
Hepatoprotective Effects of a Natural Flavanol 3,3′-Diindolylmethane against CCl4-Induced Chronic Liver Injury in Mice and TGFβ1-Induced EMT in Mouse Hepatocytes via Activation of Nrf2 Cascade

Abstract
:1. Introduction
2. Results
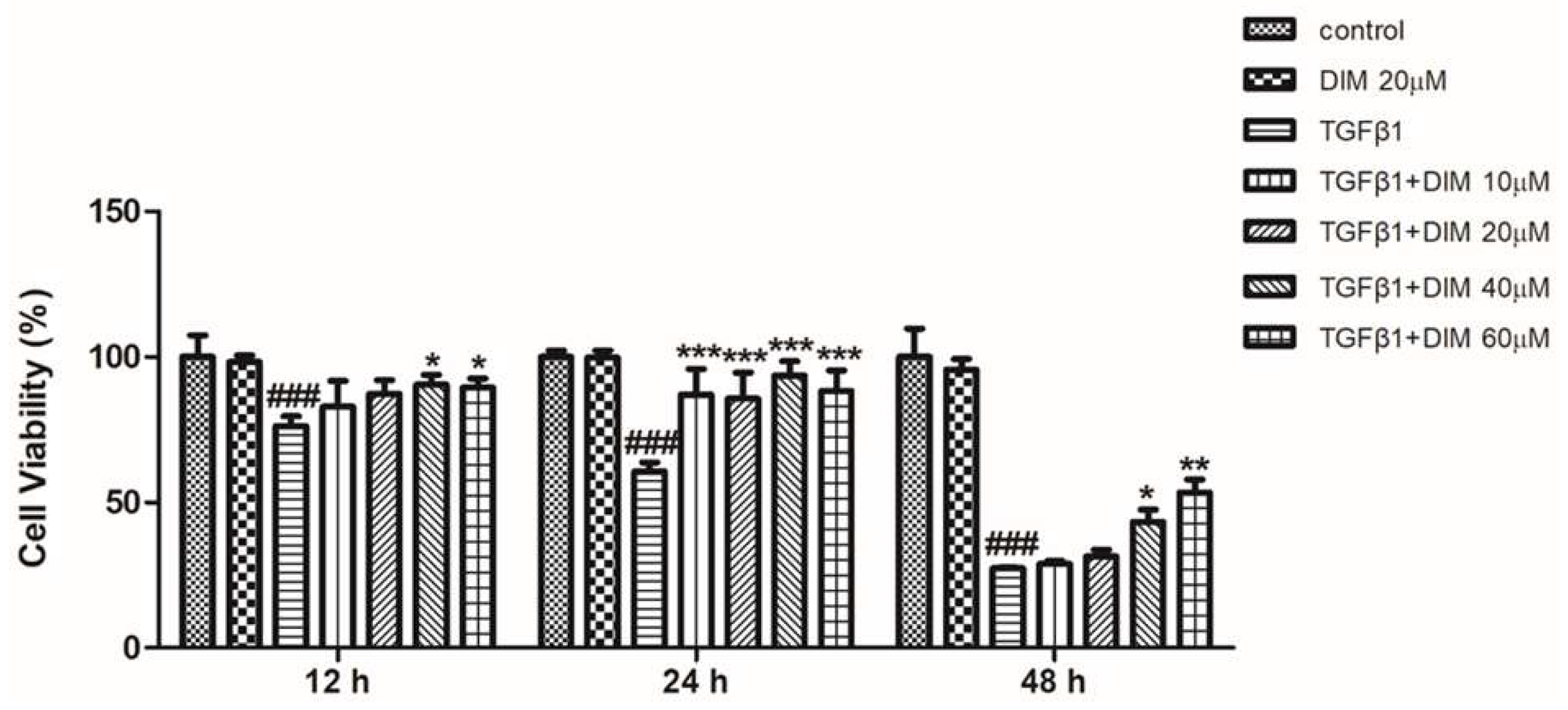
2.1. DIM Ameliorates TGF-β1 Induced Hepatotoxicity in AML12 Cells
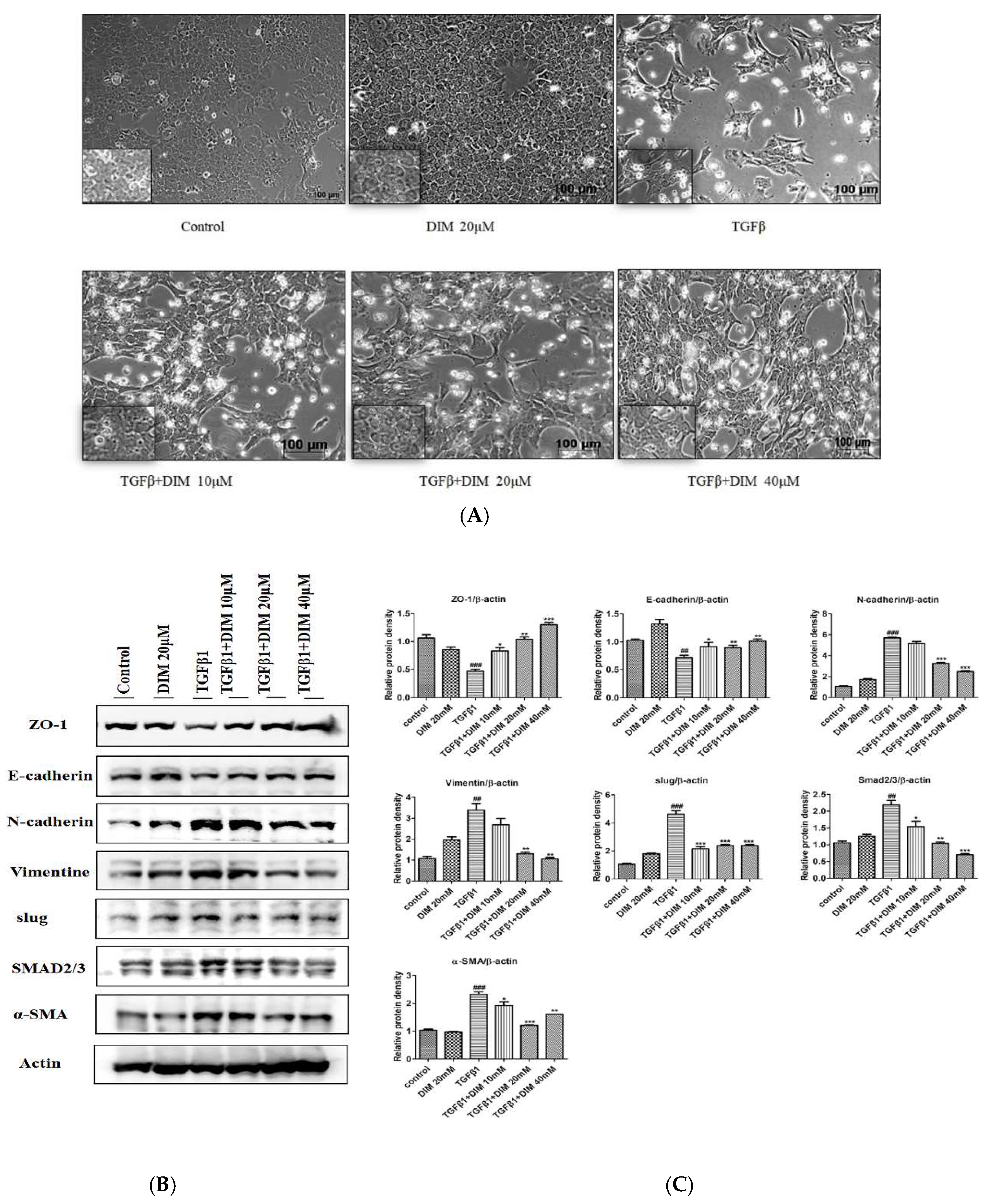
2.2. DIM Treatment Restores TGF-β1-Induced Mesenchymal Phenotype and Inhibits EMT in AML12 Cells
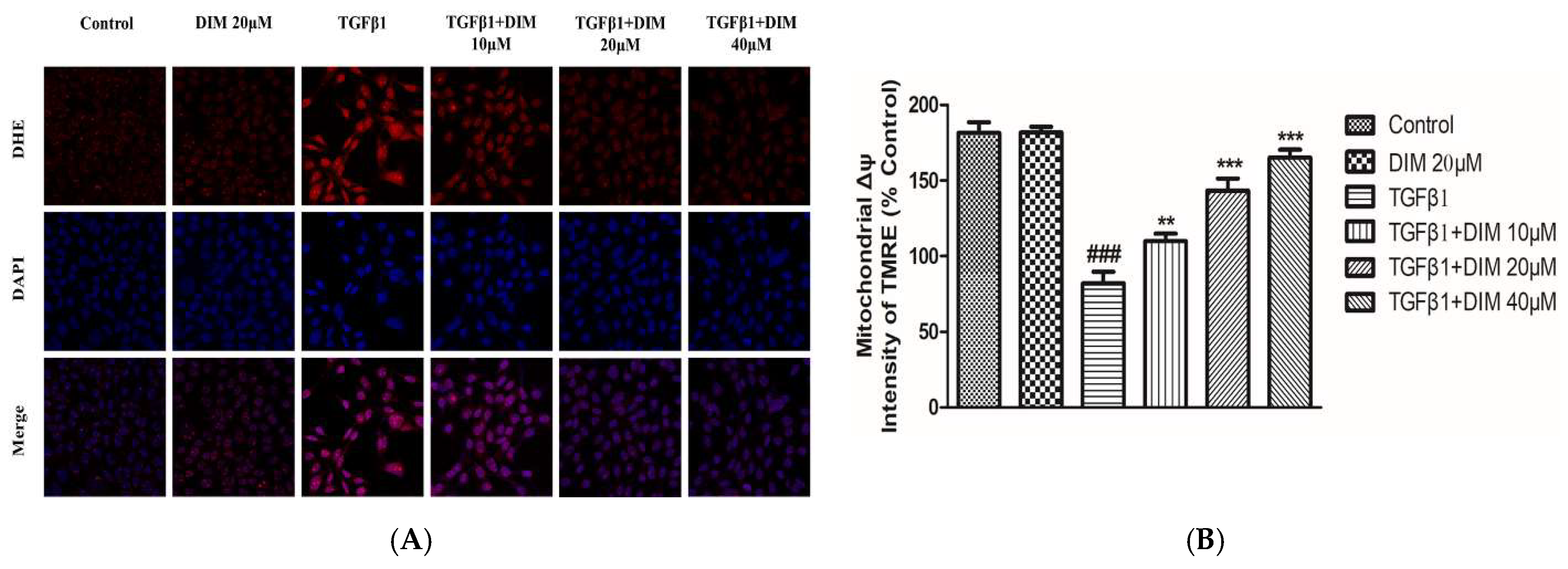
2.3. DIM Treatment Mitigates TGF-β-Induced ROS and Mitochondrial Dysfunction in AML12 Cells
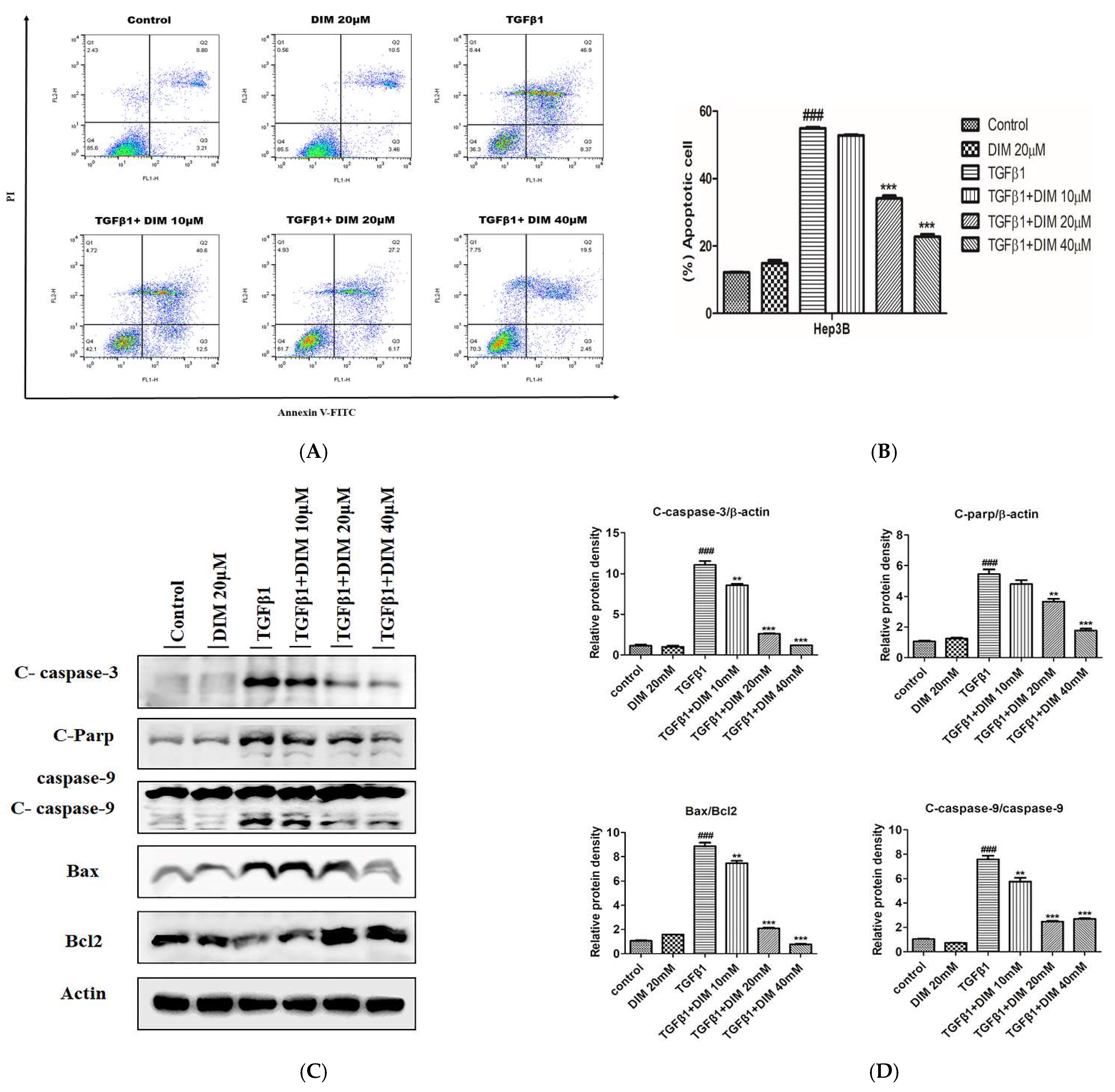
2.4. DIM Suppresses TGF-β1-Induced Apoptosis in AML12 Cells
2.5. DIM Suppresses TGF-β1-Induced Hepatocytes Apoptosis in AML12 Cells
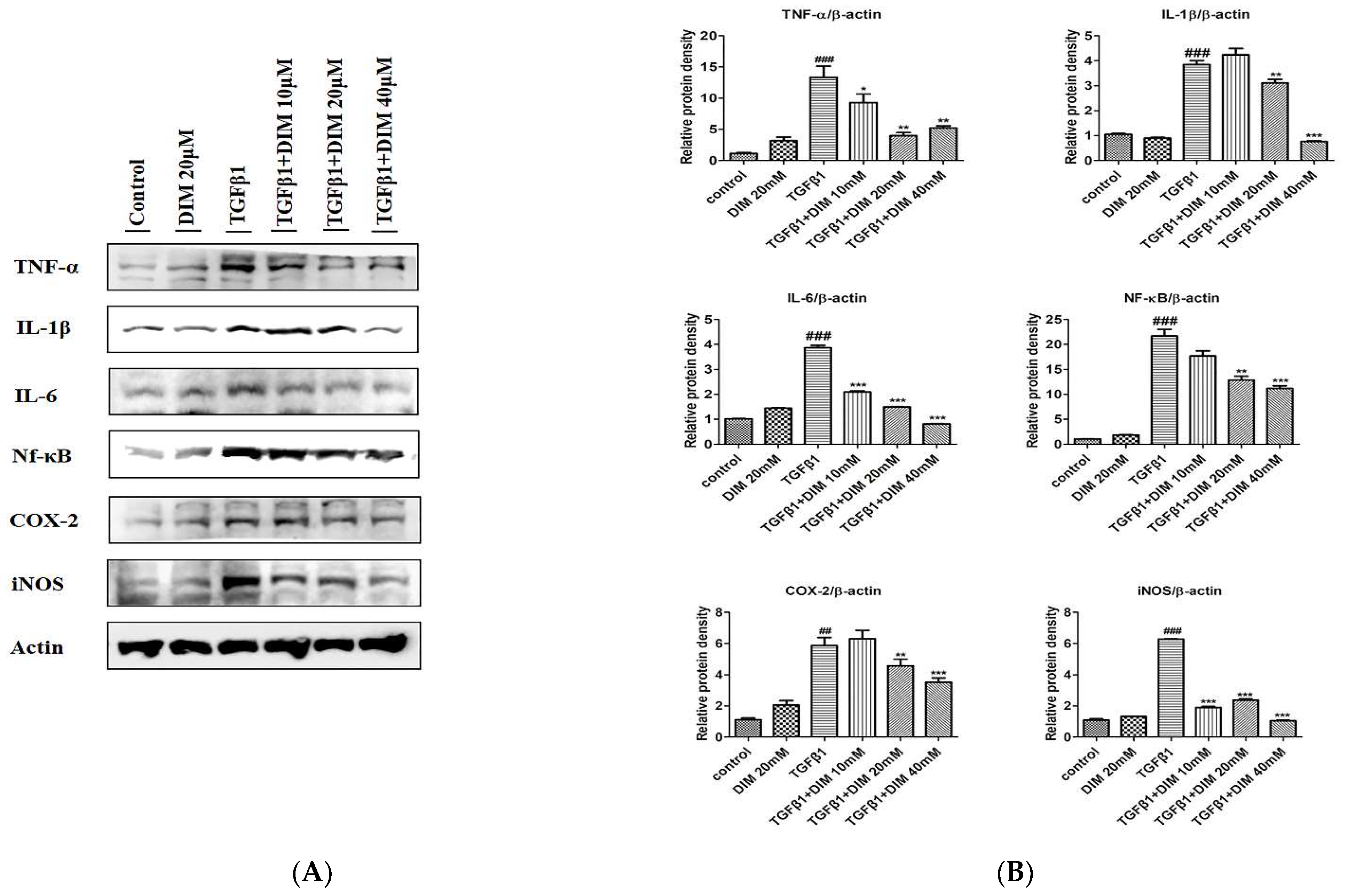
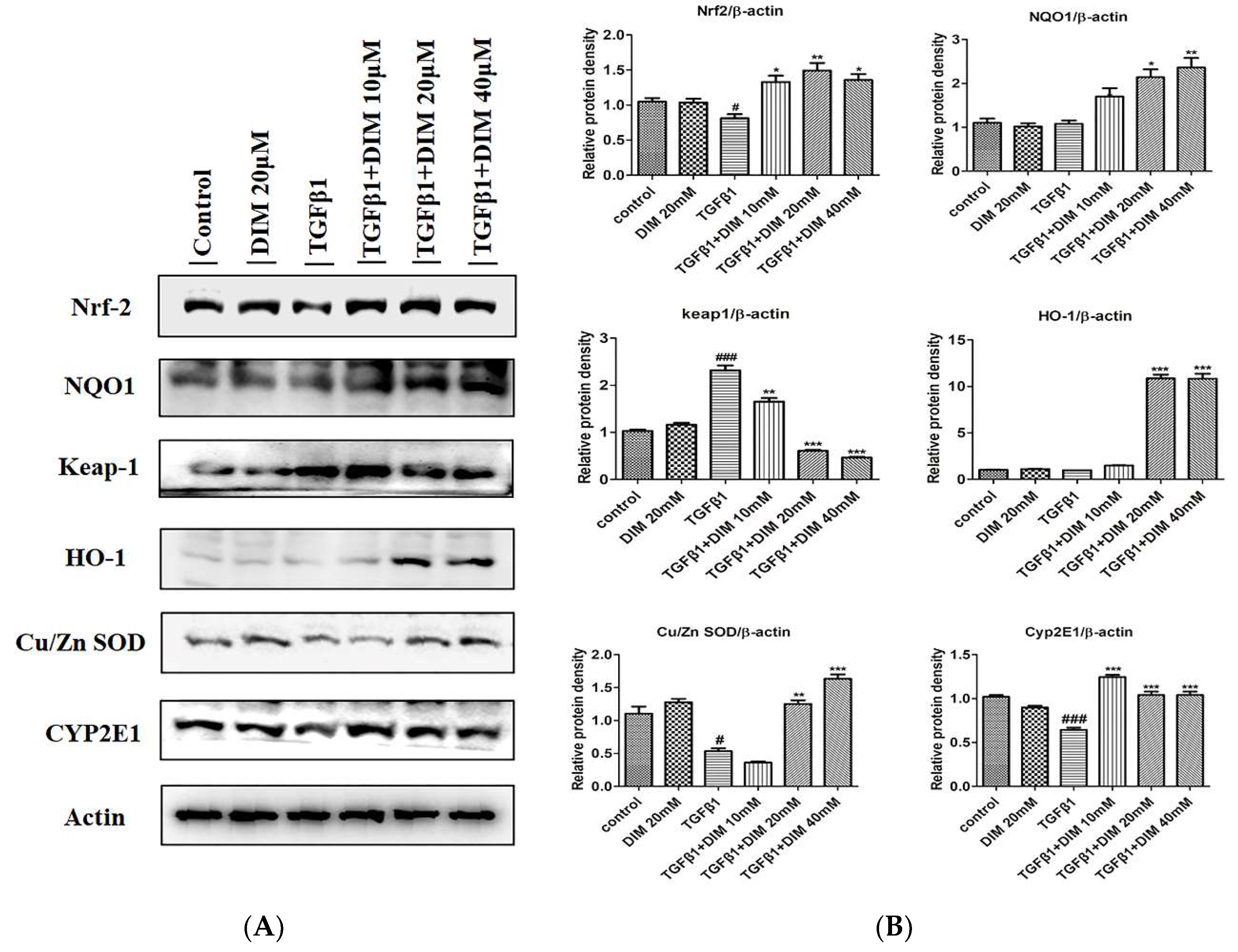
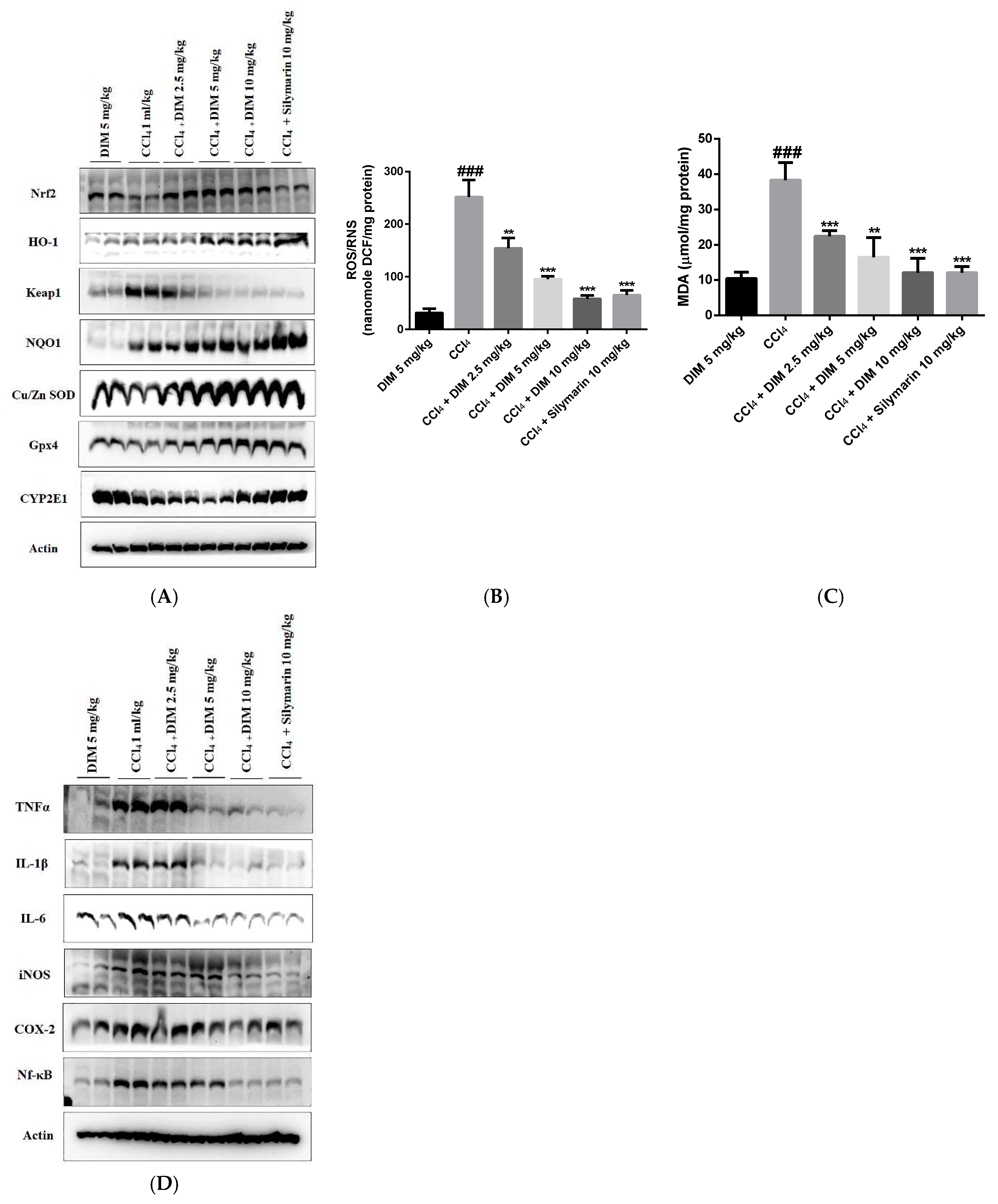
2.6. DIM Treatment Inhibits Oxidative Stress by Enhancing Antioxidant Activity by Activating the Nrf2/HO-1 Cascade
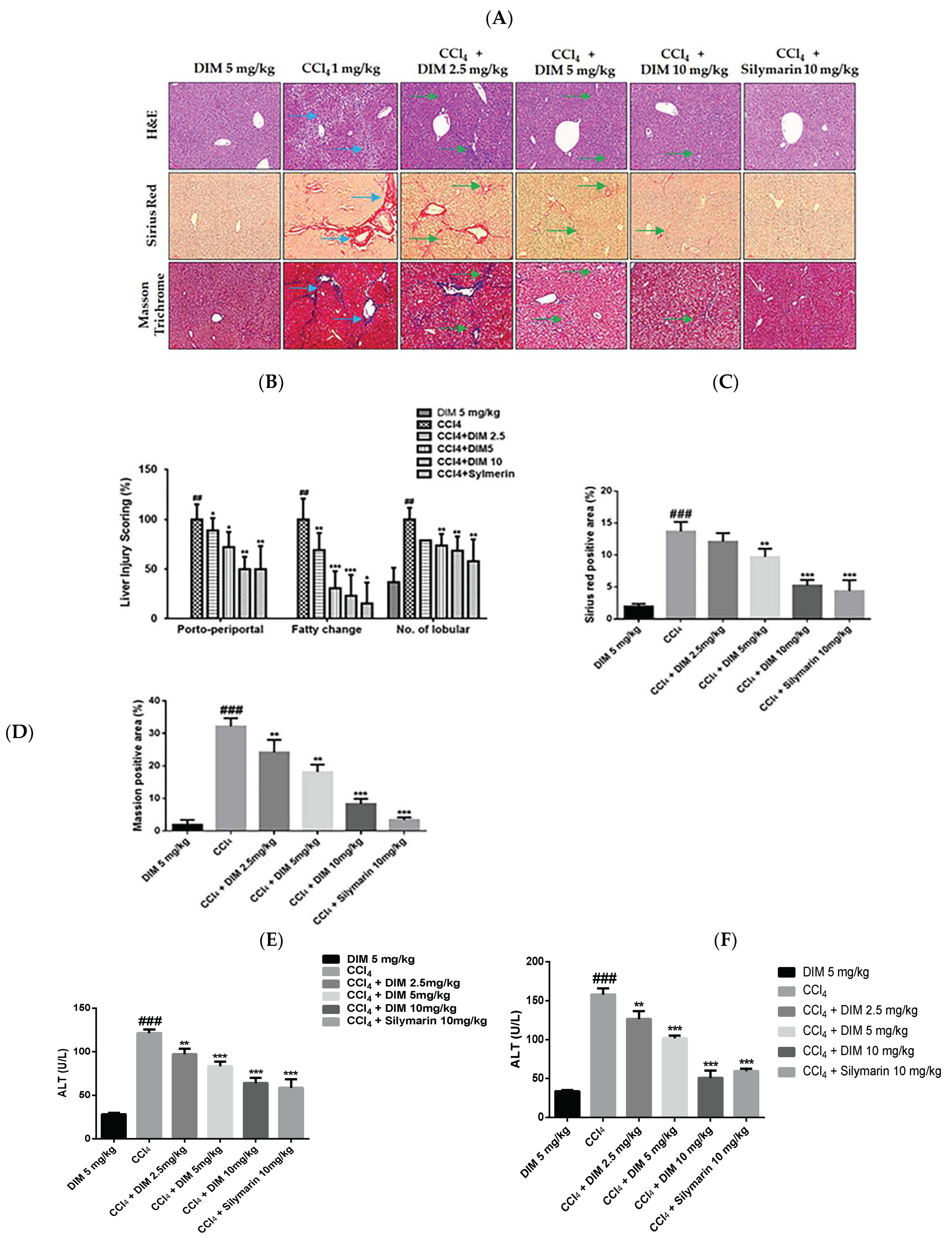
2.7. DIM Attenuates CCl4-Induced Chronic Liver Injury
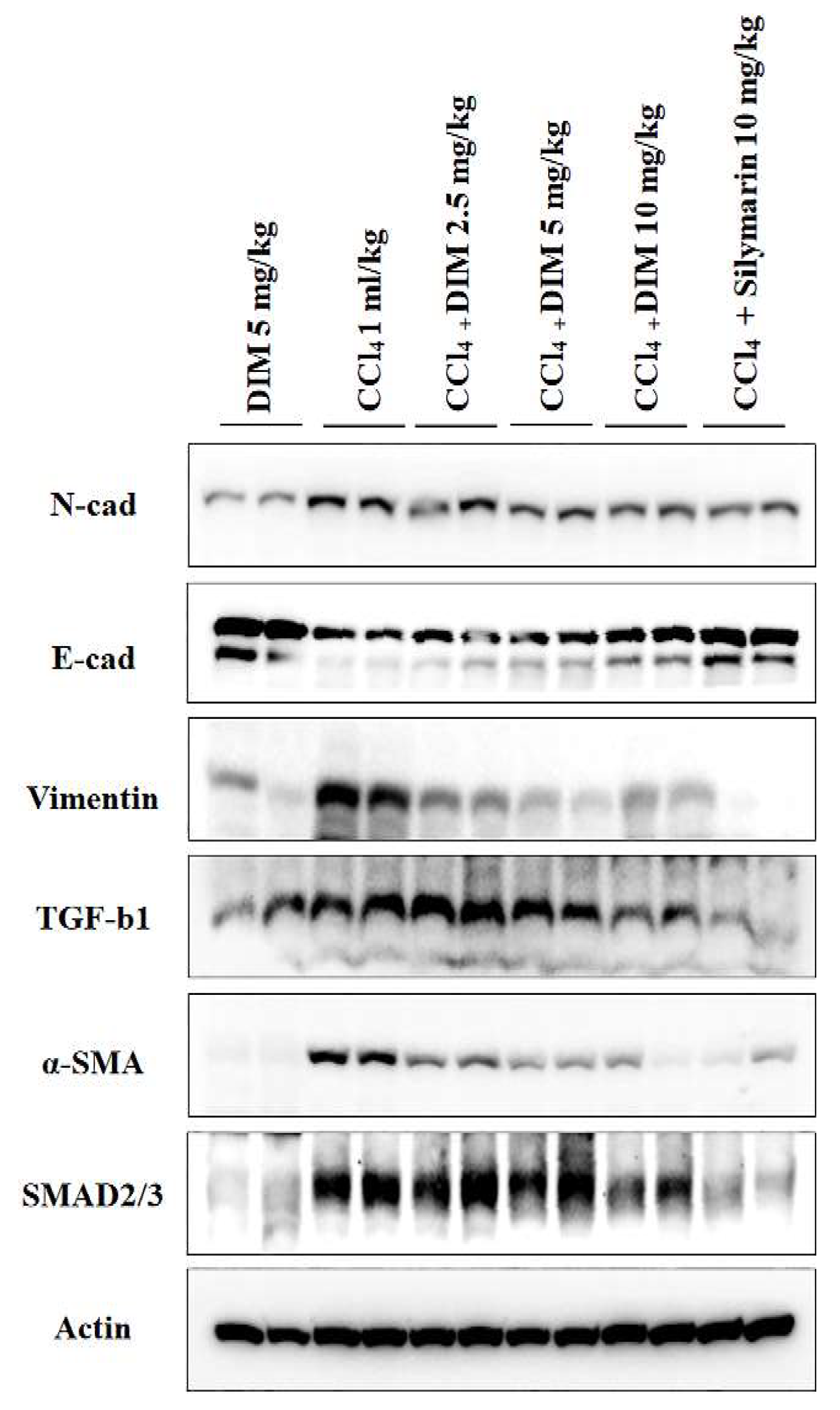
2.8. Effects of DIM on Expression Levels of EMT and TGF-β1/Smad in Mouse Liver
2.9. DIM Alleviates Oxidative Stress by Modulating Nrf2 Cascade in CCl4-Induced Liver Fibrosis
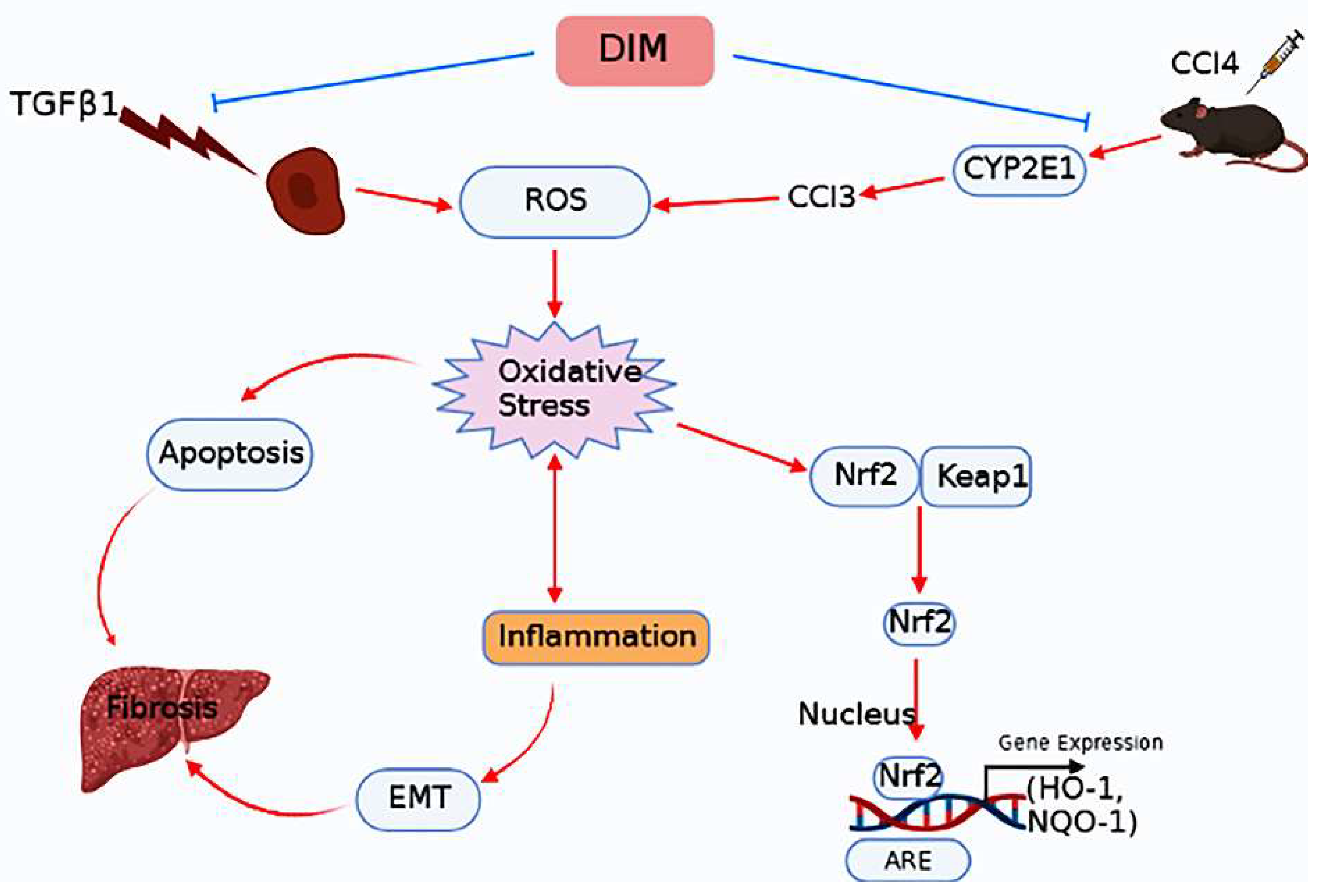
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures and Drug Treatment
4.3. Experimental Animals
4.4. Experimental Model and Drug Treatment
- (I)
- DIM group: Mice were treated with 5 mg/kg DIM subcutaneously daily for 4 weeks.
- (II)
- CCl4 group: Mice were treated with 10% CCl4 solution in mineral oil intraperitoneally with a dose of 1 mL/kg for three consecutive days for 8 weeks.
- (III)
- CCl4 + DIM 2.5 mg/kg group: Mice were i.p. injected with CCl4 for the first 4 weeks, followed by subcutaneous administration of DIM at 2.5 mg/kg for the last 4 weeks together with CCl4.
- (IV)
- CCl4 + DIM 5 mg/kg group: Mice were i.p. injected with CCl4 for the first 4 weeks, followed by subcutaneous administration of DIM at 5 mg/kg for the last 4 weeks together with CCl4.
- (V)
- CCl4 + DIM 10 mg/kg group: Mice were i.p. injected with CCl4 for the first 4 weeks, followed by subcutaneous administration of DIM at 10 mg/kg for the last 4 weeks together with CCl4.
- (VI)
- CCl4 + Silymarin 10 mg/kg group: Mice were i.p. injected with CCl4 for the first 4 weeks, followed by subcutaneous administration of DIM at 10 mg/kg for the last 4 weeks together with CCl4.
4.5. FACS Analysis
4.6. Detection of Reactive Oxygen Species (ROS)
4.7. Analysis of Mitochondrial Membrane Potential (ΔΨm)
4.8. Assessment of Biochemical Parameters
4.9. Measurements of Lipid Peroxidation
4.10. Histological Assessment
4.11. Immunoblotting
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gan, D.; Zhang, W.; Huang, C.; Chen, J.; He, W.; Wang, A.; Li, B.; Zhu, X. Ursolic acid ameliorates CCl4-induced liver fibrosis through the NOXs/ROS pathway. J. Cell Physiol. 2018, 233, 6799–6813. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Hamilton, J.P. Drug-induced Liver Injury. US Gastroenterol. Hepatol. Rev. 2010, 6, 73–80. [Google Scholar] [PubMed]
- Malhi, H.; Gores, G.J. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 2020, 11, 603926. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38 (Suppl. S1), S38–S53. [Google Scholar] [CrossRef]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.-J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Streckert, M.; Delvoux, B.; Gressner, A.M. Expression of Smads during in Vitro Transdifferentiation of Hepatic Stellate Cells to Myofibroblasts. Biochem. Biophys. Res. Commun. 2001, 283, 554–562. [Google Scholar] [CrossRef]
- Pan, X.; Wang, X.; Lei, W.; Min, L.; Yang, Y.; Wang, X.; Song, J. Nitric oxide suppresses transforming growth factor-β1–induced epithelial-to-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2009, 50, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, Z.; Yu, D.; Lin, J.; Cai, W. RUNX1 regulates TGF-β induced migration and EMT in colorectal cancer. Pathol. Res. Pract. 2020, 216, 153142. [Google Scholar] [CrossRef]
- Recknagel, R.O.; Glende, E.A.; Dolak, J.A.; Waller, R.L. Mechanisms of carbon tetrachloride toxicity. Pharmacol. Ther. 1989, 43, 139–154. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, B.; Han, X.; Xu, L.; Qi, Y.; Yin, L.; Xu, Y.; Zhao, Y.; Liu, K.; Peng, J. Protection of the flavonoid fraction from Rosa laevigata Michx fruit against carbon tetrachloride-induced acute liver injury in mice. Food Chem. Toxicol. 2013, 55, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Pomacu, M.M.; Trașcă, M.D.; Pădureanu, V.; Bugă, A.M.; Andrei, A.M.; Stănciulescu, E.C.; Baniță, I.M.; Rădulescu, D.; Pisoschi, C.G. Interrelation of inflammation and oxidative stress in liver cirrhosis. Exp. Ther. Med. 2021, 21, 602. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Siow, R.C.M.; Sugden, D.; Gao, L.; Cheng, X.; Mann, G.E. Induction of HO-1 and redox signaling in endothelial cells by advanced glycation end products: A role for Nrf2 in vascular protection in diabetes. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, X.; Zhang, M.; Jia, Y.; Yu, B.; Tian, J. Revisiting Tumors and the Cardiovascular System: Mechanistic Intersections and Divergences in Ferroptosis. Oxid. Med. Cell Longev. 2020, 2020, 9738143. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, W.; Wang, L.; Ma, L.; Zhai, D.; Wang, F.; Shi, R.; Liu, C.; Xu, Q.; Chen, G.; et al. Obacunone Attenuates Liver Fibrosis with Enhancing Anti-Oxidant Effects of GPx-4 and Inhibition of EMT. Molecules 2021, 26, 318. [Google Scholar] [CrossRef] [PubMed]
- Bataille, A.M.; Manautou, J.E. Nrf2: A Potential Target for New Therapeutics in Liver Disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Munakarmi, S.; Chand, L.; Shin, H.B.; Jang, K.Y.; Jeong, Y.J. Indole-3-Carbinol Derivative DIM Mitigates Carbon Tetrachloride-Induced Acute Liver Injury in Mice by Inhibiting Inflammatory Response, Apoptosis and Regulating Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 2048. [Google Scholar] [CrossRef]
- Munakarmi, S.; Shrestha, J.; Shin, H.-B.; Lee, G.-H.; Jeong, Y.-J. 3,3′-Diindolylmethane Suppresses the Growth of Hepatocellular Carcinoma by Regulating Its Invasion, Migration, and ER Stress-Mediated Mitochondrial Apoptosis. Cells 2021, 10, 1178. [Google Scholar] [CrossRef]
- Touyz, R.M. Molecular and cellular mechanisms in vascular injury in hypertension: Role of angiotensin II. Curr. Opin. Nephrol. Hypertens. 2005, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF β1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Kim, J.-Y.; An, H.-J.; Kim, W.-H.; Gwon, M.-G.; Gu, H.; Park, Y.-Y.; Park, K.-K. Anti-fibrotic Effects of Synthetic Oligodeoxynucleotide for TGF-β1 and Smad in an Animal Model of Liver Cirrhosis. Mol. Ther. Nucleic Acids 2017, 8, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Niu, J.; Ou, L.; Deng, B.; Wang, Y.; Li, S. Zerumbone Protects against Carbon Tetrachloride (CCl(4))-Induced Acute Liver Injury in Mice via Inhibiting Oxidative Stress and the Inflammatory Response: Involving the TLR4/NF-κB/COX-2 Pathway. Molecules 2019, 24, 1964. [Google Scholar] [CrossRef] [PubMed]
- Slater, T.; Cheeseman, K.; Ingold, K.U. Carbon tetrachloride toxicity as a model for studying free-radical mediated liver injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1985, 311, 633–645. [Google Scholar]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.J.; Choti, M.; Ding, Z.; Mezey, E.; Koteish, A.A. Histone deacetylase inhibition suppresses the transforming growth factor β1–induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology 2010, 52, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.H.; Kim, K.; Leem, J.; Park, K.K. Melatonin Inhibits Transforming Growth Factor-β1-Induced Epithelial-Mesenchymal Transition in AML12 Hepatocytes. Biology 2019, 8, 84. [Google Scholar] [CrossRef]
- Blick, T.; Widodo, E.; Hugo, H.; Waltham, M.; Lenburg, M.E.; Neve, R.M.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metastasis 2008, 25, 629–642. [Google Scholar] [CrossRef]
- Liu, J.; Zeng, L.; Zhao, Y.; Zhu, B.; Ren, W.; Wu, C. Selenium suppresses lipopolysaccharide-induced fibrosis in peritoneal mesothelial cells through inhibition of epithelial-to-mesenchymal transition. Biol. Trace Elem. Res. 2014, 161, 202–209. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, J.; Qian, J.; Wu, G.; Ma, Z. Emodin alleviates CCl4-induced liver fibrosis by suppressing epithelial-mesenchymal transition and transforming growth factor-β1 in rats. Mol. Med. Rep. 2018, 18, 3262–3270. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Kim, J.S.; Mohuczy, D.; Behrns, K.E. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology 2008, 48, 909–919. [Google Scholar] [CrossRef]
- Dai, C.; Yusuf, A.; Sun, H.; Shu, G.; Deng, X. A characterized saponin extract of Panax japonicus suppresses hepatocyte EMT and HSC activation in vitro and CCl4-provoked liver fibrosis in mice: Roles of its modulatory effects on the Akt/GSK3β/Nrf2 cascade. Phytomedicine 2021, 93, 153746. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Z.Y.; Jin, G.N.; Xiong, Y.X.; Yu, B.; Sun, Y.M.; Wang, W.; Liang, H.F.; Zhang, B.; Chen, X.P. Autocrine transforming growth factor-β/activin A-Smad signaling induces hepatic progenitor cells undergoing partial epithelial-mesenchymal transition states. Biochimie 2018, 148, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.; Zhang, J.; Lu, J. ENMD-1068 inhibits liver fibrosis through attenuation of TGF-β1/Smad2/3 signaling in mice. Sci. Rep. 2017, 7, 5498. [Google Scholar] [CrossRef]
- Fan, Y.-J.; Zong, W.-X. The cellular decision between apoptosis and autophagy. Chin. J. Cancer 2013, 32, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Alvarez, A.M.; Benito, M.; Fabregat, I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: Involvement of reactive oxygen intermediates. J. Biol. Chem. 1996, 271, 7416–7422. [Google Scholar] [CrossRef]
- Oberhammer, F.A.; Pavelka, M.; Sharma, S.; Tiefenbacher, R.; Purchio, A.F.; Bursch, W.; Schulte-Hermann, R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA 1992, 89, 5408–5412. [Google Scholar] [CrossRef] [PubMed]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid. Med. Cell. Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Casalena, G.; Daehn, I.; Bottinger, E. Transforming growth factor-β, bioenergetics, and mitochondria in renal disease. Semin. Nephrol. 2012, 32, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-P.; Hsieh, C.-H.; Liu, C.-Y.; Lin, K.-H.; Wu, P.-T.; Chen, K.-M.; Fang, K. Reactive oxygen species-driven mitochondrial injury induces apoptosis by teroxirone in human non-small cell lung cancer cells. Oncol. Lett. 2017, 14, 3503–3509. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Jiang, Y.-F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef]
- Levings, D.C.; Wang, X.; Kohlhase, D.; Bell, D.A.; Slattery, M. A distinct class of antioxidant response elements is consistently activated in tumors with NRF2 mutations. Redox Biol. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Jin, F.; Wan, C.; Li, W.; Yao, L.; Zhao, H.; Zou, Y.; Peng, D.; Huang, W. Formononetin protects against acetaminophen-induced hepatotoxicity through enhanced NRF2 activity. PLoS ONE 2017, 12, e0170900. [Google Scholar] [CrossRef]
- Xu, W.; Hellerbrand, C.; Köhler, U.A.; Bugnon, P.; Kan, Y.-W.; Werner, S.; Beyer, T.A. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab. Investig. 2008, 88, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Lee, K.H.; Khoo, H.E.; Hon, W.M. Expression of haem oxygenase in cirrhotic rat liver. J. Pathol. 2003, 199, 324–334. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.; Munday, M.R.; York, M.J.; Clarke, C.J.; Dare, T.; Turton, J.A. Comprehensive characterization of serum clinical chemistry parameters and the identification of urinary superoxide dismutase in a carbon tetrachloride-induced model of hepatic fibrosis in the female Hanover Wistar rat. Int. J. Exp. Pathol. 2007, 88, 361–376. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N | Target | Blocking Solution | Dilution | Secondary | Manufacturer | Catalogue Number |
|---|---|---|---|---|---|---|
| 1 | β-actin | 5% Skim Milk | 1:3000 | Mouse IgG | Sigma Aldrich | A5441 |
| 2 | COX-2 | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #12282 |
| 3 | TGF-β1 | %5 BSA | 1:2000 | Rabbit IgG | Abcam | Ab66043 |
| 4 | E-cadherin | %5 BSA | 1:2000 | Rabbit IgG | Cell Signalling | #3195 |
| 5 | C-Caspase-3 | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #9661 |
| 6 | Caspase-9 | %5 BSA | 1:1000 | Mouse IgG | Cell Signalling | #9508 |
| 7 | C-PARP | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #13669 |
| 8 | N-cadherin | %5 BSA | 1:2000 | Mouse IgG | Abcam | Ab18203 |
| 9 | Keap-1 | 5% Skim Milk | 1:1000 | Rabbit IgG | Santa Cruz | Sc-33569 |
| 10 | α-SMA | 5% Skim Milk | 1:2000 | Mouse IgG | Sigma | A2547 |
| 11 | Nrf-2 | 5% Skim Milk | 1:1000 | Rabbit IgG | Santa Cruz | sc-81342 |
| 12 | Nf-κB | 5% Skim Milk | 1:3000 | Rabbit IgG | Santa Cruz | sc-8008 |
| 13 | GpX4 | %5 BSA | 1:1000 | Mouse IgG | Santa Cruz | Sc-166570 |
| 14 | HO-1 | %5 BSA | 1:1500 | Mouse IgG | Enzo Life Science | 136960 |
| 15 | ZO-1 | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #13663 |
| 16 | iNOS | %5 BSA | 1:1500 | Rabbit IgG | Enzo Life Science | ADI-KAS-NO001 |
| 17 | IL-1β | 5% Skim Milk | 1:1000 | Rabbit IgG | Santa Cruz | sc-7884 |
| 18 | IL-6 | 5% Skim Milk | 1:1000 | Rabbit IgG | Santa Cruz | sc-57315 |
| 19 | Bax | 5% Skim Milk | 1:3000 | Rabbit IgG | Santa Cruz | sc-7480 |
| 20 | Bcl2 | 5% Skim Milk | 1:3000 | Rabbit IgG | Santa Cruz | sc-7382 |
| 21 | Snail | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #3879 |
| 22 | PARP | 5% Skim Milk | 1:2000 | Mouse IgG | Santa Cruz | sc-8007 |
| 23 | Caspase-3 | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #9661 |
| 24 | TNF-α | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #12744 |
| 25 | CYP2E1 | %5 BSA | 1:1500 | Rabbit IgG | Abcam | Ab28146 |
| 26 | Cu/Zn SOD | %5 BSA | 1:3000 | Rabbit IgG | Enzo Life Science | ADI-SOD-100 |
| 27 | NQO1 | 5% Skim Milk | 1:1500 | Mouse IgG | Santa Cruz | sc-32793 |
| 28 | Vimentin | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #5741 |
| 29 | Slug | %5 BSA | 1:1000 | Rabbit IgG | Cell Signalling | #9585 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munakarmi, S.; Gurau, Y.; Shrestha, J.; Risal, P.; Park, H.S.; Shin, H.B.; Jeong, Y.J. Hepatoprotective Effects of a Natural Flavanol 3,3′-Diindolylmethane against CCl4-Induced Chronic Liver Injury in Mice and TGFβ1-Induced EMT in Mouse Hepatocytes via Activation of Nrf2 Cascade. Int. J. Mol. Sci. 2022, 23, 11407. https://doi.org/10.3390/ijms231911407
Munakarmi S, Gurau Y, Shrestha J, Risal P, Park HS, Shin HB, Jeong YJ. Hepatoprotective Effects of a Natural Flavanol 3,3′-Diindolylmethane against CCl4-Induced Chronic Liver Injury in Mice and TGFβ1-Induced EMT in Mouse Hepatocytes via Activation of Nrf2 Cascade. International Journal of Molecular Sciences. 2022; 23(19):11407. https://doi.org/10.3390/ijms231911407
Chicago/Turabian StyleMunakarmi, Suvesh, Yamuna Gurau, Juna Shrestha, Prabodh Risal, Ho Sung Park, Hyun Beak Shin, and Yeon Jun Jeong. 2022. "Hepatoprotective Effects of a Natural Flavanol 3,3′-Diindolylmethane against CCl4-Induced Chronic Liver Injury in Mice and TGFβ1-Induced EMT in Mouse Hepatocytes via Activation of Nrf2 Cascade" International Journal of Molecular Sciences 23, no. 19: 11407. https://doi.org/10.3390/ijms231911407