
Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
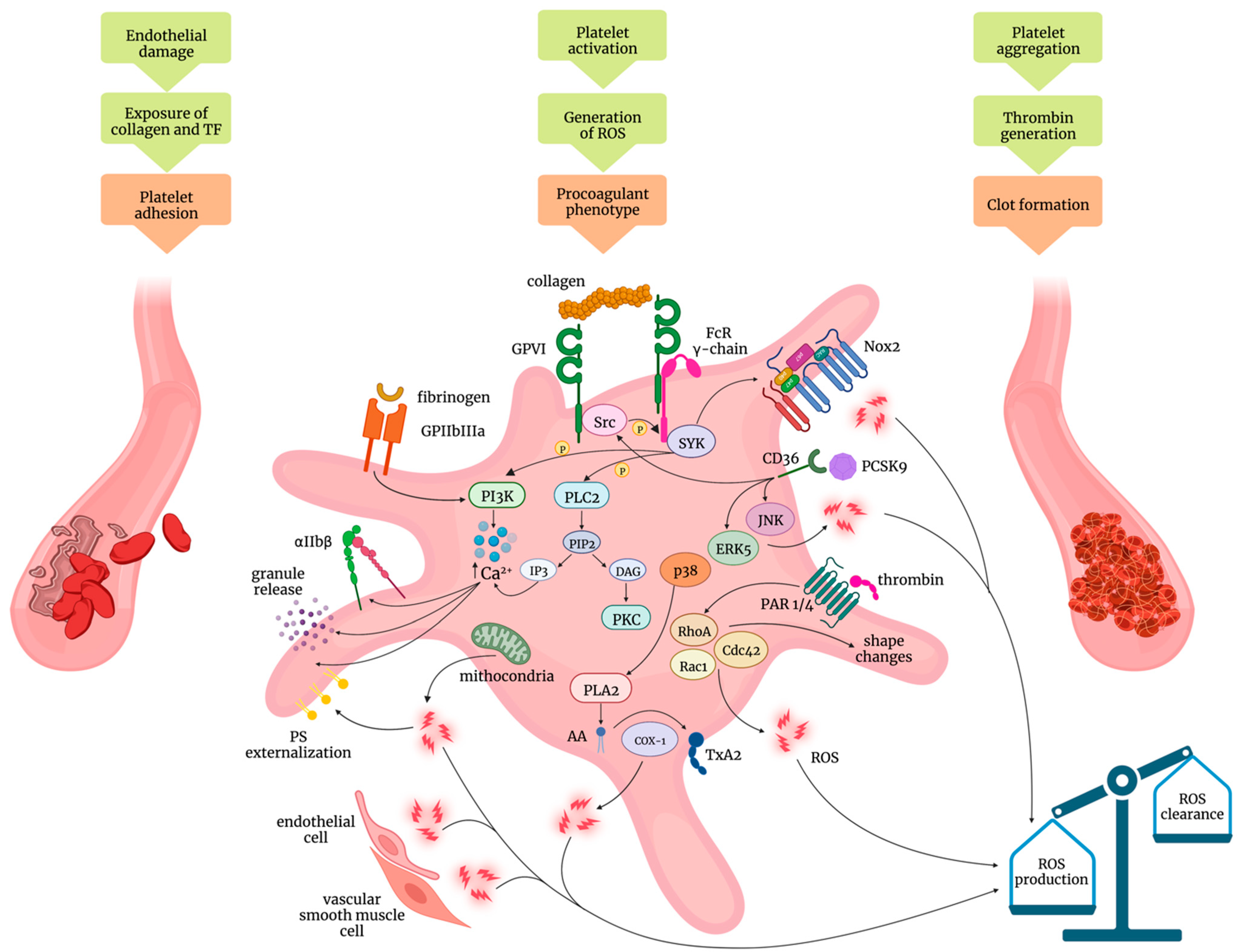
2. Mechanisms of Platelet Activation: Role of Oxidative Stress
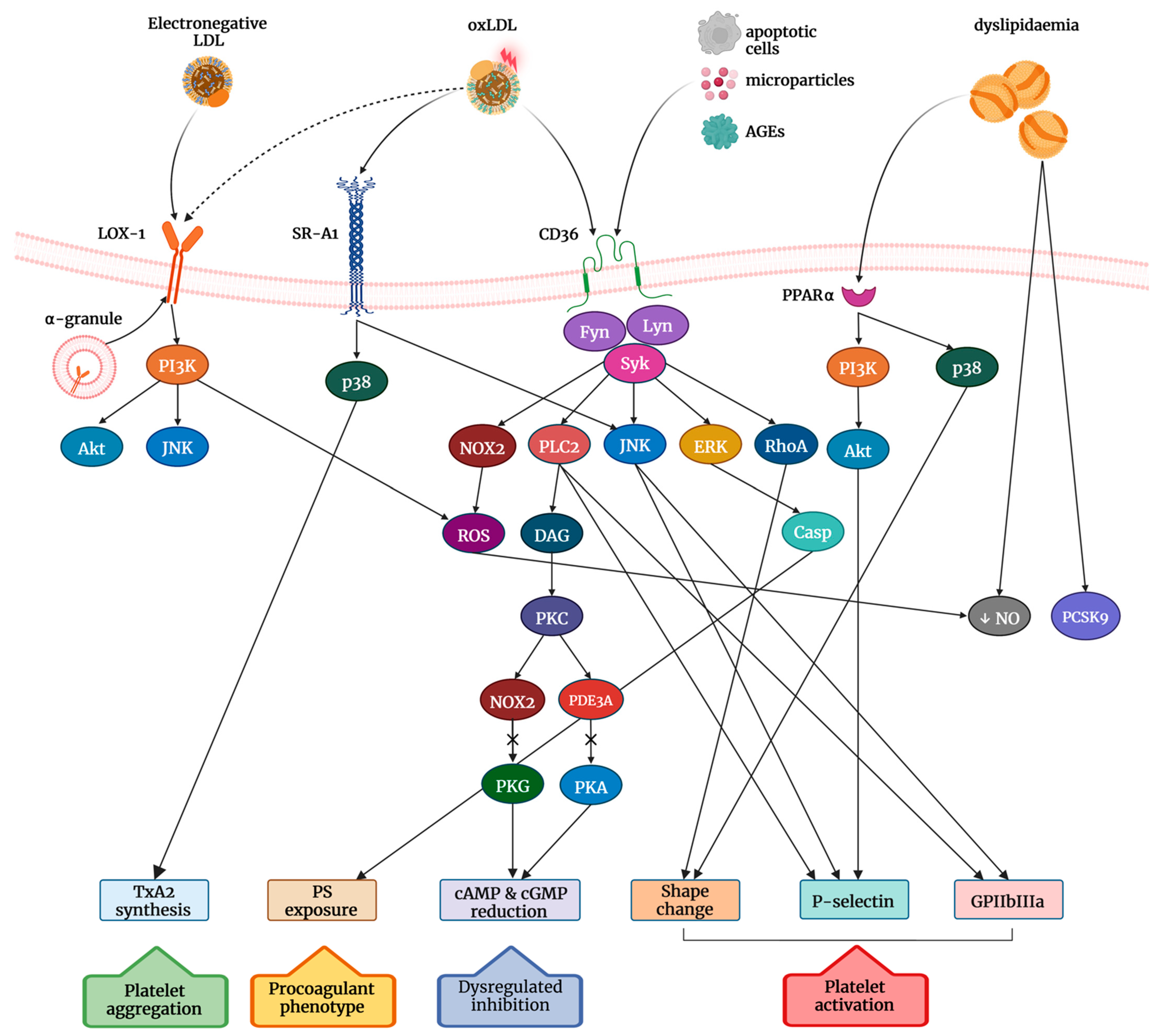
3. Platelet Reactivity in Dyslipidemia
4. Role of Scavenger Receptors in the OxLDL-Induced Signalling Transduction
5. Role of PCSK9 on Platelet Activation in Hypercholesterolemia
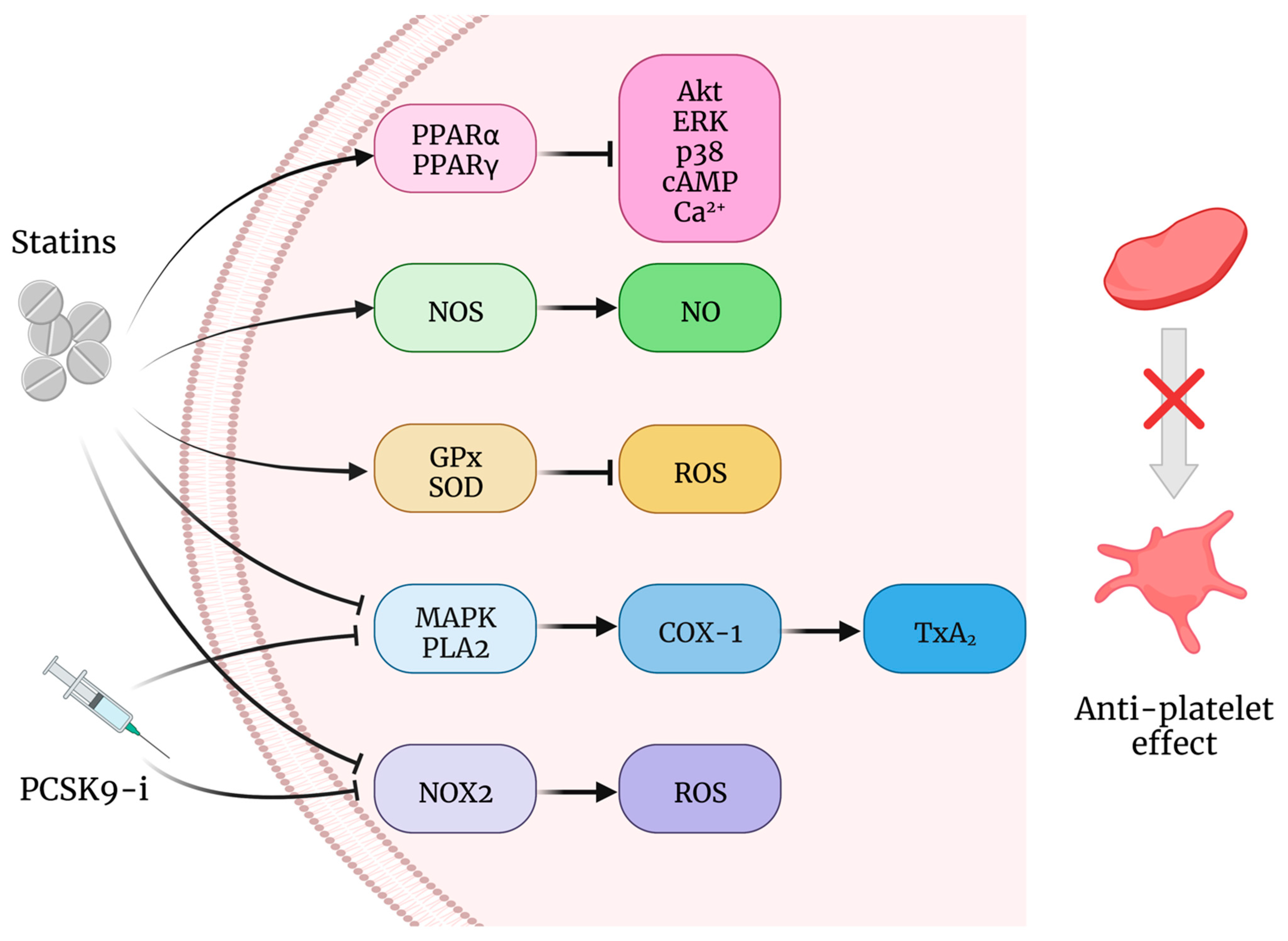
6. Current Hypolipidemic Drugs with Effects on the Redox Balance of Platelets
Statins
7. PCSK9 Inhibitors
8. Antioxidant Effects on Platelets in Hypercholesterolemia
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, L.; Lam, J.Y.; Hung, J.; Letchacovski, G.; Solymoss, C.B.; Waters, D. Hyperlipidemia and Coronary Disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation 1995, 92, 3172–3177. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.C.; Colman, R.W.; Lees, R.S. Platelet Function in Hyperlipoproteinemia. N. Engl. J. Med. 1974, 290, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Gerrard, J.M.; White, J.G. Effect of Cholesterol on Production of Thromboxane B2by Platelets in Vitro. N. Engl. J. Med. 1980, 302, 6–10. [Google Scholar] [CrossRef]
- Davì, G.; Averna, M.; Catalano, I.; Barbagallo, C.; Ganci, A.; Notarbartolo, A.; Ciabattoni, G.; Patrono, C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 1992, 85, 1792–1798. [Google Scholar] [CrossRef]
- Davì, G.; Romano, M.; Mezzetti, A.; Procopio, A.; Iacobelli, S.; Antidormi, T.; Bucciarelli, T.; Alessandrini, P.; Cuccurullo, F.; Bon, G.B. Increased Levels of Soluble P-Selectin in Hypercholesterolemic Patients. Circulation 1998, 97, 953–957. [Google Scholar] [CrossRef]
- Cipollone, F.; Mezzetti, A.; Porreca, E.; Di Febbo, C.; Nutini, M.; Fazia, M.; Falco, A.; Cuccurullo, F.; Davì, G. Association between Enhanced Soluble CD40L and Prothrombotic State in Hypercholesterolemia: Effects of Statin Therapy. Circulation 2002, 106, 399–402. [Google Scholar] [CrossRef]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, S.S.; Watkins, M.W.; Ashikaga, T.; Terrien, E.F.; Sobel, B.E.; Schneider, D.J. Usefulness of platelet reactivity before percutaneous coronary intervention in determining cardiac risk one year later. Am. J. Cardiol. 2003, 91, 876–878. [Google Scholar] [CrossRef]
- Kabbani, S.S.; Watkins, M.W.; Ashikaga, T.; Terrien, E.F.; Holoch, P.A.; Sobel, B.E.; Schneider, D.J. Platelet Reactivity Characterized Prospectively: A Determinant of Outcome 90 Days after Percutaneous Coronary Intervention. Circulation 2001, 104, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Trip, M.D.; Cats, V.M.; van Capelle, F.J.; Vreeken, J. Platelet Hyperreactivity and Prognosis in Survivors of Myocardial Infarction. N. Engl. J. Med. 1990, 322, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Vanschoonbeek, K.; Feijge, M.A.H.; Keuren, J.F.W.; Hemker, H.C.; Lodder, J.J.; Hamulyák, K.; Van Pampus, E.C.M.; Heemskerk, J.W.M. Thrombin-induced hyperactivity of platelets of young stroke patients: Involvement of thrombin receptors in the subject-dependent variability in Ca2+ signal generation. Thromb. Haemost. 2002, 88, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Icli, A.; Aksoy, F.; Nar, G.; Kaymaz, H.; Alpay, M.F.; Nar, R.; Guclu, A.; Arslan, A.; Dogan, A. Increased Mean Platelet Volume in Familial Hypercholesterolemia. Angiology 2016, 67, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin Effects on Inflammation and Platelet Activation Markers in Hypercholesterolemia. BioMed Res. Int. 2018, 2018, 6508709. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Seidu, S.; Khunti, K. Statins and primary prevention of venous thromboembolism: A systematic review and meta-analysis. Lancet Haematol. 2017, 4, e83–e93. [Google Scholar] [CrossRef]
- Ramcharan, A.S.; Van Stralen, K.J.; Snoep, J.D.; Mantel-Teeuwisse, A.K.; Rosendaal, F.R.; Doggen, C.J.M. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J. Thromb. Haemost. 2009, 7, 514–520. [Google Scholar] [CrossRef]
- Wereski, R.; Kimenai, D.M.; Bularga, A.; Taggart, C.; Lowe, D.J.; Mills, N.L.; Chapman, A.R. Risk factors for type 1 and type 2 myocardial infarction. Eur. Heart J. 2022, 43, 127–135. [Google Scholar] [CrossRef]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A Specific CD36-Dependent Signaling Pathway Is Required for Platelet Activation by Oxidized Low-Density Lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef] [Green Version]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2–mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Wraith, K.; Woodward, C.; Aburima, A.; Raslan, Z.; Hindle, M.S.; Moellmann, J.; Febbraio, M.; Naseem, K.M. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase Cγ2-dependent reactive oxygen species generation. Platelets 2019, 30, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P. Statins as Regulators of Redox Signaling in Platelets. Antioxid. Redox Signal. 2014, 20, 1300–1312. [Google Scholar] [CrossRef]
- Haramaki, N.; Ikeda, H.; Takenaka, K.; Katoh, A.; Sugano, R.; Yamagishi, S.-I.; Matsuoka, H.; Imaizumi, T. Fluvastatin Alters Platelet Aggregability in Patients with Hypercholesterolemia: Possible Improvement of Intraplatelet Redox Imbalance via HMG-CoA Reductase. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1471–1477. [Google Scholar] [CrossRef]
- Violi, F.; Carnevale, R.; Pastori, D.; Pignatelli, P. Antioxidant and antiplatelet effects of atorvastatin by Nox2 inhibition. Trends Cardiovasc. Med. 2014, 24, 142–148. [Google Scholar] [CrossRef]
- Davì, G.; Patrono, C. Platelet Activation and Atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef]
- Van Der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Takayama, H.; Ezumi, Y.; Arai, M.; Yamamoto, N.; Takahashi, H.; Okuma, M. Collagen-stimulated Activation of Syk but Not c-Src Is Severely Compromised in Human Platelets Lacking Membrane Glycoprotein VI. J. Biol. Chem. 1997, 272, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Yanaga, F.; Poole, A.; Asselin, J.; Blake, R.; Schieven, G.L.; Clark, E.A.; Law, C.L.; Watson, S.P. Syk interacts with tyrosine-phosphorylated proteins in human platelets activated by collagen and cross-linking of the Fc γ-IIA receptor. Biochem. J. 1995, 311 Pt 2, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.-J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost. 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Rozenvayn, N.; Flaumenhaft, R. Phosphatidylinositol 4,5-Bisphosphate Mediates Ca2+-induced Platelet α-Granule Secretion: Evidence for Type II Phosphatidylinositol 5-Phosphate 4-Kinase Function. J. Biol. Chem. 2001, 276, 22410–22419. [Google Scholar] [CrossRef]
- Ahmed, M.U.; Kaneva, V.; Loyau, S.; Nechipurenko, D.; Receveur, N.; Le Bris, M.; Janus-Bell, E.; Didelot, M.; Rauch, A.; Susen, S.; et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2127–2142. [Google Scholar] [CrossRef]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen Peroxide Is Involved in Collagen-Induced Platelet Activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef]
- Krötz, F.; Sohn, H.-Y.; Pohl, U. Reactive Oxygen Species: Players in the Platelet Game. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar] [CrossRef]
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Cavalot, F.; Trovati, M. N-acetyl-L-cysteine exerts direct anti-aggregating effect on human platelets. Eur. J. Clin. Investig. 2001, 31, 452–461. [Google Scholar] [CrossRef]
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J.; Fasano, M.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019, 3, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Akbar, H.; Duan, X.; Piatt, R.; Saleem, S.; Davis, A.K.; Tandon, N.N.; Bergmeier, W.; Zheng, Y. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J. Thromb. Haemost. 2018, 16, 2083–2096. [Google Scholar] [CrossRef]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Chlopicki, S.; Olszanecki, R.; Janiszewski, M.; Laurindo, F.R.M.; Panz, T.; Miedzobrodzki, J. Functional Role of NADPH Oxidase in Activation of Platelets. Antioxid. Redox Signal. 2004, 6, 691–698. [Google Scholar] [CrossRef]
- Choo, H.-J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial Calcium and Reactive Oxygen Species Regulate Agonist-Initiated Platelet Phosphatidylserine Exposure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef]
- Yang, M.; Kholmukhamedov, A.; Schulte, M.L.; Cooley, B.C.; Scoggins, N.O.; Wood, J.P.; Cameron, S.J.; Morrell, C.N.; Jobe, S.M.; Silverstein, R.L. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv. 2018, 2, 2848–2861. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Sanguigni, V.; Lenti, L.; Finocchi, A.; Mendolicchio, L.; Soresina, A.R.; Plebani, A.; et al. Inherited Human gp91 phox Deficiency Is Associated with Impaired Isoprostane Formation and Platelet Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 423–434. [Google Scholar] [CrossRef]
- Begonja, A.J.; Gambaryan, S.; Geiger, J.; Aktas, B.; Pozgajova, M.; Nieswandt, B.; Walter, U. Platelet NAD(P)H-oxidase–generated ROS production regulates αIIbβ3-integrin activation independent of the NO/cGMP pathway. Blood 2005, 106, 2757–2760. [Google Scholar] [CrossRef]
- Rae, J.; Newburger, P.E.; Dinauer, M.C.; Noack, D.; Hopkins, P.J.; Kuruto, R.; Curnutte, J.T. X-Linked Chronic Granulomatous Disease: Mutations in the CYBB Gene Encoding the gp91-phox Component of Respiratory-Burst Oxidase. Am. J. Hum. Genet. 1998, 62, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Tarafdar, A.; Celikag, M.; Patinha, D.; Gulacsy, C.E.; Hounslea, E.; Warren, Z.; Ferreira, B.; Koeners, M.P.; Caggiano, L.; et al. NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J. 2020, 34, 13959–13977. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, S.; Ding, Y.; Tong, H.; Xu, X.; Wei, G.; Chen, Y.; Ju, W.; Fu, C.; Qi, K.; et al. p47phox deficiency impairs platelet function and protects mice against arterial and venous thrombosis. Redox Biol. 2020, 34, 101569. [Google Scholar] [CrossRef] [PubMed]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose Reductase–Mediated Phosphorylation of p53 Leads to Mitochondrial Dysfunction and Damage in Diabetic Platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef]
- Marjanovic, J.A.; Li, Z.; Stojanovic, A.; Du, X. Stimulatory Roles of Nitric-oxide Synthase 3 and Guanylyl Cyclase in Platelet Activation. J. Biol. Chem. 2005, 280, 37430–37438. [Google Scholar] [CrossRef]
- Marjanovic, J.A.; Stojanovic, A.; Brovkovych, V.M.; Skidgel, R.A.; Du, X. Signaling-mediated Functional Activation of Inducible Nitric-oxide Synthase and Its Role in Stimulating Platelet Activation. J. Biol. Chem. 2008, 283, 28827–28834. [Google Scholar] [CrossRef]
- Kumar, S.; Vikram, A.; Kim, Y.-R.; Jacobs, J.S.; Irani, K. P66Shc mediates increased platelet activation and aggregation in hypercholesterolemia. Biochem. Biophys. Res. Commun. 2014, 449, 496–501. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of Platelet Function through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Heemskerk, J.W.M.; Baaten, C.C.F.M.J. Platelet Membrane Receptor Proteolysis: Implications for Platelet Function. Front. Cardiovasc. Med. 2021, 7, 608391. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New Fundamentals in Hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Nagy, M.; Heemskerk, J.W.M.; Nieswandt, B.; Braun, A. Store-operated calcium entry in thrombosis and thrombo-inflammation. Cell Calcium 2019, 77, 39–48. [Google Scholar] [CrossRef]
- Fernández, D.I.; Kuijpers, M.J.E.; Heemskerk, J.W.M. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021, 32, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.Z.; Daniel, J.L.; Kunapuli, S.P. Platelet Shape Change Is Mediated by both Calcium-dependent and -independent Signaling Pathways. Role of p160 Rho-associated coiled-coil-containing protein kinase in platelet shape change. J. Biol. Chem. 1999, 274, 28293–28300. [Google Scholar] [CrossRef]
- Jin, J.; Mao, Y.; Thomas, D.; Kim, S.; Daniel, J.L.; Kunapuli, S.P. RhoA downstream of Gq and G12/13 pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem. Pharmacol. 2009, 77, 835–844. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Han, J.-S.; Jee, Y.; Lee, S.-H.; Kim, S. Pyk2 downstream of G12/13 pathways regulates platelet shape change through RhoA/p160ROCK. Biochem. Biophys. Res. Commun. 2020, 526, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Foster, C.; Lecchi, A.; Quinton, T.M.; Prosser, D.M.; Jin, J.; Cattaneo, M.; Kunapuli, S.P. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood 2002, 99, 3629–3636. [Google Scholar] [CrossRef]
- Moers, A.; Nieswandt, B.; Massberg, S.; Wettschureck, N.; Grüner, S.; Konrad, I.; Schulte, V.; Aktas, B.; Gratacap, M.-P.; Simon, M.I.; et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 2003, 9, 1418–1422. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Cai, F.; Chen, X.; Luo, M.; Hu, L.; Lu, Y. The Role of Mitochondria-Derived Reactive Oxygen Species in Hyperthermia-Induced Platelet Apoptosis. PLoS ONE 2013, 8, e75044. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, N.; Williams, M.S. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic. Biol. Med. 2008, 45, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Ravi, S.; Chacko, B.; Sawada, H.; Kramer, P.A.; Johnson, M.S.; Benavides, G.A.; O’Donnell, V.; Marques, M.B.; Darley-Usmar, V.M. Metabolic Plasticity in Resting and Thrombin Activated Platelets. PLoS ONE 2015, 10, e0123597. [Google Scholar] [CrossRef]
- Matarrese, P.; Straface, E.; Palumbo, G.; Anselmi, M.; Gambardella, L.; Ascione, B.; Del Principe, D.; Malorni, W. Mitochondria regulate platelet metamorphosis induced by opsonized zymosan A–Activation and long-term commitment to cell death. FEBS J. 2009, 276, 845–856. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Fontana, P.; Zufferey, A.; Daali, Y.; Reny, J.-L. Antiplatelet Therapy: Targeting the TxA2 Pathway. J. Cardiovasc. Transl. Res. 2014, 7, 29–38. [Google Scholar] [CrossRef]
- Krötz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase–dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef]
- Loscalzo, J. Oxidant stress: A key determinant of atherothrombosis. Biochem. Soc. Trans. 2003, 31, 1059–1061. [Google Scholar] [CrossRef]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J. A Series of Prostaglandin F2-like Compounds Are Produced in Vivo in Humans by a Non-Cyclooxygenase, Free Radical-Catalyzed Mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef]
- Morrow, J.D. Quantification of Isoprostanes as Indices of Oxidant Stress and the Risk of Atherosclerosis in Humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Praticò, D.; Smyth, E.M.; Violi, F.; FitzGerald, G.A. Local Amplification of Platelet Function by 8-Epi Prostaglandin F2α Is Not Mediated by Thromboxane Receptor Isoforms. J. Biol. Chem. 1996, 271, 14916–14924. [Google Scholar] [CrossRef] [PubMed]
- Ni Liu, Y.; Davidson, B.P.; Yue, Q.; Belcik, T.; Xie, A.; Inaba, Y.; Mccarty, O.J.T.; Tormoen, G.W.; Zhao, Y.; Ruggeri, Z.M.; et al. Molecular Imaging of Inflammation and Platelet Adhesion in Advanced Atherosclerosis Effects of Antioxidant Therapy with NADPH Oxidase Inhibition. Circ. Cardiovasc. Imaging 2013, 6, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J.; Chauhan, A.K.; Lentz, S.R. Hydrogen Peroxide Promotes Aging-Related Platelet Hyperactivation and Thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.C.; Mahoney, C.E.; Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.-Y.; Tang, S.-S.; Handy, D.E.; Loscalzo, J. Glutathione Peroxidase-3 Deficiency Promotes Platelet-Dependent Thrombosis In Vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Wen, J.; Huang, Y.; Lu, Y.; Yuan, H. Associations of non-high-density lipoprotein cholesterol, triglycerides and the total cholesterol/HDL-c ratio with arterial stiffness independent of low-density lipoprotein cholesterol in a Chinese population. Hypertens. Res. 2019, 42, 1223–1230. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Salonen, J.T.; Salonen, R.; Seppänen, K.; Rinta-Kiikka, S.; Kuukka, M.; Korpela, H.; Alfthan, G.; Kantola, M.; Schalch, W. Effects of antioxidant supplementation on platelet function: A randomized pair-matched, placebo-controlled, double-blind trial in men with low antioxidant status. Am. J. Clin. Nutr. 1991, 53, 1222–1229. [Google Scholar] [CrossRef]
- Véricel, E.; Januel, C.; Carreras, M.; Moulin, P.; Lagarde, M. Diabetic Patients without Vascular Complications Display Enhanced Basal Platelet Activation and Decreased Antioxidant Status. Diabetes 2004, 53, 1046–1051. [Google Scholar] [CrossRef]
- Morita, H.; Ikeda, H.; Haramaki, N.; Eguchi, H.; Imaizumi, T. Only two-week smoking cessation improves platelet aggregability and intraplatelet redox imbalance of long-term smokers. J. Am. Coll. Cardiol. 2005, 45, 589–594. [Google Scholar] [CrossRef]
- Berliner, J.A.; Watson, A.D. A Role for Oxidized Phospholipids in Atherosclerosis. N. Engl. J. Med. 2005, 353, 9–11. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J.; Cooper, M.B.; Saggerson, E.D.; Prichard, B.N.C.; Tan, K.C.B.; Ling, E.; Barbera, G.; McCarthy, S.; Smith, C.C.T. Platelet function in patients with hypercholesterolaemia. Eur. J. Clin. Investig. 1994, 24 (Suppl. 1), 30–33. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, A.; Kakishita, E.; Nagai, K. The role of platelet hyperfunction in thrombus formation in hyperlipidemia. Thromb. Res. 1982, 25, 331–339. [Google Scholar] [CrossRef]
- Van Geffen, J.P.; Swieringa, F.; Van Kuijk, K.; Tullemans, B.M.E.; Solari, F.A.; Peng, B.; Clemetson, K.J.; Farndale, R.W.; Dubois, L.J.; Sickmann, A.; et al. Mild hyperlipidemia in mice aggravates platelet responsiveness in thrombus formation and exploration of platelet proteome and lipidome. Sci. Rep. 2020, 10, 21407. [Google Scholar] [CrossRef]
- Stellos, K.; Sauter, R.; Fahrleitner, M.; Grimm, J.; Stakos, D.; Emschermann, F.; Panagiota, V.; Gnerlich, S.; Perk, A.; Schönberger, T.; et al. Binding of Oxidized Low-Density Lipoprotein on Circulating Platelets Is increased in Patients with Acute Coronary Syndromes and Induces Platelet Adhesion to Vascular Wall In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2017–2020. [Google Scholar] [CrossRef]
- Dardik, R.; Varon, D.; Tamarin, I.; Zivelin, A.; Salomon, O.; Shenkman, B.; Savion, N. Homocysteine and Oxidized Low Density Lipoprotein Enhanced Platelet Adhesion to Endothelial Cells under Flow Conditions: Distinct Mechanisms of Thrombogenic Modulation. Thromb. Haemost. 2000, 83, 338–344. [Google Scholar] [CrossRef]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef]
- Hartwich, J.; Dembinska-Kieć, A.; Gruca, A.; Motyka, M.; Partyka, L.; Skrzeczyńska, J.; Bzowska, M.; Pryjma, J.; Huber, J.; Leitinger, N.; et al. Regulation of platelet adhesion by oxidized lipoproteins and oxidized phospholipids. Platelets 2002, 13, 141–151. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fuda, H.; Yanai, H.; Akita, H.; Shuping, H.; Chiba, H.; Matsuno, K. Significance of Membrane Glycoproteins in Platelet Interaction with Oxidized Low-Density Lipoprotein. Semin. Thromb. Hemost. 1998, 24, 251–253. [Google Scholar] [CrossRef]
- Daub, K.; Seizer, P.; Stellos, K.; Krämer, B.F.; Bigalke, B.; Schaller, M.; Fateh-Moghadam, S.; Gawaz, M.; Lindemann, S. Oxidized LDL-Activated Platelets Induce Vascular Inflammation. Semin. Thromb. Hemost. 2010, 36, 146–156. [Google Scholar] [CrossRef]
- Murohara, T.; Scalia, R.; Lefer, A.M. Lysophosphatidylcholine Promotes P-Selectin Expression in Platelets and Endothelial Cells. Possible Involvement of Protein Kinase C Activation and Its Inhibition by Nitric Oxide Donors. Circ. Res. 1996, 78, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.-H.; Kong, J.; Yang, M.-Y.; Jiang, G.-H.; Wang, X.-P.; Zhong, M.; Zhang, Y.; Deng, J.-T.; Zhang, W. Oxidized Low-Density Lipoprotein-Dependent Platelet-Derived Microvesicles Trigger Procoagulant Effects and Amplify Oxidative Stress. Mol. Med. 2012, 18, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.; Ludwig, A.; Petersen, F.; Flad, H.-D. Platelet-derived CXC chemokines: Old players in new games. Immunol. Rev. 2000, 177, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Coleman, L.G.; Polanowska-Grabowska, R.K.; Marcinkiewicz, M.; Gear, A.R.L. LDL oxidized by hypochlorous acid causes irreversible platelet aggregation when combined with low levels of ADP, thrombin, epinephrine, or macrophage-derived chemokine (CCL22). Blood 2004, 104, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-T.; Wang, Z.; Hu, Y.-W. Possible roles of platelet-derived microparticles in atherosclerosis. Atherosclerosis 2016, 248, 10–16. [Google Scholar] [CrossRef]
- Holvoet, P.; Mertens, A.; Verhamme, P.; Bogaerts, K.; Beyens, G.; Verhaeghe, R.; Collen, D.; Muls, E.; Van de Werf, F. Circulating Oxidized LDL Is a Useful Marker for Identifying Patients with Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 844–848. [Google Scholar] [CrossRef]
- Holvoet, P.; Collen, D.; Van De Werf, F. Malondialdehyde-modified LDL as a marker of acute coronary syndromes. JAMA 1999, 281, 1718–1721. [Google Scholar] [CrossRef]
- Tsimikas, S.; Bergmark, C.; Beyer, R.W.; Patel, R.; Pattison, J.; Miller, E.; Juliano, J.; Witztum, J.L. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 360–370. [Google Scholar] [CrossRef]
- Miyazaki, A.; Uehara, T.; Usami, Y.; Ishimine, N.; Sugano, M.; Tozuka, M. Highly oxidized low-density lipoprotein does not facilitate platelet aggregation. J. Int. Med. Res. 2020, 48, 300060520958960. [Google Scholar] [CrossRef]
- Berger, M.; Naseem, K.M. Oxidised Low-Density Lipoprotein-Induced Platelet Hyperactivity—Receptors and Signalling Mechanisms. Int. J. Mol. Sci. 2022, 23, 9199. [Google Scholar] [CrossRef]
- Naseem, K.M.; Goodall, A.H.; Bruckdorfer, K.R. Differential effects of native and oxidatively modified low-density lipoproteins on platelet function. Platelets 1997, 8, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Weidtmann, A.; Scheithe, R.; Hrboticky, N.; Pietsch, A.; Lorenz, R.; Siess, W. Mildly Oxidized LDL Induces Platelet Aggregation Through Activation of Phospholipase A2. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, J.; Wang, S.; Jiang, L.; Chen, X.; Zhou, Y.; Li, J.; Shi, J.; Liu, P.; Shu, Z.; et al. Critical role of peroxisome proliferator-activated receptor α in promoting platelet hyperreactivity and thrombosis under hyperlipidemia. Haematologica 2022, 107, 1358–1373. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Edelstein, D.; Du, X.-L.; Brownlee, M. Hyperglycemia Potentiates Collagen-Induced Platelet Activation through Mitochondrial Superoxide Overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Gyulkhandanyan, A.V.; Allen, D.J.; Mykhaylov, S.; Lyubimov, E.; Ni, H.; Freedman, J.; Leytin, V. Mitochondrial Inner Membrane Depolarization as a Marker of Platelet Apoptosis: Disclosure of Nonapoptotic Membrane Depolarization. Clin. Appl. Thromb. Hemost. 2017, 23, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Murugesan, G.; Chen, K.; Zhang, L.; Wang, Q.; Febbraio, M.; Anselmo, R.M.; Marchant, K.; Barnard, J.; Silverstein, R.L. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011, 117, 6355–6366. [Google Scholar] [CrossRef]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Febbraio, M.; Hajjar, D.P.; et al. A Novel Family of Atherogenic Oxidized Phospholipids Promotes Macrophage Foam Cell Formation via the Scavenger Receptor CD36 and Is Enriched in Atherosclerotic Lesions. J. Biol. Chem. 2002, 277, 38517–38523. [Google Scholar] [CrossRef]
- Berger, M.; Raslan, Z.; Aburima, A.; Magwenzi, S.; Wraith, K.S.; Spurgeon, B.E.J.; Hindle, M.S.; Law, R.; Febbraio, M.; Naseem, K.M. Atherogenic lipid stress induces platelet hyperactivity through CD36-mediated hyposensitivity to prostacyclin: The role of phosphodiesterase 3A. Haematologica 2020, 105, 808–819. [Google Scholar] [CrossRef]
- Rukoyatkina, N.; Walter, U.; Friebe, A.; Gambaryan, S. Differentiation of cGMP-dependent and -independent nitric oxide effects on platelet apoptosis and reactive oxygen species production using platelets lacking soluble guanylyl cyclase. Thromb. Haemost. 2011, 106, 922–933. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.-C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Salido, G.M.; Gómez-Arteta, E.; Rosado, J.A.; Pariente, J.A. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J. Thromb. Haemost. 2007, 5, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Silverstein, R.L. CD36 and ERK5 link dyslipidemia to apoptotic-like platelet procoagulant function. Curr. Opin. Hematol. 2019, 26, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.; Gwozdz, A.M.; Wang, H.; Bang, K.W.A.; Packham, M.A.; Freedman, J.; Rand, M.L. Persistence of procoagulant surface expression on activated human platelets: Involvement of apoptosis and aminophospholipid translocase activity. J. Thromb. Haemost. 2007, 5, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a Novel Family of Oxidized Phospholipids That Serve as Ligands for the Macrophage Scavenger Receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [PubMed]
- Unsworth, A.J.; Bye, A.P.; Tannetta, D.S.; Desborough, M.J.R.; Kriek, N.; Sage, T.; Allan, H.E.; Crescente, M.; Yaqoob, P.; Warner, T.D.; et al. Farnesoid X Receptor and Liver X Receptor Ligands Initiate Formation of Coated Platelets. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1482–1493. [Google Scholar] [CrossRef]
- Combescure, C.; Fontana, P.; Mallouk, N.; Berdagué, P.; Labruyere, C.; Barazer, I.; Gris, J.-C.; Laporte, S.; Fabbro-Peray, P.; Reny, J.L.; et al. Clinical implications of clopidogrel non-response in cardiovascular patients: A systematic review and meta-analysis. J. Thromb. Haemost. 2010, 8, 923–933. [Google Scholar] [CrossRef]
- Sofi, F.; Marcucci, R.; Gori, A.M.; Giusti, B.; Abbate, R.; Gensini, G.F. Clopidogrel non-responsiveness and risk of cardiovascular morbidity. An Updated Meta-Analysis. Thromb. Haemost. 2010, 103, 841–848. [Google Scholar] [CrossRef]
- Stone, G.W.; Witzenbichler, B.; Weisz, G.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; Cox, D.A.; Duffy, P.L.; Mazzaferri, E.; et al. Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): A prospective multicentre registry study. Lancet 2013, 382, 614–623. [Google Scholar] [CrossRef]
- Valiyaveettil, M.; Podrez, E.A. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 218–221. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Ashraf, M.Z.; Podrez, E.A. Scavenger receptor BI modulates platelet reactivity and thrombosis in dyslipidemia. Blood 2010, 116, 1932–1941. [Google Scholar] [CrossRef]
- Akkerman, J.W.N. From low-density lipoprotein to platelet activation. Int. J. Biochem. Cell Biol. 2008, 40, 2374–2378. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Simeone, P.; Liani, R.; Davì, G. Platelets and diabetes mellitus. Prostaglandins Other Lipid Mediat. 2015, 120, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 8267234. [Google Scholar] [CrossRef]
- Pawlowska, Z.; Swiatkowska, M.; Krzeslowska, J.; Pawlicki, L.; Cierniewski, C.S. Increased platelet-fibrinogen interaction in patients with hypercholesterolemia and hypertriglyceridemia. Atherosclerosis 1993, 103, 13–20. [Google Scholar] [CrossRef]
- Relou, I.A.M.; Hackeng, C.M.; Akkerman, J.-W.N.; Malle, E. Low-density lipoprotein and its effect on human blood platelets. Cell Mol. Life Sci. 2003, 60, 961–971. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kakino, A.; Takeshita, H.; Hayashi, N.; Li, L.; Nakano, A.; Hanasaki-Yamamoto, H.; Fujita, Y.; Imaizumi, Y.; Toyama-Yokoyama, S.; et al. Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J. 2015, 29, 3342–3356. [Google Scholar] [CrossRef]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-Dependent Surface Expression of LOX-1 in Human Platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef]
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Asp. Med. 2005, 26, 33–65. [Google Scholar] [CrossRef]
- Barale, C.; Frascaroli, C.; Cavalot, F.; Russo, I. Hypercholesterolemia impairs the Glucagon-like peptide 1 action on platelets: Effects of a lipid-lowering treatment with simvastatin. Thromb. Res. 2019, 180, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, S.R.; Stewart, S.; Holmes, A.S.; Chirkov, Y.Y.; Horowitz, J.D. Platelet Nitric Oxide Responsiveness: A Novel Prognostic Marker in Acute Coronary Syndromes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Riba, R.; Nicolaou, A.; Troxler, M.; Homer-Vaniasinkam, S.; Naseem, K.M. Altered platelet reactivity in peripheral vascular disease complicated with elevated plasma homocysteine levels. Atherosclerosis 2004, 175, 69–75. [Google Scholar] [CrossRef]
- Russo, I.; Traversa, M.; Bonomo, K.; De Salve, A.; Mattiello, L.; Del Mese, P.; Doronzo, G.; Cavalot, F.; Trovati, M.; Anfossi, G. In Central Obesity, Weight Loss Restores Platelet Sensitivity to Nitric Oxide and Prostacyclin. Obesity 2010, 18, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Buracco, S.; Cavalot, F.; Frascaroli, C.; Guerrasio, A.; Russo, I. Glucagon-like peptide 1-related peptides increase nitric oxide effects to reduce platelet activation. Thromb. Haemost. 2017, 117, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Trovati, M. Platelet Resistance to the Anti-Aggregating Agents in the Insulin Resistant States. Curr. Diabetes Rev. 2006, 2, 409–430. [Google Scholar] [CrossRef]
- Russo, I.; Del Mese, P.; Doronzo, G.; De Salve, A.; Secchi, M.; Trovati, M.; Anfossi, G. Platelet Resistance to the Antiaggregatory Cyclic Nucleotides in Central Obesity Involves Reduced Phosphorylation of Vasodilator-Stimulated Phosphoprotein. Clin. Chem. 2007, 53, 1053–1060. [Google Scholar] [CrossRef]
- Chu, F.; Wang, M.; Ma, H.; Zhu, J. Simvastatin Modulates Interaction between Vascular Smooth Muscle Cell/Macrophage and TNF-α–Activated Endothelial Cell. J. Cardiovasc. Pharmacol. 2018, 71, 268–274. [Google Scholar] [CrossRef]
- Kanshana, J.S.; Khanna, V.; Singh, V.; Jain, M.; Misra, A.; Kumar, S.; Farooqui, M.; Barthwal, M.K.; Dikshit, M. Progression and Characterization of the Accelerated Atherosclerosis in Iliac Artery of New Zealand White Rabbits: Effect of Simvastatin. J. Cardiovasc. Pharmacol. 2017, 69, 314–325. [Google Scholar] [CrossRef]
- Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef]
- Kinlay, S.; Selwyn, A.P. Effects of statins on inflammation in patients with acute and chronic coronary syndromes. Am. J. Cardiol. 2003, 91, 9B–13B. [Google Scholar] [CrossRef]
- Sadowitz, B.; Maier, K.G.; Gahtan, V. Basic Science Review: Statin Therapy-Part I: The Pleiotropic Effects of Statins in Cardiovascular Disease. Vasc. Endovasc. Surg. 2010, 44, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.-D. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res. Cardiol. 2017, 112, 32. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.-C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and Its Natural Mutants: Zymogen Cleavage and Effects on the Low Density Lipoprotein (LDL) Receptor and LDL Cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef]
- McNutt, M.C.; Lagace, T.A.; Horton, J.D. Catalytic Activity Is Not Required for Secreted PCSK9 to Reduce Low Density Lipoprotein Receptors in HepG2 Cells. J. Biol. Chem. 2007, 282, 20799–20803. [Google Scholar] [CrossRef]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Petersen-Uribe, A.; Kremser, M.; Rohlfing, A.-K.; Castor, T.; Kolb, K.; Dicenta, V.; Emschermann, F.; Li, B.; Borst, O.; Rath, D.; et al. Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease. Int. J. Mol. Sci. 2021, 22, 11179. [Google Scholar] [CrossRef]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Asia Pacific Cohort Studies Collaboration. Serum Triglycerides as a Risk Factor for Cardiovascular Diseases in the Asia-Pacific Region. Circulation 2004, 110, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.J.; Rachlis, B.; Wu, P.; Devereaux, P.J.; Arora, P.; Perri, D. Primary Prevention of Cardiovascular Mortality and Events With Statin Treatments: A Network Meta-Analysis Involving More Than 65,000 Patients. J. Am. Coll. Cardiol. 2008, 52, 1769–1781. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic effects of statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Pleiotropic Effects of Statins—Basic Research and Clinical Perspectives—. Circ. J. 2010, 74, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, L.; Santilli, F.; Pasqui, A.L.; Lattanzio, S.; Liani, R.; Ciani, F.; Ferrante, E.; Ciabattoni, G.; Scarpini, F.; Ghezzi, A.; et al. Effects of atorvastatin and rosuvastatin on thromboxane-dependent platelet activation and oxidative stress in hypercholesterolemia. Atherosclerosis 2011, 214, 122–128. [Google Scholar] [CrossRef]
- Roberto, C.; Pasquale, P.; Serena, D.S.; Simona, B.; Valerio, S.; Laura, N.; Gaetano, T.; Stefania, B.; Francesco, V. Atorvastatin inhibits oxidative stress via adiponectin-mediated NADPH oxidase down-regulation in hypercholesterolemic patients. Atherosclerosis 2010, 213, 225–234. [Google Scholar] [CrossRef]
- De Caterina, R.; Cipollone, F.; Filardo, F.P.; Zimarino, M.; Bernini, W.; Lazzerini, G.; Bucciarelli, T.; Falco, A.; Marchesani, P.; Muraro, R.; et al. Low-Density Lipoprotein Level Reduction by the 3-Hydroxy-3-Methylglutaryl Coenzyme-A Inhibitor Simvastatin Is Accompanied by a Related Reduction of F 2 -Isoprostane Formation in Hypercholesterolemic Subjects: No Further Effect of Vitamin E. Circulation 2002, 106, 2543–2549. [Google Scholar] [CrossRef]
- Sugiyama, M.; Ohashi, M.; Takase, H.; Sato, K.; Ueda, R.; Dohi, Y. Effects of atorvastatin on inflammation and oxidative stress. Heart Vessel. 2005, 20, 133–136. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Pastori, D.; Cangemi, R.; Napoleone, L.; Bartimoccia, S.; Nocella, C.; Basili, S.; Violi, F. Immediate Antioxidant and Antiplatelet Effect of Atorvastatin via Inhibition of Nox2. Circulation 2012, 126, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Desideri, G.; Croce, G.; Tucci, M.; Passacquale, G.; Broccoletti, S.; Valeri, L.; Santucci, A.; Ferri, C. Effects of Bezafibrate and Simvastatin on Endothelial Activation and Lipid Peroxidation in Hypercholesterolemia: Evidence of Different Vascular Protection by Different Lipid-Lowering Treatments. J. Clin. Endocrinol. Metab. 2003, 88, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hayashi, M.; Takayanagi, K.; Morooka, S. Lipid-lowering therapy with fluvastatin inhibits oxidative modification of low density lipoprotein and improves vascular endothelial function in hypercholesterolemic patients. Atherosclerosis 2002, 160, 369–376. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Brennan, M.-L.; Aviles, R.J.; Fu, X.; Penn, M.S.; Sprecher, D.L.; Hazen, S.L. Statins Promote Potent Systemic Antioxidant Effects through Specific Inflammatory Pathways. Circulation 2003, 108, 426–431. [Google Scholar] [CrossRef]
- Pirro, M.; Schillaci, G.; Mannarino, M.R.; Savarese, G.; Vaudo, G.; Siepi, D.; Paltriccia, R.; Mannarino, E. Effects of rosuvastatin on 3-nitrotyrosine and aortic stiffness in hypercholesterolemia. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 436–441. [Google Scholar] [CrossRef]
- Tavridou, A.; Efthimiadis, A.; Efthimiadis, I.; Manolopoulos, V.G. Simvastatin-induced changes in circulating oxidized low-density lipoprotein in different types of dyslipidemia. Heart Vessel. 2010, 25, 288–293. [Google Scholar] [CrossRef]
- Thompson, W.; Morin, L.; Jarbøl, D.E.; Andersen, J.H.; Ernst, M.T.; Nielsen, J.B.; Haastrup, P.; Schmidt, M.; Pottegård, A. Statin Discontinuation and Cardiovascular Events among Older People in Denmark. JAMA Netw. Open 2021, 4, e2136802. [Google Scholar] [CrossRef]
- Puccetti, L.; Pasqui, A.L.; Pastorelli, M.; Bova, G.; Di Renzo, M.; Leo, A.; Cercignani, M.; Palazzuoli, A.; Auteri, A.; Bruni, F. Platelet hyperactivity after statin treatment discontinuation. Thromb. Haemost. 2003, 90, 476–482. [Google Scholar] [CrossRef]
- Moscardó, A.; Vallés, J.; Latorre, A.; Madrid, I.; Santos, M.T. Reduction of platelet cytosolic phospholipase A2 activity by atorvastatin and simvastatin: Biochemical regulatory mechanisms. Thromb. Res. 2013, 131, e154–e159. [Google Scholar] [CrossRef]
- Liao, J.K. Effects of Statins on 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibition Beyond Low-Density Lipoprotein Cholesterol. Am. J. Cardiol. 2005, 96, 24F–33F. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, P.K.; Hughes, S.M.T.; Plumb, R.D.; Devine, A.; Leahey, W.; Lyons, K.S.; Johnston, D.; McVeigh, G.E. Statins have beneficial effects on platelet free radical activity and intracellular distribution of GTPases in hyperlipidaemia. Clin. Sci. 2009, 118, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Nenna, A.; Nappi, F.; Lusini, M.; Satriano, U.M.; Schilirò, D.; Spadaccio, C.; Chello, M. Effect of Statins on Platelet Activation and Function: From Molecular Pathways to Clinical Effects. BioMed Res. Int. 2021, 2021, 6661847. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hu, H.; Zheng, H.; Hao, J.; Yang, J.; Cui, W. Effects of peroxisome proliferator-activated receptor γ in simvastatin antiplatelet activity: Influences on cAMP and mitogen-activated protein kinases. Thromb. Res. 2014, 134, 111–120. [Google Scholar] [CrossRef]
- Ali, F.Y.; Armstrong, P.C.J.; Dhanji, A.-R.A.; Tucker, A.T.; Paul-Clark, M.J.; Mitchell, J.A.; Warner, T.D. Antiplatelet Actions of Statins and Fibrates Are Mediated by PPARs. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Gertz, K.; Huang, P.; Nickenig, G.; Böhm, M.; Dirnagl, U.; Endres, M. Atorvastatin Upregulates Type III Nitric Oxide Synthase in Thrombocytes, Decreases Platelet Activation, and Protects from Cerebral Ischemia in Normocholesterolemic Mice. Stroke 2000, 31, 2442–2449. [Google Scholar] [CrossRef]
- Gocmen, A.Y.; Ocak, G.A.; Ozbilim, G.; Delibas, N.; Gumuslu, S. Effect of atorvastatin on atherosclerotic plaque formation and platelet activation in hypercholesterolemic rats. Can. J. Physiol. Pharmacol. 2013, 91, 680–685. [Google Scholar] [CrossRef]
- Bea, F.; Blessing, E.; Shelley, M.I.; Shultz, J.M.; Rosenfeld, M.E. Simvastatin inhibits expression of tissue factor in advanced atherosclerotic lesions of apolipoprotein E deficient mice independently of lipid lowering: Potential role of simvastatin-mediated inhibition of Egr-1 expression and activation. Atherosclerosis 2003, 167, 187–194. [Google Scholar] [CrossRef]
- Gertz, K.; Laufs, U.; Lindauer, U.; Nickenig, G.; Böhm, M.; Dirnagl, U.; Endres, M. Withdrawal of Statin Treatment Abrogates Stroke Protection in Mice. Stroke 2003, 34, 551–557. [Google Scholar] [CrossRef]
- Tannous, M.; Cheung, R.; Vignini, A.; Mutus, B. Atorvastatin Increases ecNOS Levels in Human Platelets of Hyperlipidemic Subjects. Thromb. Haemost. 1999, 82, 1390–1394. [Google Scholar] [CrossRef]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Loffredo, L.; Carnevale, R.; Sorge, R.; Violi, F. Oxidative stress-mediated platelet CD40 ligand upregulation in patients with hypercholesterolemia: Effect of atorvastatin. J. Thromb. Haemost. 2007, 5, 1170–1178. [Google Scholar] [CrossRef] [Green Version]
- Serebruany, V.L.; Miller, M.; Pokov, A.N.; Malinin, A.I.; Lowry, D.R.; Tanguay, J.-F.; Hennekens, C.H. Effect of Statins on Platelet PAR-1 Thrombin Receptor in Patients with the Metabolic Syndrome (from the PAR-1 Inhibition by Statins [PARIS] Study). Am. J. Cardiol. 2006, 97, 1332–1336. [Google Scholar] [CrossRef] [PubMed]
- Sommeijer, D.W.; Joop, K.; Leyte, A.; Reitsma, P.H.; ten Cate, H. Pravastatin reduces fibrinogen receptor gpIIIa on platelet-derived microparticles in patients with type 2 diabetes. J. Thromb. Haemost. 2005, 3, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Alber, H.F.; Frick, M.; Suessenbacher, A.; Doerler, J.; Schirmer, M.; Stocker, E.-M.; Dichtl, W.; Pachinger, O.; Weidinger, F. Effect of atorvastatin on circulating proinflammatory T-lymphocyte subsets and soluble CD40 ligand in patients with stable coronary artery disease—A randomized, placebo-controlled study. Am. Heart J. 2006, 151, 139.e1–139.e7. [Google Scholar] [CrossRef]
- Blann, A.D.; Gurney, D.; Hughes, E.; Buggins, P.; Silverman, S.H.; Lip, G.Y. Influence of pravastatin on lipoproteins, and on endothelial, platelet, and inflammatory markers in subjects with peripheral artery disease. Am. J. Cardiol. 2001, 88, 89–92. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Cangemi, R.; Loffredo, L.; Sanguigni, V.; Stefanutti, C.; Basili, S.; Violi, F. Atorvastatin Inhibits gp91 phox Circulating Levels in Patients with Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, L.; Pasqui, A.L.; Pastorelli, M.; Bova, G.; Cercignani, M.; Palazzuoli, A.; Angori, P.; Auteri, A.; Bruni, F. Time-dependent effect of statins on platelet function in hypercholesterolaemia. Eur. J. Clin. Investig. 2002, 32, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J. Rosuvastatin: A Review of Its Use in the Prevention of Cardiovascular Disease in Apparently Healthy Women or Men with Normal LDL-C Levels and Elevated HsCRP Levels. Am. J. Cardiovasc. Drugs 2010, 10, 383–400. [Google Scholar] [CrossRef]
- Jula, A.; Marniemi, J.; Rönnemaa, T.; Virtanen, A.; Huupponen, R. Effects of Diet and Simvastatin on Fatty Acid Composition in Hypercholesterolemic Men: A Randomized Controlled Trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1952–1959. [Google Scholar] [CrossRef]
- Nyalala, J.O.; Wang, J.; Dang, A.; Faas, F.H.; Smith, W.G. Hypertriglyceridemia and hypercholesterolemia: Effects of drug treatment on fatty acid composition of plasma lipids and membranes. Prostaglandins Leukot. Essent. Fat. Acids 2008, 78, 271–280. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.-B.; Wiest, M.M.; Nguyen, U.T.; Watkins, S.M.; Krauss, R.M. Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics 2010, 6, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [PubMed]
- Garshick, M.S.; Block, R.; Drenkova, K.; Tawil, M.; James, G.; Brenna, J.T. Statin therapy upregulates arachidonic acid status via enhanced endogenous synthesis in patients with plaque psoriasis. Prostaglandins Leukot. Essent. Fat. Acids 2022, 180, 102428. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Mangoni, A.A. A Systematic Review and Meta-Analysis of the Effect of Statins on Glutathione Peroxidase, Superoxide Dismutase, and Catalase. Antioxidants 2021, 10, 1841. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Seidah, N.G. Proprotein Convertase Subtilisin Kexin 9 (PCSK9) Inhibitors in the Treatment of Hypercholesterolemia and other Pathologies. Curr. Pharm. Des. 2013, 19, 3161–3172. [Google Scholar] [CrossRef]
- Gitt, A.K.; Lautsch, D.; Ferrieres, J.; Kastelein, J.; Drexel, H.; Horack, M.; Brudi, P.; Vanneste, B.; Bramlage, P.; Chazelle, F.; et al. Low-density lipoprotein cholesterol in a global cohort of 57,885 statin-treated patients. Atherosclerosis 2016, 255, 200–209. [Google Scholar] [CrossRef]
- De Backer, G.; Jankowski, P.; Kotseva, K.; Mirrakhimov, E.; Reiner, Ž.; Rydén, L.; Tokgözoğlu, L.; Wood, D.; De Bacquer, D.; EUROASPIRE V Collaborators; et al. Management of dyslipidaemia in patients with coronary heart disease: Results from the ESC-EORP EUROASPIRE V survey in 27 countries. Atherosclerosis 2019, 285, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; De Backer, G.; Fras, Z.; Kotseva, K.; Tokgözoglu, L.; Wood, D.; De Bacquer, D.; EUROASPIRE Investigators. Lipid lowering drug therapy in patients with coronary heart disease from 24 European countries—Findings from the EUROASPIRE IV survey. Atherosclerosis 2016, 246, 243–250. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association between Lowering LDL-C and Cardiovascular Risk Reduction among Different Therapeutic Interventionss: A Systematic Review and Meta-Analysis. JAMA: J. Am. Med. Assoc. 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Nedergaard, B.S.; Rogers, W.J.; Fialkow, J.; Neutel, J.M.; Ramstad, D.; Somaratne, R.; Legg, J.C.; Nelson, P.; Scott, R.; et al. Effect of Evolocumab or Ezetimibe Added to Moderate- or High-Intensity Statin Therapy on LDL-C Lowering in Patients With Hypercholesterolemia: The LAPLACE-2 Randomized Clinical Trial. JAMA 2014, 311, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Gipe, D.; Bergeron, J.; Gaudet, D.; Weiss, R.; Dufour, R.; Wu, R.; Pordy, R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A phase 2 randomised controlled trial. Lancet 2012, 380, 29–36. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations inPCSK9,Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Zhao, Z.; Tuakli-Wosornu, Y.; Lagace, T.A.; Kinch, L.; Grishin, N.V.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular Characterization of Loss-of-Function Mutations in PCSK9 and Identification of a Compound Heterozygote. Am. J. Hum. Genet. 2006, 79, 514–523. [Google Scholar] [CrossRef]
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef]
- Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’Erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Dyrbuś, K.; Gąsior, M.; Penson, P.; Ray, K.K.; Banach, M. Inclisiran—New hope in the management of lipid disorders? J. Clin. Lipidol. 2020, 14, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Brandts, J.; Ray, K.K. Small interfering RNA to proprotein convertase subtilisin/kexin type 9: Transforming LDL-cholesterol-lowering strategies. Curr. Opin. Lipidol. 2020, 31, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Muñoz Estrella, A.; Sourlas, A.; Silverio, D.; Hilario, E.; Montan, P.D.; Guzman, E. Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases 2018, 6, 63. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Pećin, I.; Reiner, Ž. Novel Experimental Agents for the Treatment of Hypercholesterolemia. J. Exp. Pharmacol. 2021, 13, 91–100. [Google Scholar] [CrossRef]
- Oliveira, H.C.F.; Vercesi, A.E. Mitochondrial bioenergetics and redox dysfunctions in hypercholesterolemia and atherosclerosis. Mol. Asp. Med. 2020, 71, 100840. [Google Scholar] [CrossRef]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox Control of Endothelial Function and Dysfunction: Molecular Mechanisms and Therapeutic Opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef]
- Li, H.; Horke, S.; Förstermann, U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Barale, C.; Cavalot, F.; Frascaroli, C.; Bonomo, K.; Morotti, A.; Guerrasio, A.; Russo, I. Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. Int. J. Mol. Sci. 2020, 21, 4983. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.T.; Boffetta, P.; Greenwood, D.C.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: A systematic review and dose-response meta-analysis of prospective studies. Am. J. Clin. Nutr. 2018, 108, 1069–1091. [Google Scholar] [CrossRef]
- Nyakudya, T.T.; Tshabalala, T.; Dangarembizi, R.; Erlwanger, K.H.; Ndhlala, A.R. The Potential Therapeutic Value of Medicinal Plants in the Management of Metabolic Disorders. Molecules 2020, 25, 2669. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xie, J.; Zhang, H.; Pang, J.; Li, Q.; Wang, X.; Xu, H.; Sun, X.; Zhao, H.; Yang, Y.; et al. Anthocyanin supplementation at different doses improves cholesterol efflux capacity in subjects with dyslipidemia—A randomized controlled trial. Eur. J. Clin. Nutr. 2021, 75, 345–354. [Google Scholar] [CrossRef]
- Thompson, K.; Hosking, H.; Pederick, W.; Singh, I.; Santhakumar, A.B. The effect of anthocyanin supplementation in modulating platelet function in sedentary population: A randomised, double-blind, placebo-controlled, cross-over trial. Br. J. Nutr. 2017, 118, 368–374. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, Z.; Reheman, A.; Jin, J.W.; Li, C.; Wang, Y.; Andrews, M.C.; Chen, P.; Zhu, G.; Ling, W.; et al. Plant Food Delphinidin-3-Glucoside Significantly Inhibits Platelet Activation and Thrombosis: Novel Protective Roles against Cardiovascular Diseases. PLoS ONE 2012, 7, e37323. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, Y.; Adili, R.; McKeown, T.; Chen, P.; Zhu, G.; Li, D.; Ling, W.; Ni, H.; Yang, Y. Plant-based Food Cyanidin-3-Glucoside Modulates Human Platelet Glycoprotein VI Signaling and Inhibits Platelet Activation and Thrombus Formation. J. Nutr. 2017, 147, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.-H.; Deng, X.-J.; Chen, Y.-Q.; Ya, F.-L.; Zhang, X.-D.; Song, F.; Li, D.; Yang, Y. Anthocyanin Cyanidin-3-Glucoside Attenuates Platelet Granule Release in Mice Fed High-Fat Diets. J. Nutr. Sci. Vitaminol. 2017, 63, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Zhu, Y.; Shi, Z.; Tian, J.; Deng, X.; Ren, J.; Andrews, M.C.; Ni, H.; Ling, W.; Yang, Y. Plant food anthocyanins inhibit platelet granule secretion in hypercholesterolaemia: Involving the signalling pathway of PI3K–Akt. Thromb. Haemost. 2014, 112, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Li, K.; Fan, D.; Zhao, Y.; Gao, X.; Ma, X.; Xu, L.; Shi, Y.; Ya, F.; Zou, J.; et al. Dose-dependent effects of anthocyanin supplementation on platelet function in subjects with dyslipidemia: A randomized clinical trial. eBioMedicine 2021, 70, 103533. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.-G.; Xiao, H.-B.; Lu, X.-Y.; Sun, Z.-L. Naringin Activates AMPK Resulting in Altered Expression of SREBPs, PCSK9, and LDLR to Reduce Body Weight in Obese C57BL/6J Mice. J. Agric. Food Chem. 2018, 66, 8983–8990. [Google Scholar] [CrossRef] [PubMed]
- Cavia-Saiz, M.; Busto, M.D.; Pilar-Izquierdo, M.C.; Ortega, N.; Perez-Mateos, M.; Muñiz, P. Antioxidant properties, radical scavenging activity and biomolecule protection capacity of flavonoid naringenin and its glycoside naringin: A comparative study. J. Sci. Food Agric. 2010, 90, 1238–1244. [Google Scholar] [CrossRef]
- Viswanatha, G.L.; Shylaja, H.; Keni, R.; Nandakumar, K.; Rajesh, S. A systematic review and meta-analysis on the cardio-protective activity of naringin based on pre-clinical evidences. Phytother. Res. 2022, 36, 1064–1092. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morotti, A.; Barale, C.; Melchionda, E.; Russo, I. Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell. Int. J. Mol. Sci. 2022, 23, 11446. https://doi.org/10.3390/ijms231911446
Morotti A, Barale C, Melchionda E, Russo I. Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell. International Journal of Molecular Sciences. 2022; 23(19):11446. https://doi.org/10.3390/ijms231911446
Chicago/Turabian StyleMorotti, Alessandro, Cristina Barale, Elena Melchionda, and Isabella Russo. 2022. "Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell" International Journal of Molecular Sciences 23, no. 19: 11446. https://doi.org/10.3390/ijms231911446