
Pathological Features in Paediatric Patients with TK2 Deficiency
 , , , , , , , , , and add
Show full author list
, , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
2.1. Clinical, Biochemical, and Genetic Data of the Patients
2.2. Pathological Characteristics of Patients
2.3. Immunofluorescence Studies for Mitochondrial Complexes
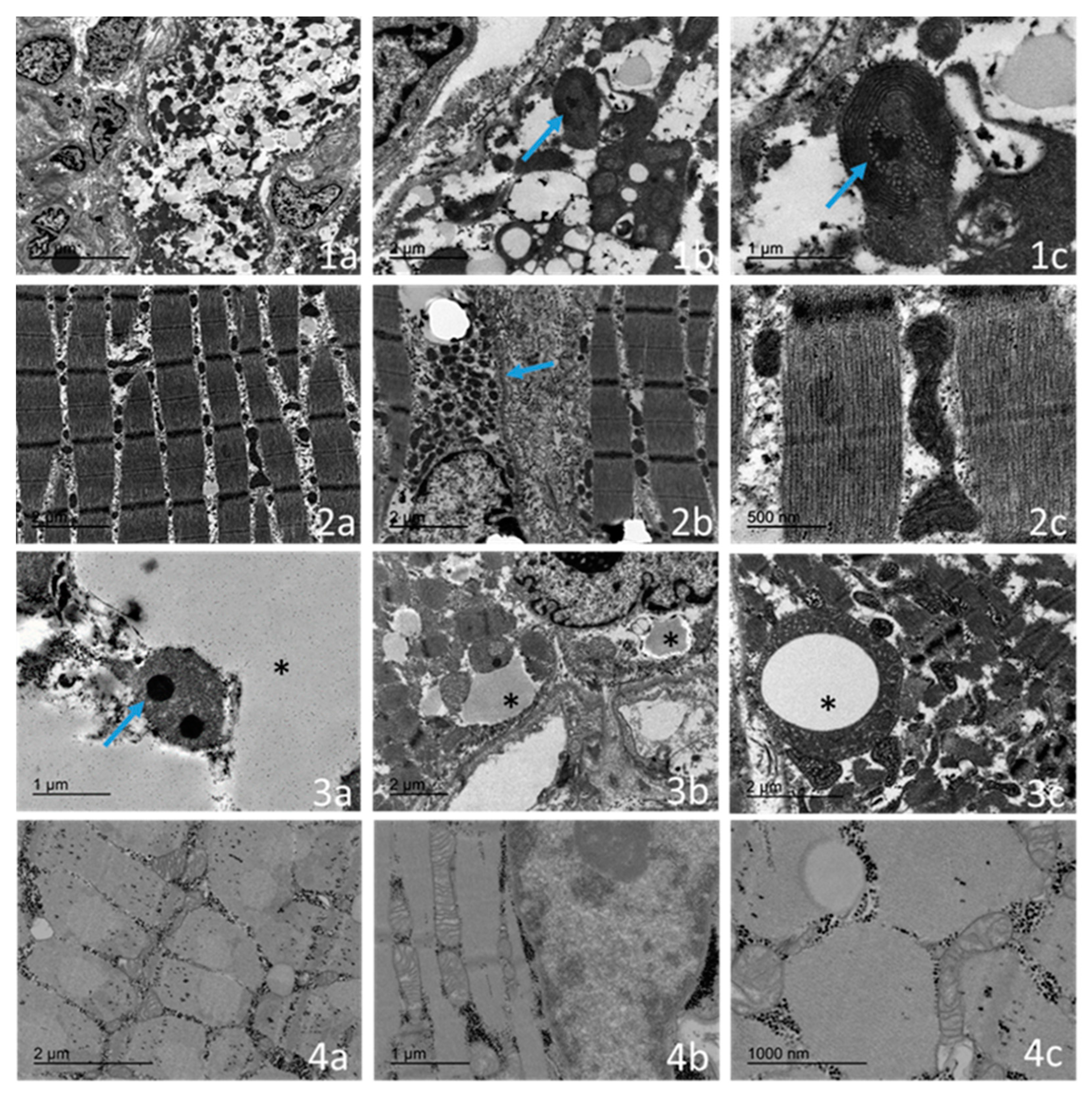
2.4. Ultrastructural Examination
3. Discussion
4. Materials and Methods
4.1. Patients and Control Subjects
4.2. Pathological Studies
4.3. Other Laboratory Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Saada, A.; Shaag, A.; Mandel, H.; Nevo, Y.; Eriksson, S.; Elpeleg, O. Mutant mitochondrial thymidine kinase in mitochondri-al DNA depletion myopathy. Nat. Genet. 2001, 29, 342–344. [Google Scholar] [CrossRef]
- Chanprasert, S.; Wang, J.; Weng, S.W.; Enns, G.M.; Boué, D.R.; Wong, B.L.; Mendell, J.R.; Perry, D.A.; Sahenk, Z.; Craigen, W.J.; et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol. Genet. Metab. 2013, 110, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Taylor, R.W.; Nascimento, A.; Poulton, J.; Fratter, C.; Domínguez-González, C.; Evans, J.C.; Loos, M.; Isohanni, P.; Suomalainen, A.; et al. Retrospective natural history of thymidine kinase 2 deficiency. J. Med. Genet. 2018, 55, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, E.; Dai, H.; Stefans, V.; Vogel, H.; Al Jasmi, F.; Schrier Vergano, S.A.; Castro, D.; Bernes, S.; Bhambhani, V.; et al. Clinical and molecular spectrum of thymidine kinase 2-related mtDNA maintenance defect. Mol. Genet. Metab. 2018, 124, 124–130. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Hernández-Laín, A.; Rivas, E.; Hernández-Voth, A.; Sayas Catalán, J.; Fernández-Torrón, R.; Fui-za-Luces, C.; García García, J.; Morís, G.; Olivé, M.; et al. Late-onset thymidine kinase 2 deficiency: A review of 18 cases. Orphanet J. Rare Dis. 2019, 14, 100. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Klein-steuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2-Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A.; Fiorini, A.C.; de Almeida, A.G.; Scorza, C.A. TK2-related mitochondrial disorder is not restricted to the skeletal muscle. Mol. Genet. Metab. Rep. 2018, 16, 13–14. [Google Scholar] [CrossRef]
- Phadke, R. Myopathology of Adult and Paediatric Mitochondrial Diseases. J. Clin. Med. 2017, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodríguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Tran-scriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics 2014, 15, 91. [Google Scholar] [CrossRef]
- Martí, R.; Nascimento, A.; Colomer, J.; Lara, M.C.; López-Gallardo, E.; Ruiz-Pesini, E.; Montoya, J.; Andreu, A.L.; Briones, P.; Pineda, M. Hearing loss in a patient with the myopathic form of mitochondrial DNA depletion syndrome and a novel mu-tation in the TK2 gene. Pediatr. Res. 2010, 68, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Gonzalez, C.; Badosa, C.; Madruga-Garrido, M.; Martí, I.; Paradas, C.; Ortez, C.; Diaz-Manera, J.; Berardo, A.; Alonso-Pérez, J.; Trifunov, S.; et al. Growth Differentiation Factor 15 is a potential biomarker of therapeutic response for TK2 deficient myopathy. Sci. Rep. 2020, 10, 10111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saada, A.; Eriksson, S. Kinetic properties of mutant human thymidine kinase 2 suggest a mechanism for mito-chondrial DNA depletion myopathy. J. Biol. Chem. 2003, 278, 6963–6968. [Google Scholar] [CrossRef]
- Cámara, Y.; Carreño-Gago, L.; Martín, M.A.; Melià, M.J.; Blázquez, A.; Delmiro, A.; Garrabou, G.; Morén, C.; Díaz-Manera, J.; Gallardo, E.; et al. Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology 2015, 84, 2286–2288. [Google Scholar] [CrossRef]
- Roos, S.; Lindgren, U.; Ehrstedt, C.; Moslemi, A.R.; Oldfors, A. Mitochondrial DNA depletion in single fibers in a patient with novel TK2 mutations. Neuromuscul. Disord. 2014, 24, 713–720. [Google Scholar] [CrossRef]
- Sewry, C.A. Muscular dystrophies: An update on pathology and diagnosis. Acta Neuropathol. 2010, 120, 343–358. [Google Scholar] [CrossRef]
- Collins, J.; Bove, K.E.; Dimmock, D.; Morehart, P.; Wong, L.J.; Wong, B. Progressive myofiber loss with extensive fibro-fatty replacement in a child with mitochondrial DNA depletion syndrome and novel thymidine kinase 2 gene mutations. Neuromuscul. Disord. 2009, 19, 784–787. [Google Scholar] [CrossRef]
- Romero, N.B.; Clarke, N.F. Congenital myopathies. Handb. Clin. Neurol. 2013, 113, 1321–1336. [Google Scholar]
- De Paepe, B.; Smet, J.; Lammens, M.; Seneca, S.; Martin, J.J.; De Bleecker, J.; De Meirleir, L.; Lissens, W.; Van Coster, R. Im-munohistochemical analysis of the oxidative phosphorylation complexes in skeletal muscle from patients with mitochondrial DNA encoded tRNA gene defects. J. Clin. Pathol. 2009, 62, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; Grady, J.P.; Grünewald, A.; Vincent, A.; Dobson, P.F.; Taylor, R.W.; Turnbull, D.M.; Rygiel, K.A. A novel im-munofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: Understanding mechanisms and improving diagnosis. Sci. Rep. 2015, 15, 15037. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; Ng, Y.S.; White, K.; Davey, T.; Mannella, C.; Falkous, G.; Feeney, C.; Schaefer, A.M.; McFarland, R.; Gorman, G.S.; et al. The Spectrum of Mitochondrial Ultrastructural Defects in Mitochondrial Myopathy. Sci. Rep. 2016, 6, 30610. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef]
- Sabouny, R.; Shutt, T.E. The role of mitochondrial dynamics in mtDNA maintenance. J. Cell Sci. 2021, 134, jcs258944. [Google Scholar]
- Dubowitz, V.; Sewry, C.; Oldfors, A. Muscle Biopsy: A Practical Approach, 5th ed.; Elsevier: London, UK, 2020; 530p. [Google Scholar]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodríguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. GDF-15 Is Elevated in Children with Mitochondrial Diseases and Is Induced by Mitochondrial Dysfunction. PLoS ONE 2016, 11, e0148709. [Google Scholar] [CrossRef]
- Casado, M.; Sierra, C.; Batllori, M.; Artuch, R.; Ormazabal, A. A targeted metabolomic procedure for amino acid analysis in different biological specimens by ultra-high-performance liquid chromatography-tandem mass spectrometry. Metabolomics 2018, 14, 76. [Google Scholar] [CrossRef]
- Marcuello, A.; Gonzalez-Alonso, J.; Calbet, J.A.; Damsgaard, R.; López-Pérez, M.J.; Díez-Sánchez, C. Skeletal muscle mito-chondrial DNA content in exercising humans. J. Appl. Physiol. 2005, 99, 1372–1377. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Garcia-Villoria, J.; Pons, M.R.; Nascimento, A.; Colomer, J.; Campistol, J.; Yoldi, M.E.; López-Gallardo, E.; Montoya, J.; et al. Mitochondrial DNA depletion syndrome: New descriptions and the use of citrate synthase as a helpful tool to better characterise the patients. Mol. Genet. Metab. 2012, 31, 409–415. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| % RRF | 70% | 30% | 40% | 40% | 25% | 10% | 8% | 5% |
| % SDH | 20% | 12% | 15% | 5% | 10% | 10% | 3% | 8% |
| % COX negative | 90% | 65% | 60% | 90% | 70% | 25% | 14% | 5% |
| Increased lipid content | Yes | Yes | Yes | N.A. | Yes | Yes | Yes | No |
| Cytoplasmic bodies | No | No | Yes | Yes | Yes | No | No | No |
| Expression of neonatal myosin | 20% | 25% | 20% | 25% | N.A. | N.A. | 4% | 2% |
| Sarcolemmal vacuoles | Yes | Yes | No | Yes | Yes | Yes | Yes | No |
| Inflammation | Severe | Severe | Mild | Severe | Moderate | Mild | Mild | Mild |
| Macrophages infiltration | Yes | Yes | Yes | Yes | N.A. | N.A. | Yes | Yes |
| T-lymphocyte infiltration | Isolated | Isolated | Isolated | No | N.A. | N.A. | No | Isolated |
| B-lymphocyte infiltration | No | Isolated | No | Yes | N.A. | N.A. | Yes | No |
| MHC I overexpression | Yes | Yes | Yes | Yes | N.A. | N.A. | Yes | Yes |
| Myofagocytosis | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Variation size fibres | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Predominate type I fibres | Yes | No | Yes | Yes | N.A. | N.A. | Yes | Yes |
| Case | 1 | 4 | 7 | 8 |
|---|---|---|---|---|
| Electron microscopy | ||||
| Increase in mitochondrial number | Yes | Yes | Yes | Yes |
| Changes in size and shape | Yes | Yes | Yes | Yes |
| Anomalous cristae | Yes | Yes | Yes | Yes |
| Presence of electron-dense inclusions | No | Yes | No | No |
| Lipid accumulation | Yes | Yes | Yes | No |
| Atrophic fibres | Yes | No | Yes | No |
| Cytoplasmatic bodies | Yes | Yes | No | No |
| Autophagic vacuoles | Yes | No | Yes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jou, C.; Nascimento, A.; Codina, A.; Montoya, J.; López-Gallardo, E.; Emperador, S.; Ruiz-Pesini, E.; Montero, R.; Natera-de Benito, D.; Ortez, C.I.; et al. Pathological Features in Paediatric Patients with TK2 Deficiency. Int. J. Mol. Sci. 2022, 23, 11002. https://doi.org/10.3390/ijms231911002
Jou C, Nascimento A, Codina A, Montoya J, López-Gallardo E, Emperador S, Ruiz-Pesini E, Montero R, Natera-de Benito D, Ortez CI, et al. Pathological Features in Paediatric Patients with TK2 Deficiency. International Journal of Molecular Sciences. 2022; 23(19):11002. https://doi.org/10.3390/ijms231911002
Chicago/Turabian StyleJou, Cristina, Andres Nascimento, Anna Codina, Julio Montoya, Ester López-Gallardo, Sonia Emperador, Eduardo Ruiz-Pesini, Raquel Montero, Daniel Natera-de Benito, Carlos I. Ortez, and et al. 2022. "Pathological Features in Paediatric Patients with TK2 Deficiency" International Journal of Molecular Sciences 23, no. 19: 11002. https://doi.org/10.3390/ijms231911002