
Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects
Abstract
:1. Introduction
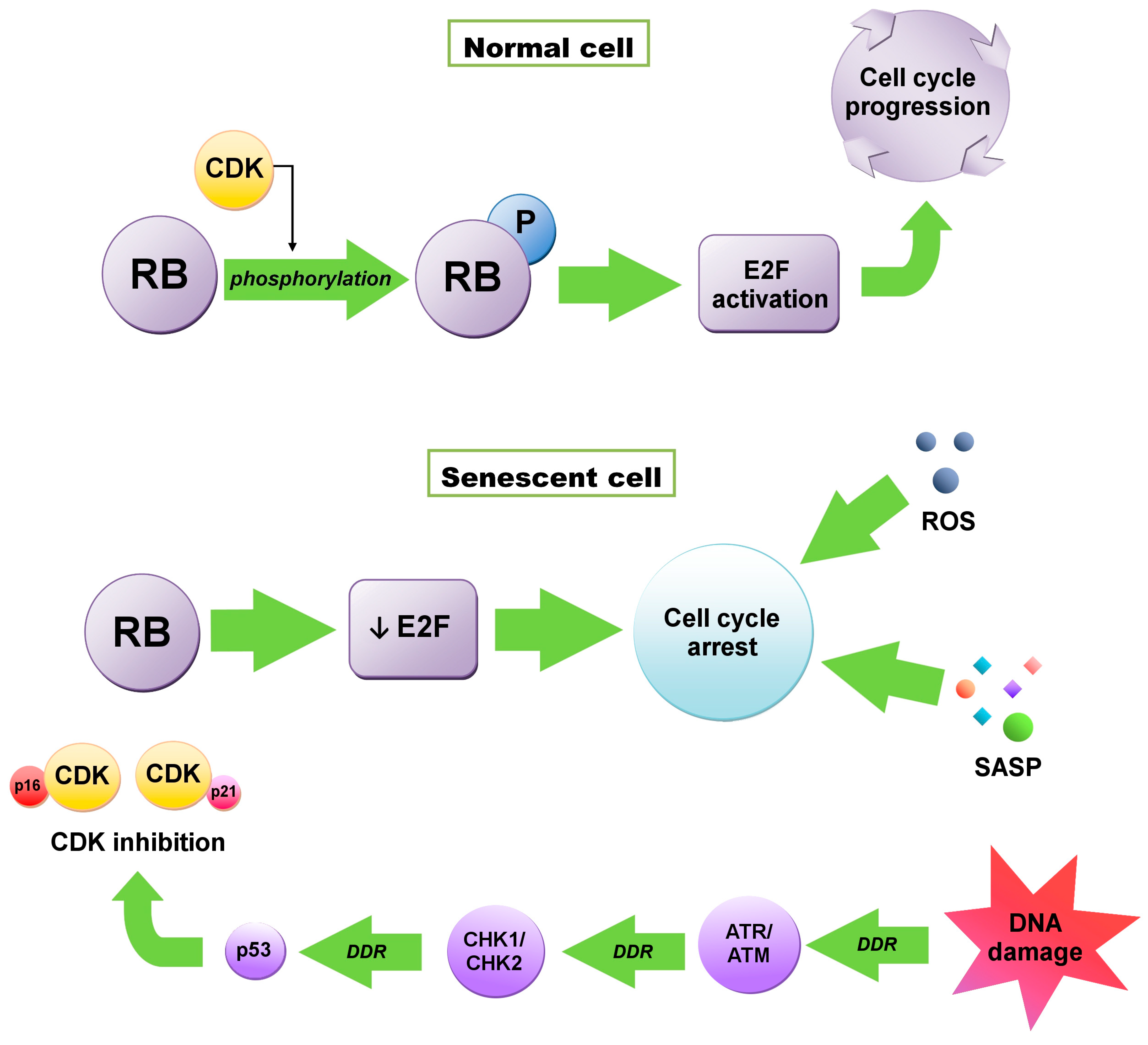
2. Senescence Induction
2.1. Replicative Senescence
2.2. Stress–Induced Senescence

2.2.1. Oncogene–Induced Senescence
2.2.2. Oxidative Stress
2.2.3. Metabolic Stress
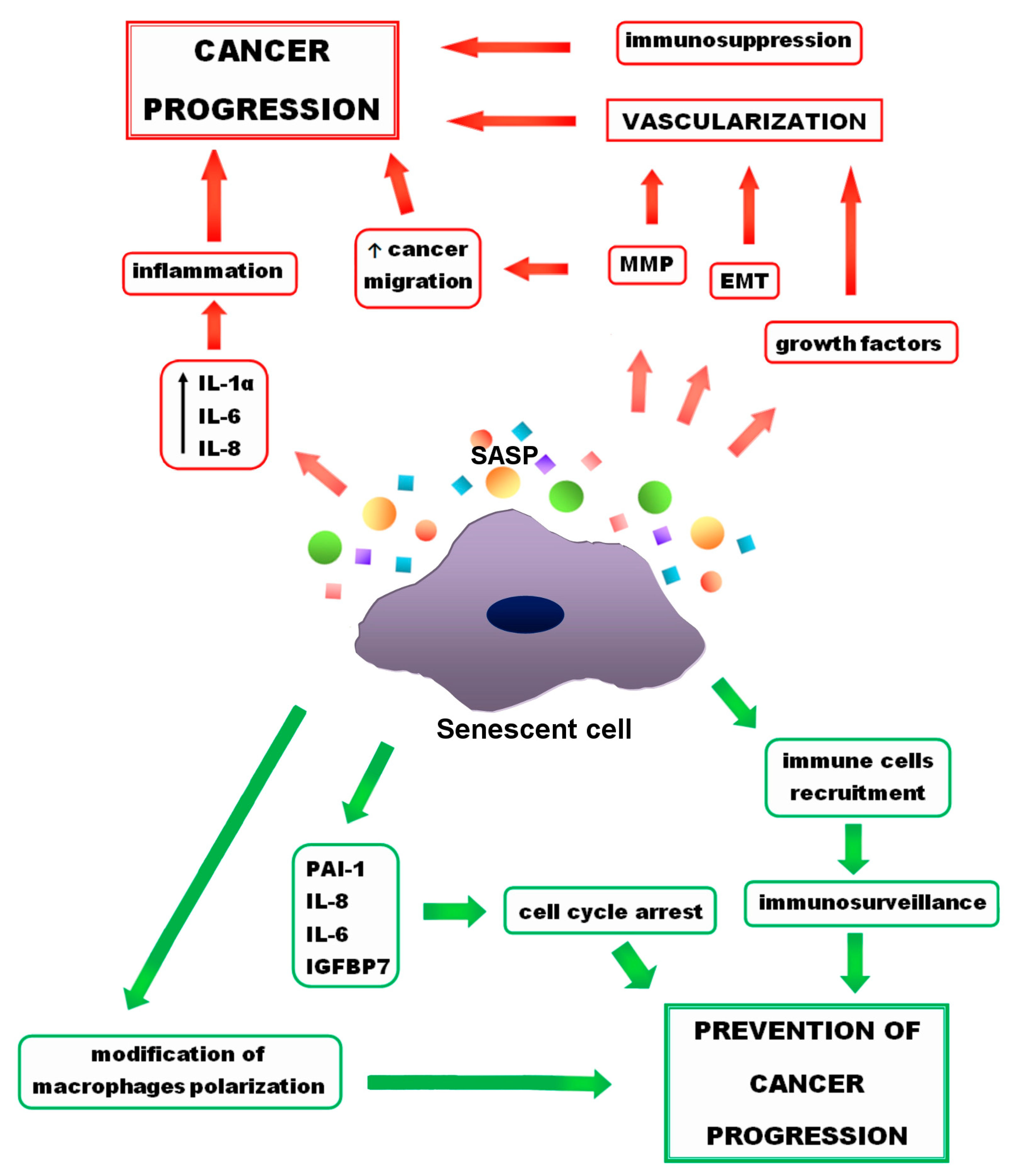
3. The SASP
3.1. The SASP Composition
3.2. The SASP Beneficial Effects
3.3. The Role of the SASP in Tumor-Promoting Environment
3.4. The SASP Regulation
3.4.1. Epigenetic Regulation
3.4.2. Post-Transcriptional Control
3.4.3. DDR and the SASP
4. Senescence and Cancer
4.1. Tumor Suppression
Immunosurveillance
4.2. Tumor Promotion
4.2.1. Invasiveness
4.2.2. EMT
4.2.3. Immunosenescence
5. Anticancer Therapies
5.1. Chemotherapy
5.2. Radiotherapy
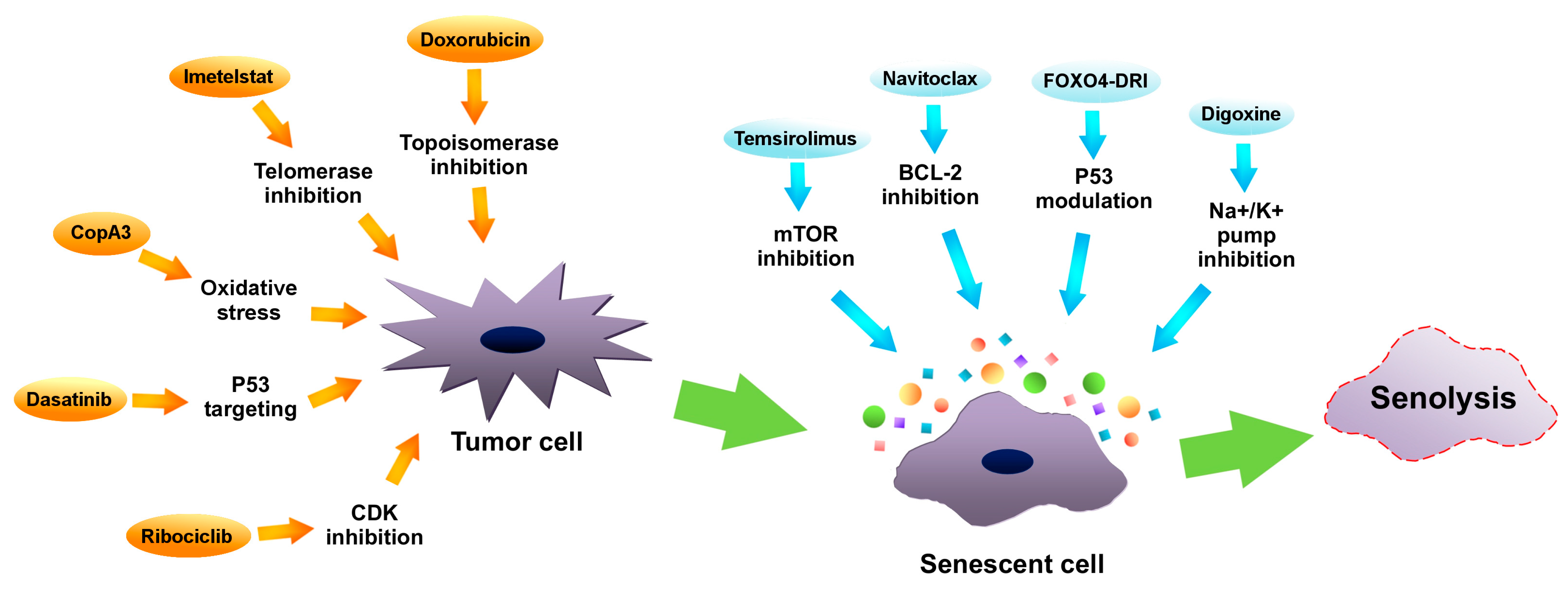
5.3. Prosenescence Therapies
5.3.1. Telomerase Inhibition
5.3.2. Topoisomerase Inhibition
5.3.3. Modulation of Cell Cycle Machinery
5.3.4. P53 Targeting
5.3.5. Oxidative Stress
5.3.6. SASP Reprogramming
5.4. Elimination of Senescent Cells
5.4.1. Senotherapies

5.4.2. Immunotherapy
5.5. Combination of Prosenescence Therapy and Senotherapy
6. Conclusions
- The composition of SASP depends on different agents such as cell type, senescence inducer, and time lapse. However, some molecules are present regardless of the cell type and the senescence inducer. This suggests that SASP has core molecules.
- The SASP can be regulated by a plethora of factors, and many of them have not been recognized yet. Their discovery could positively influence actual cancer treatment strategies.
- Senescence can support cancer treatment by arresting the cell cycle and activating immunosurveillance.
- Senescence may promote tumor progression by enhancing angiogenesis, providing growth factors, and protecting cancer cells from the immune system.
- Senescence and SASP can promote tumor resistance to therapy and lead to disease relapse.
- Telomerase inhibition, topoisomerase inhibition, cell cycle modulation, P53 regulation, oxidative stress induction and SASP reprogramming are effective in prosenescence therapy. It is advisable to look for more prosenescence compounds.
- Senotherapy is an effective method of neutralization and elimination of senescent tumor cells. Searching for more senolytic and senostatic compounds is recommended. Moreover, more research is needed on the currently known senolytics.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | 5′AMP-activated protein kinase |
| ARF | ADP ribosylation factor |
| ATM | ataxia telangiectasia mutated kinase; serine-protein kinase ATM |
| ATR | ataxia telangiectasia and Rad3-related kinase |
| BRD4 | bromodomain-containing protein 4 |
| C/EBPβ | CCAAT/enhancer-binding protein-β |
| CCF | cytosolic chromatin fragments |
| CCL2 | C-C motif chemokine ligand 2 |
| CCL5 | C-C motif chemokine ligand 5 |
| CCN1 | cellular communication network factor 1 |
| CDK | cyclin-dependent kinase |
| cGAMP | cyclic GMP-AMP |
| cGAS | cyclic GMP-AMP synthase |
| CHK1 | checkpoint kinase 1 |
| CHK2 | checkpoint kinase 2 |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| CXCL5 | C-X-C motif chemokine ligand 5 |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| DAMP | damage-associated molecular patterns |
| DDR | DNA damage response |
| DSB | double-strand break |
| ECM | extracellular matrix |
| EMT | epithelial-to-mesenchymal transformation |
| ERK | extracellular signal-regulated kinase |
| FGFR4 | fibroblast growth factor receptor 4 |
| GATA4 | GATA Binding Protein 4 |
| GDNF | glial cell line-derived neurotrophic factor |
| H2AX | H2A histone family member X |
| γH2AX | phosphorylated H2A histone family member X |
| HCC | hepatocellular carcinoma |
| HDAC | histone deacetylase |
| HMEC | human normal mammary epithelial cells |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A reductase |
| HNSCC | head and neck squamous carcinoma |
| HSP | heat shock proteins |
| IFN | interferon |
| IFN-I | type-I interferon |
| IGFBP | insulin-like growth factor-binding protein |
| IL-1 α | interleukin 1 α |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| IR | ionizing radiation |
| JAK2 | janus kinase 2 |
| MAPK | mitogen-activated protein kinases |
| MAPKAPK2 | MAPK activated protein kinase 2 |
| MDC1 | mediator of DNA damage checkpoint 1 |
| MDM2 | mouse double minute 2 homolog |
| MDSC | myeloid-derived suppressor cells |
| MICA | MHC class I chain-related protein A |
| MICB | MHC class I chain-related protein B |
| MiDAS | mitochondrial dysfunction-associated senescence |
| MMP | metalloproteinase |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NAD+ | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAMPT | nicotinamide phosphoribosyltransferase |
| NBS1 | Nijmegen breakage syndrome 1 protein |
| NF-κB | nuclear factor kappa B |
| NFBD1 | nuclear factor with BRCT domains protein 1NK—natural killer |
| NKG2D | natural killer group 2D |
| NOTCH1 | neurogenic locus notch homolog protein 1 |
| NSCLC | non-small-cell lung carcinoma |
| OIS | oncogene-induced senescence |
| OPN | osteoponin |
| PAI-1 | plasminogen activator inhibitor |
| PDGF-AA | platelet-derived growth factor AA |
| PI3K | phosphoinositide 3-kinase |
| PICS | PTEN loss-induced cellular senescence |
| POT1 | protection of telomeres 1 |
| PTB1 | polypyrimidine tract-binding protein 1 |
| PTEN | phosphatase and tensin homolog deleted on chromosome ten |
| RB | retinoblastoma |
| ROS | reactive oxygen species |
| SAL | supraphysiological androgen level |
| SASP | senescence-associated secretory phenotype |
| SA-β-galactosidase | senescence-associated beta-galactosidase |
| SIRT1 | sirtuin 1 |
| STAT3 | signal transducer and activator of transcription |
| STING | stimulator of interferon genes |
| TASCC | TOR-autophagy spatial coupling compartment |
| TBK1 | TANK-binding kinase 1 |
| TERT | telomerase reverse transcriptase |
| TGF | transforming growth factor |
| Th1 | T helper type 1 |
| TIS | therapy-induced senescence |
| TLR2 | toll-like receptor 2 |
| TNF α | tumor necrosis factor α |
| TRF2 | telomeric repeat-binding factor 2 |
| TSC2 | tuberous sclerosis complex 2 |
| ULBP2 | UL16 binding protein 2 |
| UPR | unfolded protein response |
| VEGF | vascular endothelial growth factor |
References
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Kuliman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Davaapil, H.; Brockes, J.P.; Yun, M.H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 2017, 144, 106–114. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Yang, J.; Liu, M.; Hong, D.; Zeng, M.; Zhang, X. The Paradoxical Role of Cellular Senescence in Cancer. Front. Cell Dev. Biol. 2021, 9, 722205. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.H.; de Lange, T. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004, 2, E240. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Sławińska, N.; Krupa, R. Molecular Aspects of Senescence and Organismal Ageing-DNA Damage Response, Telomeres, Inflammation and Chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Parisotto, M.; Grelet, E.; El Bizri, R.; Dai, Y.; Terzic, J.; Eckert, D.; Gargowitsch, L.; Bornert, J.M.; Metzger, D. PTEN deletion in luminal cells of mature prostate induces replication stress and senescence in vivo. J. Exp. Med. 2018, 215, 1749–1763. [Google Scholar] [CrossRef] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Brown, N.J.; Pockley, A.G.; Wyld, L. The unfolded protein response and cancer: A brighter future unfolding? J. Mol. Med. 2007, 85, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Xue, W.; Zender, L.; Yon, M.; Hernando, E.; Lowe, S.W. Implications of cellular senescence in tissue damage response, tumor suppression, and stem cell biology. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 513–522. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Tong, X.; O’Kelly, J.; Xie, D.; Mori, A.; Lemp, N.; McKenna, R.; Miller, C.W.; Koeffler, H.P. Cyr61 suppresses the growth of non-small-cell lung cancer cells via the beta-catenin-c-myc-p53 pathway. Oncogene 2004, 23, 4847–4855. [Google Scholar] [CrossRef]
- Li, X.; Yuan, N.; Lin, L.; Yin, L.; Qu, Y. Targeting cysteine-rich angiogenic inducer-61 by antibody immunotherapy suppresses growth and migration of non-small cell lung cancer. Exp. Ther. Med. 2018, 16, 730–738. [Google Scholar] [CrossRef]
- Zhu, Y.; Almuntashiri, S.; Han, Y.; Wang, X.; Somanath, P.R.; Zhang, D. The Roles of CCN1/CYR61 in Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 7810. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Meng, J.; Kou, S.; Jiang, Z.; Huang, X.; Lu, Z.; Zhao, H.; Lau, L.F.; Zhou, B.; Zhang, H. CCN1-Induced Cellular Senescence Promotes Heart Regeneration. Circulation 2019, 139, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Won, J.H.; Cheng, N.; Lau, L.F. The matricellular protein CCN1 in tissue injury repair. J. Cell Commun. Signal. 2018, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Jandova, J.; Beyer, T.E.; Meuillet, E.J.; Watts, G.S. The matrix protein CCN1/CYR61 is required for alpha(V)beta5-mediated cancer cell migration. Cell Biochem. Funct. 2012, 30, 687–695. [Google Scholar] [CrossRef]
- Sabile, A.A.; Arlt, M.J.; Muff, R.; Bode, B.; Langsam, B.; Bertz, J.; Jentzsch, T.; Puskas, G.J.; Born, W.; Fuchs, B. Cyr61 expression in osteosarcoma indicates poor prognosis and promotes intratibial growth and lung metastasis in mice. J. Bone Miner. Res. 2012, 27, 58–67. [Google Scholar] [CrossRef]
- Fromigue, O.; Hamidouche, Z.; Vaudin, P.; Lecanda, F.; Patino, A.; Barbry, P.; Mari, B.; Marie, P.J. CYR61 downregulation reduces osteosarcoma cell invasion, migration, and metastasis. J. Bone Miner. Res. 2011, 26, 1533–1542. [Google Scholar] [CrossRef]
- Huang, Y.T.; Lan, Q.; Lorusso, G.; Duffey, N.; Ruegg, C. The matricellular protein CYR61 promotes breast cancer lung metastasis by facilitating tumor cell extravasation and suppressing anoikis. Oncotarget 2017, 8, 9200–9215. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghosh, A.; VonHoff, D.D.; Banerjee, S.K. Cyr61/CCN1 targets for chemosensitization in pancreatic cancer. Oncotarget 2019, 10, 3579–3580. [Google Scholar] [CrossRef]
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Curnow, S.J.; Scheel-Toellner, D.; Jenkinson, W.; Raza, K.; Durrani, O.M.; Faint, J.M.; Rauz, S.; Wloka, K.; Pilling, D.; Rose-John, S.; et al. Inhibition of T cell apoptosis in the aqueous humor of patients with uveitis by IL-6/soluble IL-6 receptor trans-signaling. J. Immunol. 2004, 173, 5290–5297. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Immune surveillance: A balance between protumor and antitumor immunity. Curr. Opin. Genet. Dev. 2008, 18, 11–18. [Google Scholar] [CrossRef]
- Bakouny, Z.; Choueiri, T.K. IL-8 and cancer prognosis on immunotherapy. Nat. Med. 2020, 26, 650–651. [Google Scholar] [CrossRef]
- Ruan, W.; Xu, E.; Xu, F.; Ma, Y.; Deng, H.; Huang, Q.; Lv, B.; Hu, H.; Lin, J.; Cui, J.; et al. IGFBP7 plays a potential tumor suppressor role in colorectal carcinogenesis. Cancer Biol. Ther. 2007, 6, 354–359. [Google Scholar] [CrossRef]
- Chen, D.; Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Bhutia, S.K.; Das, S.K.; Fuller, C.; Su, Z.Z.; Fisher, P.B.; Sarkar, D. Insulin-like growth factor-binding protein-7 functions as a potential tumor suppressor in hepatocellular carcinoma. Clin. Cancer Res. 2011, 17, 6693–6701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lian, R.; Zhao, J.; Feng, X.; Ye, R.; Pan, L.; Wu, J.; Li, M.; Huang, Y.; Cai, J. IGFBP7 inhibits cell proliferation by suppressing AKT activity and cell cycle progression in thyroid carcinoma. Cell Biosci. 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhao, R.; Song, C.; Wang, H.; Rong, J.; Wang, F.; Yan, L.; Song, Y.; Xie, Y. Increased IGFBP7 Expression Correlates with Poor Prognosis and Immune Infiltration in Gastric Cancer. J. Cancer 2021, 12, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xi, Y.; Zhu, G.; Jia, J.; Huang, H.; Liu, Y.; Guo, Y.; Liu, L. Downregulated IGFBP7 facilitates liver metastasis by modulating epithelial-mesenchymal transition in colon cancer. Oncol. Rep. 2019, 42, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Takiyama, Y.; Honjyo, J.; Makino, Y.; Fujita, Y.; Tateno, M.; Haneda, M. Role of IGFBP7 in Diabetic Nephropathy: TGF-β1 Induces IGFBP7 via Smad2/4 in Human Renal Proximal Tubular Epithelial Cells. PLoS ONE 2016, 11, e0150897. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, L.; Wang, X.; Hou, F. Silence of IGFBP7 suppresses apoptosis and epithelial mesenchymal transformation of high glucose induced-podocytes. Exp. Ther. Med. 2018, 16, 1095–1102. [Google Scholar] [CrossRef]
- Wicklein, D.; Otto, B.; Suling, A.; Elies, E.; Lüers, G.; Lange, T.; Feldhaus, S.; Maar, H.; Schröder-Schwarz, J.; Brunner, G.; et al. CEACAM1 promotes melanoma metastasis and is involved in the regulation of the EMT associated gene network in melanoma cells. Sci. Rep. 2018, 8, 11893. [Google Scholar] [CrossRef]
- Tang, X.; Mu, J.; Ma, L.; Tan, Q.; Wang, J.; Tan, J.; Zhang, S. IGFBP7 overexpression promotes acquired resistance to AZD9291 in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2021, 571, 38–45. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Bhakta, N.; Liu, Q.; Yeo, F.; Baassiri, M.; Ehrhardt, M.J.; Srivastava, D.K.; Metzger, M.L.; Krasin, M.J.; Ness, K.K.; Hudson, M.M.; et al. Cumulative burden of cardiovascular morbidity in paediatric, adolescent, and young adult survivors of Hodgkin’s lymphoma: An analysis from the St Jude Lifetime Cohort Study. Lancet Oncol. 2016, 17, 1325–1334. [Google Scholar] [CrossRef]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; LeBrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of premature ageing in survivors of cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Woenne, E.C.; Lederle, W.; Zwick, S.; Palmowski, M.; Krell, H.; Semmler, W.; Mueller, M.M.; Kiessling, F. MMP inhibition blocks fibroblast-dependent skin cancer invasion, reduces vascularization and alters VEGF-A and PDGF-BB expression. Anticancer Res. 2010, 30, 703–711. [Google Scholar]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Pazolli, E.; Luo, X.; Brehm, S.; Carbery, K.; Chung, J.J.; Prior, J.L.; Doherty, J.; Demehri, S.; Salavaggione, L.; Piwnica-Worms, D.; et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009, 69, 1230–1239. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Nacarelli, T.; Fukumoto, T.; Zundell, J.A.; Fatkhutdinov, N.; Jean, S.; Cadungog, M.G.; Borowsky, M.E.; Zhang, R. NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res. 2020, 80, 890–900. [Google Scholar] [CrossRef]
- Basu, S.K.; Lee, S.; Salotti, J.; Basu, S.; Sakchaisri, K.; Xiao, Z.; Walia, V.; Westlake, C.J.; Morrison, D.K.; Johnson, P.F. Oncogenic RAS-Induced Perinuclear Signaling Complexes Requiring KSR1 Regulate Signal Transmission to Downstream Targets. Cancer Res. 2018, 78, 891–908. [Google Scholar] [CrossRef]
- Alimirah, F.; Pulido, T.; Valdovinos, A.; Alptekin, S.; Chang, E.; Jones, E.; Diaz, D.A.; Flores, J.; Velarde, M.C.; Demaria, M.; et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 2020, 80, 3606–3619. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, N.; Banito, A.; Roe, J.S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.H.; Aksoy, O.; Bolden, J.E.; Chen, C.C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef]
- Hari, P.; Millar, F.R.; Tarrats, N.; Birch, J.; Quintanilla, A.; Rink, C.J.; Fernández-Duran, I.; Muir, M.; Finch, A.J.; Brunton, V.G.; et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci. Adv. 2019, 5, eaaw0254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102. [Google Scholar] [CrossRef]
- Fujita, M.; Sasada, M.; Eguchi, M.; Iyoda, T.; Okuyama, S.; Osawa, T.; Tsuzuranuki, K.; Sakamoto, M.; Hagihara, Y.; Matsumura, M.; et al. Induction of cellular senescence in fibroblasts through β1-integrin activation by tenascin-C-derived peptide and its protumor effect. Am. J. Cancer Res. 2021, 11, 4364–4379. [Google Scholar]
- Wiley, C.D. Bubble Bubble, Senescent Cells Are a Cauldron of Tumor Trouble. Cancer Res. 2020, 80, 3193–3194. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Miao, J.W.; Liu, L.J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [CrossRef]
- Vicente, R.; Mausset-Bonnefont, A.L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B.; Dagarag, M.; Valenzuela, H.F. In vitro senescence of immune cells. Exp. Gerontol. 2003, 38, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef]
- Ramello, M.C.; Tosello Boari, J.; Canale, F.P.; Mena, H.A.; Negrotto, S.; Gastman, B.; Gruppi, A.; Acosta Rodríguez, E.V.; Montes, C.L. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014, 5, e1507. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Chen, S.; Liu, H.; Su, N.; Zhang, G.; Wang, L. Myeloid-derived suppressor cells promote age-related increase of lung cancer growth via B7-H1. Exp. Gerontol. 2015, 61, 84–91. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lam, E.W.; Sun, Y. Senescent cells: A new Achilles’ heel to exploit for cancer medicine? Aging Cell 2019, 18, e12875. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef]
- Chen, C.; Xu, Z.Q.; Zong, Y.P.; Ou, B.C.; Shen, X.H.; Feng, H.; Zheng, M.H.; Zhao, J.K.; Lu, A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178. [Google Scholar] [CrossRef]
- Zhao, J.; Ou, B.; Han, D.; Wang, P.; Zong, Y.; Zhu, C.; Liu, D.; Zheng, M.; Sun, J.; Feng, H.; et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3β/β-catenin pathways. Mol. Cancer 2017, 16, 70. [Google Scholar] [CrossRef]
- Zhu, F.; Liu, P.; Li, J.; Zhang, Y. Eotaxin-1 promotes prostate cancer cell invasion via activation of the CCR3-ERK pathway and upregulation of MMP-3 expression. Oncol. Rep. 2014, 31, 2049–2054. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Pellegrini, M.; Pallini, R.; Levi, A.; Falchetti, M.L. Cancer Response to Therapy-Induced Senescence: A Matter of Dose and Timing. Cancers 2021, 13, 484. [Google Scholar] [CrossRef]
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Jallepalli, P.V.; Waizenegger, I.C.; Bunz, F.; Langer, S.; Speicher, M.R.; Peters, J.M.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Securin is required for chromosomal stability in human cells. Cell 2001, 105, 445–457. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, B.C.; Park, M.J.; Lee, Y.S.; Kim, Y.N.; Lee, B.L.; Lee, J.S. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011, 18, 666–677. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yang, A.; McDonald, D.G.; Riemer, E.C.; Vanek, K.N.; Schulte, B.A.; Wang, G.Y. MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget 2017, 8, 69797–69807. [Google Scholar] [CrossRef]
- Wennerberg, E.; Vanpouille-Box, C.; Bornstein, S.; Yamazaki, T.; Demaria, S.; Galluzzi, L. Immune recognition of irradiated cancer cells. Immunol. Rev. 2017, 280, 220–230. [Google Scholar] [CrossRef]
- He, Y.; Thummuri, D.; Zheng, G.; Okunieff, P.; Citrin, D.E.; Vujaskovic, Z.; Zhou, D. Cellular senescence and radiation-induced pulmonary fibrosis. Transl. Res. 2019, 209, 14–21. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576. [Google Scholar] [CrossRef]
- Shammas, M.A.; Simmons, C.G.; Corey, D.R.; Shmookler Reis, R.J. Telomerase inhibition by peptide nucleic acids reverses ‘immortality’ of transformed human cells. Oncogene 1999, 18, 6191–6200. [Google Scholar] [CrossRef]
- Hájek, M.; Matulová, N.; Votruba, I.; Holý, A.; Tloust’ová, E. Inhibition of human telomerase by diphosphates of acyclic nucleoside phosphonates. Biochem. Pharmacol. 2005, 70, 894–900. [Google Scholar] [CrossRef]
- Ji, X.M.; Xie, C.H.; Fang, M.H.; Zhou, F.X.; Zhang, W.J.; Zhang, M.S.; Zhou, Y.F. Efficient inhibition of human telomerase activity by antisense oligonucleotides sensitizes cancer cells to radiotherapy. Acta Pharmacol. Sin. 2006, 27, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kondo, S. Telomerase RNA inhibition using antisense oligonucleotide against human telomerase RNA linked to a 2′,5′-oligoadenylate. Methods Mol. Biol. 2007, 405, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Burchett, K.M.; Yan, Y.; Ouellette, M.M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE 2014, 9, e85155. [Google Scholar] [CrossRef] [PubMed]
- Djojosubroto, M.W.; Chin, A.C.; Go, N.; Schaetzlein, S.; Manns, M.P.; Gryaznov, S.; Harley, C.B.; Rudolph, K.L. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 2005, 42, 1127–1136. [Google Scholar] [CrossRef]
- Marian, C.O.; Cho, S.K.; McEllin, B.M.; Maher, E.A.; Hatanpaa, K.J.; Madden, C.J.; Mickey, B.E.; Wright, W.E.; Shay, J.W.; Bachoo, R.M. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res. 2010, 16, 154–163. [Google Scholar] [CrossRef]
- Relitti, N.; Saraswati, P.A.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Milczarek, M. The Premature Senescence in Breast Cancer Treatment Strategy. Cancers 2020, 12, 1815. [Google Scholar] [CrossRef]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef]
- Jackson, J.G.; Pereira-Smith, O.M. Primary and Compensatory Roles for RB Family Members at Cell Cycle Gene Promoters That Are Deacetylated and Downregulated in Doxorubicin-Induced Senescence of Breast Cancer Cells. Mol. Cell Biol. 2006, 26, 2501–2510. [Google Scholar] [CrossRef]
- Inao, T.; Kotani, H.; Iida, Y.; Kartika, I.D.; Okimoto, T.; Tanino, R.; Harada, M. Different sensitivities of senescent breast cancer cells to immune cell-mediated cytotoxicity. Cancer Sci. 2019, 110, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Blagosklonny, M.V. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: Duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013, 12, 3063–3069. [Google Scholar] [CrossRef]
- Tao, Y.F.; Wang, N.N.; Xu, L.X.; Li, Z.H.; Li, X.L.; Xu, Y.Y.; Fang, F.; Li, M.; Qian, G.H.; Li, Y.H.; et al. Molecular mechanism of G1 arrest and cellular senescence induced by LEE011, a novel CDK4/CDK6 inhibitor, in leukemia cells. Cancer Cell Int. 2017, 17, 35. [Google Scholar] [CrossRef]
- Yoshida, A.; Diehl, J.A. CDK4/6 inhibitor: From quiescence to senescence. Oncoscience 2015, 2, 896–897. [Google Scholar] [CrossRef]
- Maskey, R.S.; Wang, F.; Lehman, E.; Wang, Y.; Emmanuel, N.; Zhong, W.; Jin, G.; Abraham, R.T.; Arndt, K.T.; Myers, J.S.; et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle 2021, 20, 65–80. [Google Scholar] [CrossRef]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- van Leeuwen, I.; Lain, S. Sirtuins and p53. Adv. Cancer Res. 2009, 102, 171–195. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Jin, T.; Yang, Q.; Liu, W.; Liu, S.; Ji, M.; He, N.; Chen, C.; Shi, B.; Hou, P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1249–1258. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2017, 292, 19607. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, D. An update on gene therapy in China. Curr. Opin. Mol. Ther. 2009, 11, 547–553. [Google Scholar]
- Dos Santos, C.; McDonald, T.; Ho, Y.W.; Liu, H.; Lin, A.; Forman, S.J.; Kuo, Y.H.; Bhatia, R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 2013, 122, 1900–1913. [Google Scholar] [CrossRef]
- Dey, D.K.; Kang, S.C. CopA3 peptide induces permanent cell-cycle arrest in colorectal cancer cells. Mech. Ageing Dev. 2021, 196, 111497. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.Y.; Campisi, J.; Kapahi, P. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib + Quercetin (D + Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, A.S.; Wu, L.; Tannock, I.F. Concurrent and sequential administration of chemotherapy and the Mammalian target of rapamycin inhibitor temsirolimus in human cancer cells and xenografts. Clin. Cancer Res. 2009, 15, 5389–5395. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef]
- Lewińska, A.; Przybylski, P.; Adamczyk-Grochala, J.; Błoniarz, D.; Litwinienko, G.; Wnuk, M. Senolysis-Based Elimination of Chemotherapy-Induced Senescent Breast Cancer Cells by Quercetin Derivative with Blocked Hydroxy Groups. Cancers 2022, 14, 605. [Google Scholar] [CrossRef]
- Sasaki, N.; Gomi, F.; Yoshimura, H.; Yamamoto, M.; Matsuda, Y.; Michishita, M.; Hatakeyama, H.; Kawano, Y.; Toyoda, M.; Korc, M.; et al. FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer. Cancers 2020, 12, 2976. [Google Scholar] [CrossRef] [PubMed]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Galiana, I.; Lozano-Torres, B.; Sancho, M.; Alfonso, M.; Bernardos, A.; Bisbal, V.; Serrano, M.; Martínez-Máñez, R.; Orzáez, M. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanoSenolytic. J. Control. Release 2020, 323, 624–634. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| SASP Component | Beneficial Effects | Detrimental Effects |
|---|---|---|
| CCN1 | p53 pathway activation → tumor suppression [49] | EMT promotion → cancer progression [50,51] |
| Tissue regeneration [52,53] | Cancer cells migration and metastasis [54,55,56,57] | |
| Wound healing [52,53] | Chemoresistance [58] | |
| IL-6 | Epithelial memory of inflammation → tissue regeneration in pancreas [59] | MDSC activation → immunosuppression → lack of response to checkpoint inhibitor therapy [60] |
| ↑ CCL4, CCL5, CCL17, CXCL10 → T-cells activation [61] Prevention of T-cells apoptosis [62,63] Promotion of T-cells proliferation [64] | Cancer development [59] Inhibition of cancer cells apoptosis [65] Promotion of tumor cells proliferation, angiogenesis and metastasis [65] | |
| IL-8 | Response to infection (phagocytosis, oxidative burst, neutrophil extracellular trap [66] | Altering composition of immune cells in tumor microenvironment [66] |
| Activation of vascular endothelial cells → cells angiogenesis → ↑ proliferation, migration [67] | Activation of vascular endothelial cells → cancer cells angiogenesis → cancer migration [68] | |
| Infiltration of immune cells → tumor-reactive cytotoxic mechanism [69] | Infiltration of neutrophils and MDSC → immunosuppression [66,70] | |
| ↑ growth factor secretion by tumor associated macrophages [68] | ||
| Lower survival rate in tumor patients [70] | ||
| EMT induction [66] | ||
| IGFBP7 | ↑CDK inhibitors → cell cycle arrest → tumor suppression [71,72,73] | Infiltration of immune cells in gastric cancer [74] |
| ↑ E-cadherin, ↓ N-cadherin, ↓ Vimentin → EMT inhibition [75,76,77] | Lower survival rate in gastric cancer patients [74] | |
| Higher survival rate in melanoma patients [78] | Chemoresistance [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piskorz, W.M.; Cechowska-Pasko, M. Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects. Int. J. Mol. Sci. 2022, 23, 11082. https://doi.org/10.3390/ijms231911082
Piskorz WM, Cechowska-Pasko M. Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects. International Journal of Molecular Sciences. 2022; 23(19):11082. https://doi.org/10.3390/ijms231911082
Chicago/Turabian StylePiskorz, Wiktoria Monika, and Marzanna Cechowska-Pasko. 2022. "Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects" International Journal of Molecular Sciences 23, no. 19: 11082. https://doi.org/10.3390/ijms231911082