertebrates. Zool. Lett. 2019, 5, 33. [Google Scholar] [CrossRef]
- Davies, P.C.W.; Lineweaver, C.H. Cancer Tumors as Metazoa 1.0: Tapping Genes of Ancient Ancestors. Phys. Biol. 2011, 8, 015001. [Google Scholar] [CrossRef]
- Vincent, M. Cancer: A De-Repression of a Default Survival Program Common to All Cells?: A Life-History Perspective on the Nature of Cancer. BioEssays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 72–82. [Google Scholar] [CrossRef]
- Greaves, M. Evolutionary Determinants of Cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef]
- Bussey, K.J.; Davies, P.C.W. Reverting to Single-Cell Biology: The Predictions of the Atavism Theory of Cancer. Prog. Biophys. Mol. Biol. 2021, 165, 49–55. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer Progression as a Sequence of Atavistic Reversions. BioEssays News Rev. Mol. Cell. Dev. Biol. 2021, 43, e2000305. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered Interactions between Unicellular and Multicellular Genes Drive Hallmarks of Transformation in a Diverse Range of Solid Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Somatic Mutations in Early Metazoan Genes Disrupt Regulatory Links between Unicellular and Multicellular Genes in Cancer. eLife 2019, 8, e40947. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.J.; Cisneros, L.H.; Lineweaver, C.H.; Davies, P.C.W. Ancestral Gene Regulatory Networks Drive Cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 6160–6162. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary Framework of the Human Interactome: Unicellular and Multicellular Giant Clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Cell-Cycle Dependence of Transcriptome Gene Modules: Comparison of Regression Lines. FEBS J. 2020, 287, 4427–4439. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting Cancer’s Weaknesses (Not Its Strengths): Therapeutic Strategies Suggested by the Atavistic Model. BioEssays News Rev. Mol. Cell. Dev. Biol. 2014, 36, 827–835. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the Evolution of Multicellularity Set the Stage for Cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Whole-Genome Duplications in Evolution, Ontogeny, and Pathology: Complexity and Emergency Reserves. Mol. Biol. 2021, 55, 813–827. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Boddy, A.M.; Jansen, G.; Hibner, U.; Hochberg, M.E.; Maley, C.C.; Wilkinson, G.S. Cancer across the Tree of Life: Cooperation and Cheating in Multicellularity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140219. [Google Scholar] [CrossRef]
- Scudellari, M. How IPS Cells Changed the World. Nature 2016, 534, 310–312. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Shilina, M.A.; Anatskaya, O.V.; Alekseenko, L.L.; Fridlyanskaya, I.I.; Krasnenko, A.; Kim, A.; Korostin, D.; Ilynsky, V.; Elmuratov, A.; et al. Molecular Genetic Analysis of Human Endometrial Mesenchymal Stem Cells That Survived Sublethal Heat Shock. Stem Cells Int. 2017, 2017, 2362630. [Google Scholar] [CrossRef] [PubMed]
- Moroz, L.L.; Romanova, D.Y. Selective Advantages of Synapses in Evolution. Front. Cell Dev. Biol. 2021, 9, 726563. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US); Institute of Medicine (US) Committee on the Biological and Biomedical Applications of Stem Cell Research. Stem Cells and the Future of Regenerative Medicine; National Academies Press: Washington, DC, USA, 2002; ISBN 978-0-309-07630-2. [Google Scholar]
- Vinogradov, A.E.; Anatskaya, O.V. Growth of Biological Complexity from Prokaryotes to Hominids Reflected in the Human Genome. Int. J. Mol. Sci. 2021, 22, 11640. [Google Scholar] [CrossRef] [PubMed]
- Sha, Q.-Q.; Zheng, W.; Wu, Y.-W.; Li, S.; Guo, L.; Zhang, S.; Lin, G.; Ou, X.-H.; Fan, H.-Y. Dynamics and Clinical Relevance of Maternal MRNA Clearance during the Oocyte-to-Embryo Transition in Humans. Nat. Commun. 2020, 11, 4917. [Google Scholar] [CrossRef]
- Talbert, P.B.; Meers, M.P.; Henikoff, S. Old Cogs, New Tricks: The Evolution of Gene Expression in a Chromatin Context. Nat. Rev. Genet. 2019, 20, 283–297. [Google Scholar] [CrossRef]
- Irie, N.; Satoh, N.; Kuratani, S. The Phylum Vertebrata: A Case for Zoological Recognition. Zool. Lett. 2018, 4, 32. [Google Scholar] [CrossRef]
- Uesaka, M.; Kuratani, S.; Irie, N. The Developmental Hourglass Model and Recapitulation: An Attempt to Integrate the Two Models. J. Exp. Zoolog. B Mol. Dev. Evol. 2022, 338, 76–86. [Google Scholar] [CrossRef]
- Ossipova, O.; Kerney, R.; Saint-Jeannet, J.-P.; Sokol, S.Y. Regulation of Neural Crest Development by the Formin Family Protein Daam1. Genes. J. Genet. Dev. 2018, 56, e23108. [Google Scholar] [CrossRef]
- Mononen, M.M.; Leung, C.Y.; Xu, J.; Chien, K.R. Trajectory Mapping of Human Embryonic Stem Cell Cardiogenesis Reveals Lineage Branch Points and an ISL1 Progenitor-Derived Cardiac Fibroblast Lineage. Stem Cells 2020, 38, 1267–1278. [Google Scholar] [CrossRef]
- Willnow, D.; Benary, U.; Margineanu, A.; Vignola, M.L.; Konrath, F.; Pongrac, I.M.; Karimaddini, Z.; Vigilante, A.; Wolf, J.; Spagnoli, F.M. Quantitative Lineage Analysis Identifies a Hepato-Pancreato-Biliary Progenitor Niche. Nature 2021, 597, 87–91. [Google Scholar] [CrossRef]
- Court, F.; Arnaud, P. An Annotated List of Bivalent Chromatin Regions in Human ES Cells: A New Tool for Cancer Epigenetic Research. Oncotarget 2017, 8, 4110–4124. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome Chaos: Creating New Genomic Information Essential for Cancer Macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Pisco, A.O.; Huang, S. Non-Genetic Cancer Cell Plasticity and Therapy-Induced Stemness in Tumour Relapse: “What Does Not Kill Me Strengthens Me”. Br. J. Cancer 2015, 112, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. Cancer Metastases: So Close and So Far. J. Natl. Cancer Inst. 2015, 107, djv236. [Google Scholar] [CrossRef]
- Huang, S. Reconciling Non-Genetic Plasticity with Somatic Evolution in Cancer. Trends Cancer 2021, 7, 309–322. [Google Scholar] [CrossRef]
- Corradetti, B.; Dogra, P.; Pisano, S.; Wang, Z.; Ferrari, M.; Chen, S.-H.; Sidman, R.L.; Pasqualini, R.; Arap, W.; Cristini, V. Amphibian Regeneration and Mammalian Cancer: Similarities and Contrasts from an Evolutionary Biology Perspective: Comparing the Regenerative Potential of Mammalian Embryos and Urodeles to Develop Effective Strategies against Human Cancer. BioEssays News Rev. Mol. Cell. Dev. Biol. 2021, 43, e2000339. [Google Scholar] [CrossRef]
- Khyeam, S.; Lee, S.; Huang, G.N. Genetic, Epigenetic, and Post-Transcriptional Basis of Divergent Tissue Regenerative Capacities among Vertebrates. Adv. Genet. 2021, 2, e10042. [Google Scholar] [CrossRef]
- Nguyen, P.D.; de Bakker, D.E.M.; Bakkers, J. Cardiac Regenerative Capacity: An Evolutionary Afterthought? Cell. Mol. Life Sci. 2021, 78, 5107–5122. [Google Scholar] [CrossRef]
- Postovit, L.-M.; Margaryan, N.V.; Seftor, E.A.; Kirschmann, D.A.; Lipavsky, A.; Wheaton, W.W.; Abbott, D.E.; Seftor, R.E.B.; Hendrix, M.J.C. Human Embryonic Stem Cell Microenvironment Suppresses the Tumorigenic Phenotype of Aggressive Cancer Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4329–4334. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.W. On Growth and Form; Dover: New York, NY, USA, 1917; ISBN 978-0-486-67135-2. [Google Scholar]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Sarkans, U.; Füllgrabe, A.; Ali, A.; Athar, A.; Behrangi, E.; Diaz, N.; Fexova, S.; George, N.; Iqbal, H.; Kurri, S.; et al. From ArrayExpress to BioStudies. Nucleic Acids Res. 2021, 49, D1502–D1506. [Google Scholar] [CrossRef]
- Chu, L.-F.; Leng, N.; Zhang, J.; Hou, Z.; Mamott, D.; Vereide, D.T.; Choi, J.; Kendziorski, C.; Stewart, R.; Thomson, J.A. Single-Cell RNA-Seq Reveals Novel Regulators of Human Embryonic Stem Cell Differentiation to Definitive Endoderm. Genome Biol. 2016, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.R.; Farran, C.A.E.; Zeng, Y.Y.; Yi, Y.; Warrier, T.; Gautam, P.; Collins, J.J.; Xu, J.; Dröge, P.; Koh, C.-G.; et al. Parallel Bimodal Single-Cell Sequencing of Transcriptome and Chromatin Accessibility. Genome Res. 2020, 30, 1027–1039. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-Cell RNA-Seq Profiling of Human Preimplantation Embryos and Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Plaza Reyes, A.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage Communication Regulates Human Liver Bud Development from Pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Vinogradov, A.E. “Genome Design” Model and Multicellular Complexity: Golden Middle. Nucleic Acids Res. 2006, 34, 5906–5914. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An Updated Literature-Based Knowledgebase for Tumor Suppressor Genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A Comprehensive Resource for Annotation and Prediction of Animal Transcription Factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Singh, P.P.; Isambert, H. OHNOLOGS v2: A Comprehensive Resource for the Genes Retained from Whole Genome Duplication in Vertebrates. Nucleic Acids Res. 2020, 48, D724–D730. [Google Scholar] [CrossRef]
- Assou, S.; Le Carrour, T.; Tondeur, S.; Ström, S.; Gabelle, A.; Marty, S.; Nadal, L.; Pantesco, V.; Réme, T.; Hugnot, J.-P.; et al. A Meta-Analysis of Human Embryonic Stem Cells Transcriptome Integrated into a Web-Based Expression Atlas. Stem Cells 2007, 25, 961–973. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical Significance for Genomewide Studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinogradov, A.E.; Anatskaya, O.V. Cellular Biogenetic Law and Its Distortion by Protein Interactions: A Possible Unified Framework for Cancer Biology and Regenerative Medicine. Int. J. Mol. Sci. 2022, 23, 11486. https://doi.org/10.3390/ijms231911486
Vinogradov AE, Anatskaya OV. Cellular Biogenetic Law and Its Distortion by Protein Interactions: A Possible Unified Framework for Cancer Biology and Regenerative Medicine. International Journal of Molecular Sciences. 2022; 23(19):11486. https://doi.org/10.3390/ijms231911486
Chicago/Turabian StyleVinogradov, Alexander E., and Olga V. Anatskaya. 2022. "Cellular Biogenetic Law and Its Distortion by Protein Interactions: A Possible Unified Framework for Cancer Biology and Regenerative Medicine" International Journal of Molecular Sciences 23, no. 19: 11486. https://doi.org/10.3390/ijms231911486