
Micro-Players of Great Significance—Host microRNA Signature in Viral Infections in Humans and Animals

Abstract
:1. Introduction
MicroRNAs and Viral Infections in Humans and Animals
2. MicroRNA Signature in Human Viral Infections
2.1. MicroRNA-155 (miR-155)
2.1.1. Hepatitis Viruses
2.1.2. Hemorrhagic Viruses
2.1.3. Respiratory Viruses
2.2. MicroRNA-223 (miR-223)
2.2.1. Hepatitis Viruses
2.2.2. Hemorrhagic Viruses
2.2.3. Respiratory Viruses
2.2.4. Human Immunodeficiency Viruses
2.3. MicroRNA-146a (miR-146a)
2.3.1. Hepatitis Viruses
2.3.2. Hemorrhagic Viruses
2.3.3. Respiratory Viruses
2.4. MicroRNA-122 (miR-122)
2.4.1. Hepatitis Viruses
2.4.2. Hemorrhagic Viruses
2.4.3. Respiratory Viruses
2.5. MicroRNA-125b (miR-125b)
2.5.1. Hepatitis Viruses
2.5.2. Respiratory Viruses
2.5.3. Human Immunodeficiency Viruses
2.5.4. Neurotropic Viruses
2.6. MicroRNA-132 (miR-132)
2.6.1. Hepatitis Viruses
2.6.2. Respiratory Viruses
2.6.3. Human Herpesviruses
2.7. MicroRNA-34a (miR-34a)
2.7.1. Hepatitis Viruses
2.7.2. Hemorrhagic Viruses
2.7.3. Respiratory Viruses
2.7.4. Human T-Lymphotropic Virus Type 1
2.8. MicroRNA-21 (miR-21)
2.8.1. Hepatitis Viruses
2.8.2. Hemorrhagic Viruses
2.8.3. Respiratory Viruses
2.8.4. Human Immunodeficiency Viruses
2.8.5. Cardiotropic Viruses
2.8.6. Vector-Borne Viruses
2.9. MicroRNA-16 (miR-16)
2.9.1. Hepatitis Viruses
2.9.2. Respiratory Viruses
2.10. MicroRNA-181 Family (miR-181)
2.10.1. Hepatitis Viruses
2.10.2. Respiratory Viruses
2.11. Let-7 Family (Let-7)
2.11.1. Hepatitis Viruses
2.11.2. Hemorrhagic Viruses
2.11.3. Respiratory Viruses
2.11.4. Human Immunodeficiency Viruses
2.11.5. Neurotropic Viruses
2.12. MicroRNA-10a (miR-10a)
2.12.1. Hepatitis Viruses
2.12.2. Respiratory Viruses
2.12.3. Cardiotropic Viruses
3. MicroRNA Signature in Animal Viral Infections
3.1. MicroRNA-155 (miR-155)
3.1.1. Hemorrhagic Viruses
3.1.2. Respiratory Viruses
3.1.3. Lymphotropic Viruses
3.2. MicroRNA-223 (miR-223)
3.2.1. Respiratory Viruses
3.2.2. Lymphotropic Viruses
3.2.3. Vector-Borne Viruses
3.3. MicroRNA-146a (miR-146a)
3.3.1. Respiratory Viruses
3.3.2. Lymphotropic Viruses
3.3.3. Vector-Borne Viruses
3.3.4. Other Viruses
3.4. MicroRNA-145 (miR-145)
3.4.1. Respiratory Viruses
3.4.2. Neurotropic Viruses
3.5. MicroRNA-21 (miR-21)
3.5.1. Respiratory Viruses
3.5.2. Neurotropic Viruses
3.6. MicroRNA-16/miR-15a Cluster (miR-16/miR-15a Cluster)
3.6.1. Hemorrhagic Viruses
3.6.2. Respiratory Viruses
3.7. MicroRNA-181 Family (miR-181 Family)
3.7.1. Respiratory Viruses
3.7.2. Lymphotropic Viruses
3.7.3. Other Viruses
3.8. Let-7 Family (Let-7)
3.8.1. Lymphotropic Viruses
3.8.2. Other Viruses
3.9. MicroRNA-122 (miR-122)
3.9.1. Hemorrhagic Viruses
3.9.2. Respiratory Viruses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Sharp, P.A. MicroRNA Functions in Stress Responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, N. MicroRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.; Cuppen, E. Phylogenetic Shadowing and Computational Identification of Human microRNA Genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef]
- Ignaz, P. Circulating microRNA in Disease Diagnostics and Their Potential Biological Relevance; Springer: Berlin/Heidelberg, Germany, 2015; Volume 106. [Google Scholar]
- Zhang, Y.; Li, Y.-K. MicroRNAs in the regulation of immune response against infections. J. Zhejiang Univ. Sci. B 2013, 14, 1–7. [Google Scholar] [CrossRef]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef]
- Barbu, M.G.; Condrat, C.E.; Thompson, D.C.; Bugnar, O.L.; Cretoiu, D.; Toader, O.D.; Suciu, N.; Voinea, S.C. MicroRNA Involvement in Signaling Pathways During Viral Infection. Front. Cell Dev. Biol. 2020, 8, 143. [Google Scholar] [CrossRef]
- Lee, Y.; Jeon, K.; Lee, J.-T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and Host Interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef]
- Chen, M.; Wang, F.; Xia, H.; Yao, S. MicroRNA-155: Regulation of Immune Cells in Sepsis. Mediat. Inflamm. 2021, 2021, 8874854. [Google Scholar] [CrossRef]
- Tam, W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 2001, 274, 157–167. [Google Scholar] [CrossRef]
- Mahesh, G.; Biswas, R. MicroRNA-155: A Master Regulator of Inflammation. J. Interf. Cytokine Res. 2019, 39, 321–330. [Google Scholar] [CrossRef]
- Vigorito, E.; Kohlhaas, S.; Lu, D.; Leyland, R. miR-155: An ancient regulator of the immune system. Immunol. Rev. 2013, 253, 146–157. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/reverse-transcribing-dna-and-rna-viruses/w/hepadnaviridae/1317/genus-orthohepadnavirus (accessed on 19 July 2022).
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Su, C.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells. Virol. J. 2011, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, N.; Panigrahi, R.; Pal, A.; Biswas, A.; Singh, S.P.; Kar, S.K.; Bandopadhyay, M.; Das, D.; Saha, D.; Kanda, T.; et al. Expression of microRNA-155 correlates positively with the expression of Toll-like receptor 7 and modulates hepatitis B virus via C/EBP-β in hepatocytes. J. Viral Hepat. 2015, 22, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-L.; Deng, H.; Li, X.-H.; Huang, Y.-X.; Xie, D.-Y.; Gao, Z.-L. Expression of MicroRNA-155 is Downregulated in Peripheral Blood Mononuclear Cells of Chronic Hepatitis B Patients. Hepat. Mon. 2016, 16, e34483. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Huang, Z.; Liu, H.; Chen, J.; Xie, Z.; Chen, Z.; Peng, J.; Sun, J.; Hou, J.; Zhang, X. Lower Expression of MicroRNA-155 Contributes to Dysfunction of Natural Killer Cells in Patients with Chronic Hepatitis B. Front. Immunol. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Fang, J.; Zhuge, L.; Rao, H.; Huang, S.; Jin, L.; Li, J. Increased Levels of miR-155 are Related to Higher T-Cell Activation in the Peripheral Blood of Patients with Chronic Hepatitis B. Genet. Test. Mol. Biomark. 2019, 23, 118–123. [Google Scholar] [CrossRef]
- Wang, W.; Bian, H.; Li, F.; Li, X.; Zhang, D.; Sun, S.; Song, S.; Zhu, Q.; Ren, W.; Qin, C.; et al. HBeAg induces the expression of macrophage miR-155 to accelerate liver injury via promoting production of inflammatory cytokines. Cell. Mol. Life Sci. 2018, 75, 2627–2641. [Google Scholar] [CrossRef]
- Chen, L.; Ming, X.; Li, W.; Bi, M.; Yan, B.; Wang, X.; Yang, P.; Yang, B. The microRNA-155 mediates hepatitis B virus replication by reinforcing SOCS1 signalling–induced autophagy. Cell Biochem. Funct. 2020, 38, 436–442. [Google Scholar] [CrossRef]
- Xiong, Y.; Chen, S.; Liu, L.; Zhao, Y.; Lin, W.; Ni, J. Increased Serum MicroRNA-155 Level Associated with Nonresponsiveness to Hepatitis B Vaccine. Clin. Vaccine Immunol. 2013, 20, 1089–1091. [Google Scholar] [CrossRef]
- Hefzy, E.M.; Hassuna, N.A.; Shaker, O.G.; Masoud, M.; Abelhameed, T.A.; Ahmed, T.I.; Hemeda, N.F.; Abdelhakeem, M.A.; Mahmoud, R.H. miR-155 T/A (rs767649) and miR-146a A/G (rs57095329) single nucleotide polymorphisms as risk factors for chronic hepatitis B virus infection among Egyptian patients. PLoS ONE 2021, 16, e0256724. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae/362/genus-hepacivirus (accessed on 19 July 2022).
- Bala, S.; Tilahun, Y.; Taha, O.; Alao, H.; Kodys, K.; Catalano, D.; Szabo, G. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J. Transl. Med. 2012, 10, 151. [Google Scholar] [CrossRef]
- Jiang, M.; Broering, R.; Trippler, M.; Wu, J.; Zhang, E.; Zhang, X.; Gerken, G.; Lu, M.; Schlaak, J.F. MicroRNA-155 controls Toll-like receptor 3- and hepatitis C virus-induced immune responses in the liver. J. Viral Hepat. 2014, 21, 99–110. [Google Scholar] [CrossRef]
- Sidorkiewicz, M.; Grek, M.; Piekarska, A.; Bartkowiak, J.; Fendler, W.; Kuydowicz, J.; Wroblewski, P.; Paradowski, M. Coordinated increase of miRNA-155 and miRNA-196b expression correlates with the detection of the antigenomic strand of hepatitis C virus in peripheral blood mononuclear cells. Int. J. Mol. Med. 2011, 28, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Ren, J.P.; Zhao, J.; Wang, J.M.; Zhou, Y.; Li, G.Y.; Moorman, J.P.; Yao, Z.Q. MicroRNA-155 regulates interferon-γ production in natural killer cells via Tim-3 signalling in chronic hepatitis C virus infection. Immunology 2015, 145, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Riad, S.E.; El-Ekiaby, N.; Mekky, R.Y.; Ahmed, R.; EL Din, M.A.M.; El-Sayed, M.; Abouelkhair, M.M.; Salah, A.; Zekri, A.R.; Esmat, G.; et al. Expression signature of microRNA-155 in hepatitis C virus genotype 4 infection. Biomed. Rep. 2015, 3, 93–97. [Google Scholar] [CrossRef]
- El-Ekiaby, N.; Hamdi, N.; Negm, M.; Ahmed, R.; Zekri, A.-R.; Esmat, G.; Abdelaziz, A. Repressed induction of interferon-related microRNAs miR-146a and miR-155 in peripheral blood mononuclear cells infected with HCV genotype 4. FEBS Open Bio 2012, 2, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012, 56, 1631–1640. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae/360/genus-flavivirus (accessed on 19 July 2022).
- Arboleda, J.F.; Fernandez, G.J.; Urcuqui-Inchima, S. Vitamin D-mediated attenuation of miR-155 in human macrophages infected with dengue virus: Implications for the cytokine response. Infect. Genet. Evol. 2019, 69, 12–21. [Google Scholar] [CrossRef]
- Costa, V.V.; Fagundes, C.T.; Souza, D.G.; Teixeira, M.M. Inflammatory and Innate Immune Responses in Dengue Infection: Protection versus disease induction. Am. J. Pathol. 2013, 182, 1950–1961. [Google Scholar] [CrossRef]
- Su, Y.; Huang, Y.; Wu, Y.; Chen, H.; Hsu, C.; Hsu, Y.; Lee, J. MicroRNA-155 inhibits dengue virus replication by inducing heme oxygenase-1-mediated antiviral interferon responses. FASEB J. 2020, 34, 7283–7294. [Google Scholar] [CrossRef]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and Transmission Dynamics of West Nile Virus Disease. Emerg. Infect. Dis. 2005, 11, 1167–1173. [Google Scholar] [CrossRef]
- Kumar, M.; Nerurkar, V.R. Integrated analysis of microRNAs and their disease related targets in the brain of mice infected with West Nile virus. Virology 2014, 452–453, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Baik, H.H.; Jin, B.K. IL-13-Induced Oxidative Stress via Microglial NADPH Oxidase Contributes to Death of Hippocampal Neurons In Vivo. J. Immunol. 2009, 183, 4666–4674. [Google Scholar] [CrossRef] [PubMed]
- Natekar, J.P.; Rothan, H.A.; Arora, K.; Strate, P.G.; Kumar, M. Cellular microRNA-155 Regulates Virus-Induced Inflammatory Response and Protects against Lethal West Nile Virus Infection. Viruses 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae (accessed on 19 July 2022).
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.S. Influenza virus-induced lung injury: Pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Shen, S.-M.; Jiang, H.; Zhao, J.-N.; Shi, Y. Down-regulation of miR-155 inhibits inflammatory response in human pulmonary microvascular endothelial cells infected with influenza A virus by targeting sphingosine-1-phosphate receptor. Chin. Med. J. 2020, 133, 2429–2436. [Google Scholar] [CrossRef]
- Izzard, L.; Dlugolenski, D.; Xia, Y.; McMahon, M.; Middleton, D.; Tripp, R.A.; Stambas, J. Enhanced immunogenicity following miR-155 incorporation into the influenza A virus genome. Virus Res. 2017, 235, 115–120. [Google Scholar] [CrossRef]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef]
- Pociask, D.A.; Robinson, K.M.; Chen, K.; McHugh, K.J.; Clay, M.; Huang, G.T.; Benos, P.V.; Janssen-Heininger, Y.M.; Kolls, J.K.; Anathy, V.; et al. Epigenetic and Transcriptomic Regulation of Lung Repair during Recovery from Influenza Infection. Am. J. Pathol. 2017, 187, 851–863. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/negative-sense-rna-viruses/w/pneumoviridae (accessed on 19 July 2022).
- Collins, P.L.; Murphy, B.R. Respiratory Syncytial Virus: Reverse Genetics and Vaccine Strategies. Virology 2002, 296, 204–211. [Google Scholar] [CrossRef]
- Arroyo, M.; Salka, K.; Chorvinsky, E.; Xuchen, X.; Abutaleb, K.; Perez, G.F.; Weinstock, J.; Gaviria, S.; Gutierrez, M.J.; Nino, G. Airway mir-155 responses are associated with TH1 cytokine polarization in young children with viral respiratory infections. PLoS ONE 2020, 15, e0233352. [Google Scholar] [CrossRef] [PubMed]
- Inchley, C.S.; Sonerud, T.; Fjærli, H.O.; Nakstad, B. Nasal mucosal microRNA expression in children with respiratory syncytial virus infection. BMC Infect. Dis. 2015, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.G.; Chen, Q.; Wang, R.; Xiong, T.T.; Yang, L.X. Relationship between the expression of miR-146a and miR-155 in peripheral blood mononuclear cells and inflammation response in infants with respiratory syncytial virus pneumonia. Chin. J. Child Health Care 2020, 28, 316–319. [Google Scholar] [CrossRef]
- Oshansky, C.M.; Zhang, W.; Moore, E.; Tripp, R.A. The host response and molecular pathogenesis associated with respiratory syncytial virus infection. Futur. Microbiol. 2009, 4, 279–297. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef]
- Lu, C.; Huang, X.; Zhang, X.; Roensch, K.; Cao, Q.; Nakayama, K.I.; Blazar, B.R.; Zeng, Y.; Zhou, X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood 2011, 117, 4293–4303. [Google Scholar] [CrossRef]
- Wang, S.; Ling, Y.; Yao, Y.; Zheng, G.; Chen, W. Luteolin inhibits respiratory syncytial virus replication by regulating the MiR-155/SOCS1/STAT1 signaling pathway. Virol. J. 2020, 17, 187. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Nemati, M.; Saha, B.; Bansode, Y.D.; Jafarzadeh, S. Protective Potentials of Type III Interferons in COVID-19 Patients: Lessons from Differential Properties of Type I- and III Interferons. Viral Immunol. 2021, 34, 307–320. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Nemati, M.; Jafarzadeh, S. Contribution of STAT3 to the pathogenesis of COVID-19. Microb. Pathog. 2021, 154, 104836. [Google Scholar] [CrossRef] [PubMed]
- Haroun, R.A.-H.; Osman, W.H.; Amin, R.E.; Hassan, A.K.; Abo-Shanab, W.S.; Eessa, A.M. Circulating plasma miR-155 is a potential biomarker for the detection of SARS-CoV-2 infection. Pathology 2022, 54, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Demirci, Y.M.; Demirci, M.D.S. Circular RNA–MicroRNA–MRNA interaction predictions in SARS-CoV-2 infection. J. Integr. Bioinform. 2021, 18, 45–50. [Google Scholar] [CrossRef]
- Donyavi, T.; Bokharaei-Salim, F.; Baghi, H.B.; Khanaliha, K.; Janat-Makan, M.A.; Karimi, B.; Nahand, J.S.; Mirzaei, H.; Khatami, A.; Garshasbi, S.; et al. Acute and post-acute phase of COVID-19: Analyzing expression patterns of miRNA-29a-3p, 146a-3p, 155–5p, and let-7b-3p in PBMC. Int. Immunopharmacol. 2021, 97, 107641. [Google Scholar] [CrossRef]
- Soni, D.K.; Cabrera-Luque, J.; Kar, S.; Sen, C.; Devaney, J.; Biswas, R. Suppression of miR-155 attenuates lung cytokine storm induced by SARS-CoV-2 infection in human ACE2-transgenic mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsiè, R.; Klironomos, F.; Koppstein, D.; Hönzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef]
- Chow, J.; Salmena, L. Prediction and Analysis of SARS-CoV-2-Targeting MicroRNA in Human Lung Epithelium. Genes 2020, 11, 1002. [Google Scholar] [CrossRef]
- Qi, M.; Liu, B.; Li, S.; Ni, Z.; Li, F. Construction and Investigation of Competing Endogenous RNA Networks and Candidate Genes Involved in SARS-CoV-2 Infection. Int. J. Gen. Med. 2021, 14, 6647–6659. [Google Scholar] [CrossRef]
- Garg, A.; Seeliger, B.; Derda, A.A.; Xiao, K.; Gietz, A.; Scherf, K.; Sonnenschein, K.; Pink, I.; Hoeper, M.M.; Welte, T.; et al. Circulating cardiovascular microRNAs in critically ill COVID-19 patients. Eur. J. Heart Fail. 2021, 23, 468–475. [Google Scholar] [CrossRef]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kaján, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef]
- Kidd, A.H.; Garwicz, D.; Oberg, M. Human and Simian Adenoviruses: Phylogenetic Inferences from Analysis of VA RNA Genes. Virology 1995, 207, 32–45. [Google Scholar] [CrossRef]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, M.; Tellgren-Roth, C.; Pettersson, U. Fluctuating expression of microRNAs in adenovirus infected cells. Virology 2015, 478, 99–111. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 Feedback Promotes Type I IFN Signaling in Antiviral Innate Immunity by Targeting Suppressor of Cytokine Signaling. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/picornaviridae (accessed on 19 July 2022).
- Bondanese, V.P.; Francisco-Garcia, A.; Bedke, N.; Davies, D.E.; Sanchez-Elsner, T. Identification of host miRNAs that may limit human rhinovirus replication. World J. Biol. Chem. 2014, 5, 437–456. [Google Scholar] [CrossRef]
- Gutierrez, M.J.; Gómez, J.L.; Perez, G.F.; Pancham, K.; Val, S.; Pillai, D.K.; Giri, M.; Ferrante, S.; Freishtat, R.; Rose, M.C.; et al. Airway Secretory microRNAome Changes during Rhinovirus Infection in Early Childhood. PLoS ONE 2016, 11, e0162244. [Google Scholar] [CrossRef]
- Taïbi, F.; Meuth, V.M.-L.; Massy, Z.A.; Metzinger, L. miR-223: An inflammatory oncomiR enters the cardiovascular field. Biochim. Biophys. Acta 2014, 1842, 1001–1009. [Google Scholar] [CrossRef]
- Jiao, P.; Wang, X.-P.; Luoreng, Z.-M.; Yang, J.; Jia, L.; Ma, Y.; Wei, D.-W. miR-223: An Effective Regulator of Immune Cell Differentiation and Inflammation. Int. J. Biol. Sci. 2021, 17, 2308–2322. [Google Scholar] [CrossRef]
- Yuan, S.; Wu, Q.; Wang, Z.; Che, Y.; Zheng, S.; Chen, Y.; Zhong, X.; Shi, F. miR-223: An Immune Regulator in Infectious Disorders. Front. Immunol. 2021, 12, 781815. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, C.; Che, X.; Wang, L.; Yu, D.; Zhang, T.; Huang, L.; Li, H.; Tan, W.; Wang, C.; et al. Circulating MicroRNAs, miR-21, miR-122, and miR-223, in patients with hepatocellular carcinoma or chronic hepatitis. Mol. Carcinog. 2011, 50, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Wong, Q.W.; Lung, R.W.; Law, P.T.; Lai, P.B.-S.; Chan, K.Y.Y.; To, K.; Wong, N. MicroRNA-223 Is Commonly Repressed in Hepatocellular Carcinoma and Potentiates Expression of Stathmin1. Gastroenterology 2008, 135, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Giray, B.G.; Emekdas, G.; Tezcan, S.; Ulger, M.; Serin, M.S.; Sezgin, O.; Altintas, E.; Tiftik, E.N. Profiles of serum microRNAs; miR-125b-5p and miR223-3p serve as novel biomarkers for HBV-positive hepatocellular carcinoma. Mol. Biol. Rep. 2014, 41, 4513–4519. [Google Scholar] [CrossRef]
- Yu, G.; Chen, X.; Chen, S.; Ye, W.; Hou, K.; Liang, M. MiR-19a, miR-122 and miR-223 are differentially regulated by hepatitis B virus X protein and involve in cell proliferation in hepatoma cells. J. Transl. Med. 2016, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Hyrina, A.; Olmstead, A.D.; Steven, P.; Krajden, M.; Tam, E.; Jean, F. Treatment-Induced Viral Cure of Hepatitis C Virus-Infected Patients Involves a Dynamic Interplay among three Important Molecular Players in Lipid Homeostasis: Circulating microRNA (miR)-24, miR-223, and Proprotein Convertase Subtilisin/Kexin Type 9. eBioMedicine 2017, 23, 68–78. [Google Scholar] [CrossRef]
- El-Guendy, N.M.; Helwa, R.; El-Halawany, M.S.; Ali, S.A.R.; Aly, M.T.; Alieldin, N.H.; Fouad, S.A.H.; Saeid, H.; Abdel-Wahab, A.-H.A. The Liver MicroRNA Expression Profiles Associated With Chronic Hepatitis C Virus (HCV) Genotype-4 Infection: A Preliminary Study. Hepat. Mon. 2016, 16, e33881. [Google Scholar] [CrossRef]
- Wu, N.; Gao, N.; Fan, D.; Wei, J.; Zhang, J.; An, J. miR-223 inhibits dengue virus replication by negatively regulating the microtubule-destabilizing protein STMN1 in EAhy926 cells. Microbes Infect. 2014, 16, 911–922. [Google Scholar] [CrossRef]
- Li, Y.; Chan, E.Y.; Li, J.; Ni, C.; Peng, X.; Rosenzweig, E.; Tumpey, T.M.; Katze, M.G. MicroRNA Expression and Virulence in Pandemic Influenza Virus-Infected Mice. J. Virol. 2010, 84, 3023–3032. [Google Scholar] [CrossRef]
- Vela, E.M.; Kasoji, M.D.; Wendling, M.Q.; Price, J.A.; Knostman, K.A.B.; Bresler, H.S.; Long, J.P. MicroRNA expression in mice infected with seasonal H1N1, swine H1N1 or highly pathogenic H5N. J. Med Microbiol. 2014, 63, 1131–1142. [Google Scholar] [CrossRef]
- Wu, Z.; Hao, R.; Li, P.; Zhang, X.; Liu, N.; Qiu, S.; Wang, L.; Wang, Y.; Xue, W.; Liu, K.; et al. MicroRNA Expression Profile of Mouse Lung Infected with 2009 Pandemic H1N1 Influenza Virus. PLoS ONE 2013, 8, e74190. [Google Scholar] [CrossRef] [PubMed]
- Tambyah, P.A.; Sepramaniam, S.; Ali, J.M.; Chai, S.C.; Swaminathan, P.; Armugam, A.; Jeyaseelan, K. microRNAs in Circulation Are Altered in Response to Influenza A Virus Infection in Humans. PLoS ONE 2013, 8, e76811. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-J.; Kim, H.B.; Baek, Y.H.; Kim, E.-H.; Pascua, P.N.Q.; Park, S.-J.; Kwon, H.-I.; Lim, G.-J.; Kim, S.; Kim, Y.-I.; et al. Differential microRNA expression following infection with a mouse-adapted, highly virulent avian H5N2 virus. BMC Microbiol. 2014, 14, 252. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Williams, A.E.; Perry, M.M.; Birrell, M.A.; Belvisi, M.G.; Lindsay, M.A. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genom. 2007, 8, 240. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Miao, Y.; Chen, R.-L.; Zhang, Y.-Q.; Wu, H.; Yang, S.-M.; Shang, L.-Q. STIM1 mediates IAV-induced inflammation of lung epithelial cells by regulating NLRP3 and inflammasome activation via targeting miR-223. Life Sci. 2021, 266, 118845. [Google Scholar] [CrossRef] [PubMed]
- Micaroni, M.; Stanley, A.C.; Khromykh, T.; Venturato, J.; Wong, C.X.F.; Lim, J.P.; Marsh, B.J.; Storrie, B.; Gleeson, P.A.; Stow, J.L. Rab6a/a’ Are Important Golgi Regulators of Pro-Inflammatory TNF Secretion in Macrophages. PLoS ONE 2013, 8, e57034. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Mallick, B.; Ghosh, Z.; Chakrabarti, J. MicroRNome Analysis Unravels the Molecular Basis of SARS Infection in Bronchoalveolar Stem Cells. PLoS ONE 2009, 4, e7837. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, X.; Jiang, X.-M.; Guo, J.; Fu, Z.; Zhou, Z.; Yang, P.; Guo, H.; Guo, X.; Liang, G.; et al. Decreased inhibition of exosomal miRNAs on SARS-CoV-2 replication underlies poor outcomes in elderly people and diabetic patients. Signal Transduct. Target. Ther. 2021, 6, 300. [Google Scholar] [CrossRef]
- Demiray, A.; Sarı, T.; Çalışkan, A.; Nar, R.; Aksoy, L.; Akbubak, I.H. Serum microRNA signature is capable of predictive and prognostic factor for SARS-COV-2 virulence. Turk. J. Biochem. 2021, 46, 245–253. [Google Scholar] [CrossRef]
- Houshmandfar, S.; Saeedi-Boroujeni, A.; Rashno, M.; Khodadadi, A.; Mahmoudian-Sani, M.-R. miRNA-223 as a regulator of inflammation and NLRP3 inflammasome, the main fragments in the puzzle of immunopathogenesis of different inflammatory diseases and COVID-19. Naunyn-Schmiedebergs Arch. Pharmakol. 2021, 394, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/reverse-transcribing-dna-and-rna-viruses/w/retroviridae (accessed on 19 July 2022).
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Rapaka, R.S.; Rutter, J.; Shurtleff, D. Do Opioids Activate Latent HIV-1 by Down-Regulating Anti-HIV microRNAs? J. Neuroimmune Pharmacol. 2012, 7, 519–523. [Google Scholar] [CrossRef]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; et al. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40, 2181–2196. [Google Scholar] [CrossRef]
- Monticelli, S.; Ansel, K.M.; Xiao, C.; Socci, N.D.; Krichevsky, A.M.; Thai, T.-H.; Rajewsky, N.; Marks, D.S.; Sander, C.; Rajewsky, K.; et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005, 6, R71. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Citarella, F.; Carissimi, C.; Goldoni, M.; Carucci, N.; Fulci, V.; Franceschini, D.; Meloni, F.; Barnaba, V.; Macino, G. An emerging player in the adaptive immune response: MicroRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood 2010, 115, 265–273. [Google Scholar] [CrossRef]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a Feedback Inhibits RIG-I-Dependent Type I IFN Production in Macrophages by Targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, X.; Ju, Y.; Zhao, B.; Yan, X.; Hu, J.; Shi, L.; Yang, L.; Ma, Z.; Chen, L.; et al. MicroRNA-146a Feedback Suppresses T Cell Immune Function by Targeting Stat1 in Patients with Chronic Hepatitis B. J. Immunol. 2013, 191, 293–301. [Google Scholar] [CrossRef]
- Fu, L.; Fu, X.; Mo, J.; Li, X.; Li, R.; Peng, S. miR-146a-5p enhances hepatitis B virus replication through autophagy to promote aggravation of chronic hepatitis B. IUBMB Life 2019, 71, 1336–1346. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y. miR-146 promotes HBV replication and expression by targeting ZEB2. Biomed. Pharmacother. 2018, 99, 576–582. [Google Scholar] [CrossRef]
- Ji, M.; Mei, X.; Jing, X.; Xu, X.; Chen, X.; Pan, W. The cooperative complex of Argonaute-2 and microRNA-146a regulates hepatitis B virus replication through flap endonuclease. Life Sci. 2020, 257, 118089. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-F.; Dai, X.-P.; Zhang, W.; Sun, S.-H.; Zeng, Y.; Zhao, G.-Y.; Kou, Z.-H.; Guo, Y.; Yu, H.; Du, L.-Y.; et al. Upregulation of MicroRNA-146a by Hepatitis B Virus X Protein Contributes to Hepatitis Development by Downregulating Complement Factor H. mBio 2015, 6, e02459–e002473. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, X.; Li, J.; Xie, Q.; Lin, J.; Chang, Y. Association of a single-nucleotide polymorphism within the miR-146a gene with susceptibility for acute-on-chronic hepatitis B liver failure. Immunogenetics 2013, 65, 257–263. [Google Scholar] [CrossRef]
- He, Q.; Li, W.; Ren, J.; Huang, Y.; Huang, Y.; Hu, Q.; Chen, J.; Chen, W. ZEB2 inhibits HBV transcription and replication by targeting its core promoter. Oncotarget 2016, 7, 16003–16011. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.Y.; Ge, X.; Shan, X.; Lei, X.; Ke, C.; Huang, A.; Hu, J. Effect of structure-specific nucleic acid enzyme FEN1 on HBV replication. Third. Mil. Med. Univ. 2012, 34, 1935–1938. [Google Scholar]
- Ren, J.P.; Ying, R.S.; Cheng, Y.Q.; Wang, L.; Elgazzar, M.A.; Li, G.Y.; Ning, S.B.; Moorman, J.P.; Yao, Z.Q. HCV-induced miR146a controls SOCS1/STAT3 and cytokine expression in monocytes to promote regulatory T-cell development. J. Viral Hepat. 2016, 23, 755–766. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Wang, H.; Tong, Q.-X.; Jie, S.-H.; Yang, D.-L.; Peng, C. Involvement of TLR2-MyD88 in abnormal expression of miR-146a in peripheral blood monocytes of patients with chronic hepatitis C. J. Huazhong Univ. Sci. Technol. 2015, 35, 219–224. [Google Scholar] [CrossRef]
- Bandiera, S.; Pernot, S.; El Saghire, H.; Durand, S.C.; Thumann, C.; Crouchet, E.; Ye, T.; Fofana, I.; Oudot, M.A.; Barths, J.; et al. Hepatitis C Virus-Induced Upregulation of MicroRNA miR-146a-5p in Hepatocytes Promotes Viral Infection and Deregulates Metabolic Pathways Associated with Liver Disease Pathogenesis. J. Virol. 2016, 90, 6387–6400. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 2017, 39, 563–574. [Google Scholar] [CrossRef]
- Wu, S.; He, L.; Li, Y.; Wang, T.; Feng, L.; Jiang, L.; Zhang, P.; Huang, X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF. J. Infect. 2013, 67, 329–341. [Google Scholar] [CrossRef]
- Pu, J.; Wu, S.; Xie, H.; Li, Y.; Yang, Z.; Wu, X.; Huang, X. miR-146a Inhibits dengue-virus-induced autophagy by targeting TRAF6. Arch. Virol. 2017, 162, 3645–3659. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, L.F.; de Andrade, A.A.S.; Pagliari, C.; de Carvalho, L.V.; Silveira, T.S.; Cardoso, J.F.; e Silva, A.L.T.; de Vasconcelos, J.M.; Moreira-Nunes, C.A.; Burbano, R.M.R.; et al. Differential expression analysis and profiling of hepatic miRNA and isomiRNA in dengue hemorrhagic fever. Sci. Rep. 2021, 11, 5554. [Google Scholar] [CrossRef]
- Ouyang, X.; Jiang, X.; Gu, D.; Zhang, Y.; Kong, S.K.; Jiang, C.; Xie, W. Dysregulated Serum MiRNA Profile and Promising Biomarkers in Dengue-infected Patients. Int. J. Med. Sci. 2016, 13, 195–205. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, X.; Zhu, Y.; Qin, W. Downregulation of miR-146a inhibits influenza A virus replication by enhancing the type I interferon response in vitro and in vivo. Biomed. Pharmacother. 2019, 111, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Buggele, W.A.; Johnson, K.E.; Horvath, C.M. Influenza A Virus Infection of Human Respiratory Cells Induces Primary MicroRNA Expression. J. Biol. Chem. 2012, 287, 31027–31040. [Google Scholar] [CrossRef]
- Terrier, O.; Textoris, J.; Carron, C.; Marcel, V.; Bourdon, J.-C.; Rosa-Calatrava, M. Host microRNA molecular signatures associated with human H1N1 and H3N2 influenza A viruses reveal an unanticipated antiviral activity for miR-146a. J. Gen. Virol. 2013, 94, 985–995. [Google Scholar] [CrossRef]
- Deng, Y.; Yan, Y.; Tan, K.S.; Liu, J.; Chow, V.T.; Tao, Z.-Z.; Wang, D.-Y. MicroRNA-146a induction during influenza H3N2 virus infection targets and regulates TRAF6 levels in human nasal epithelial cells (hNECs). Exp. Cell Res. 2017, 352, 184–192. [Google Scholar] [CrossRef]
- Eilam-Frenkel, B.; Naaman, H.; Brkic, G.; Veksler-Lublinsky, I.; Rall, G.; Shemer-Avni, Y.; Gopas, J. MicroRNA 146-5p, miR-let-7c-5p, miR-221 and miR-345-5p are differentially expressed in Respiratory Syncytial Virus (RSV) persistently infected HEp-2 cells. Virus Res. 2018, 251, 34–39. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Strońska, A.; Lalko, A. Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. Int. J. Mol. Sci. 2021, 22, 9366. [Google Scholar] [CrossRef]
- Tang, H.; Gao, Y.; Li, Z.; Miao, Y.; Huang, Z.; Liu, X.; Xie, L.; Li, H.; Wen, W.; Zheng, Y.; et al. The noncoding and coding transcriptional landscape of the peripheral immune response in patients with COVID-19. Clin. Transl. Med. 2020, 10, e200. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Giuliani, A.; Matacchione, G.; Latini, S.; Laprovitera, N.; Pomponio, G.; Ferrarini, A.; Baroni, S.S.; Pavani, M.; Moretti, M.; et al. Decreased serum levels of the inflammaging marker miR-146a are associated with clinical non-response to tocilizumab in COVID-19 patients. Mech. Ageing Dev. 2021, 193, 111413. [Google Scholar] [CrossRef] [PubMed]
- Roganović, J. Downregulation of microRNA-146a in diabetes, obesity and hypertension may contribute to severe COVID-19. Med Hypotheses 2021, 146, 110448. [Google Scholar] [CrossRef]
- Roganović, J.R. microRNA-146a and -155, upregulated by periodontitis and type 2 diabetes in oral fluids, are predicted to regulate SARS-CoV-2 oral receptor genes. J. Periodontol. 2021, 92, e35–e43. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Girard, M.; Jacquemin, E.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. miR-122, a paradigm for the role of microRNAs in the liver. J. Hepatol. 2008, 48, 648–656. [Google Scholar] [CrossRef]
- Xu, H.; He, J.-H.; Xiao, Z.-D.; Zhang, Q.-Q.; Chen, Y.-Q.; Zhou, H.; Qu, L.-H. Liver-enriched transcription factors regulate MicroRNA-122 that targets CUTL1 during liver development. Hepatology 2010, 52, 1431–1442. [Google Scholar] [CrossRef]
- Laudadio, I.; Manfroid, I.; Achouri, Y.; Schmidt, D.; Wilson, M.D.; Cordi, S.; Thorrez, L.; Knoops, L.; Jacquemin, P.; Schuit, F.; et al. A Feedback Loop Between the Liver-Enriched Transcription Factor Network and Mir-122 Controls Hepatocyte Differentiation. Gastroenterology 2012, 142, 119–129. [Google Scholar] [CrossRef]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef]
- Arataki, K.; Hayes, C.N.; Akamatsu, S.; Akiyama, R.; Abe, H.; Tsuge, M.; Miki, D.; Ochi, H.; Hiraga, N.; Imamura, M.; et al. Circulating microRNA-22 correlates with microRNA-122 and represents viral replication and liver injury in patients with chronic hepatitis B. J. Med Virol. 2013, 85, 789–798. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G1-modulated P53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, C.; Cai, S.; Xia, M.; Liao, G.; Zhang, X.; Peng, J. Circulating miR-122 Is a Predictor for Virological Response in CHB Patients with High Viral Load Treated with Nucleos(t)ide Analogs. Front. Genet. 2019, 10, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimifard, M.; Zandi, M.; Moradi, A. Evaluation of miR-122 levels in chronic HBV and liver cirrhosis patients. Arch. Med. Lab. Sci. 2013, 2, 3. [Google Scholar]
- Van der Ree, M.H.; Jansen, L.; Kruize, Z.; van Nuenen, A.C.; van Dort, K.A.; Takkenberg, R.B.; Reesink, H.W.; Kootstra, N.A. Plasma MicroRNA Levels Are Associated with Hepatitis B e Antigen Status and Treatment Response in Chronic Hepatitis B Patients. J. Infect. Dis. 2017, 215, 1421–1429. [Google Scholar] [CrossRef]
- Chen, S.; Ni, M.; Yu, B.; Lv, T.; Lu, M.; Gong, F. Construction and identification of a human liver specific microRNA eukaryotic expression vector. Cell. Mol. Immunol. 2007, 4, 473–477. [Google Scholar]
- Chen, Y.; Shen, A.; Rider, P.J.; Yu, Y.; Wu, K.; Mu, Y.; Hao, Q.; Liu, Y.; Gong, H.; Zhu, Y.; et al. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J. 2011, 25, 4511–4521. [Google Scholar] [CrossRef]
- Li, R.-F.; Fan, C.-G.; Wang, C.-M.; Tian, C.; Wang, Y.; Li, L.; Sun, W.-S.; Liu, Y.-G. miR-122 inhibits viral replication and cell proliferation in hepatitis B virus-related hepatocellular carcinoma and targets NDRG3. Oncol. Rep. 2011, 26, 1281–1286. [Google Scholar] [CrossRef]
- Wu, D.-B.; Liu, F.-W.; Li, J.; Liu, C.; Liu, L.; Chen, E.-Q.; Zhao, L.-S.; Tang, H.; Zhou, T.-Y. Intrahepatic IFN-alpha expression in liver specimens from HBV-infected patients with different outcomes. Eur. Rev. Med Pharmacol. Sci. 2013, 17, 2474–2480. [Google Scholar]
- Koeberlein, B.; Hausen, A.Z.; Bektas, N.; Zentgraf, H.; Chin, R.; Toan, N.L.; Kandolf, R.; Torresi, J.; Bock, C.T. Hepatitis B virus overexpresses suppressor of cytokine signaling-3 (SOCS3) thereby contributing to severity of inflammation in the liver. Virus Res. 2010, 148, 51–59. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Xi, Y.; Zhu, W.-N.; Zeng, C.; Zhang, Z.-Q.; Guo, Z.-C.; Hao, D.-L.; Liu, G.; Feng, L.; Chen, H.-Z.; et al. Positive regulation of hepatic miR-122 expression by HNF4α. J. Hepatol. 2011, 55, 602–611. [Google Scholar] [CrossRef]
- Song, K.; Han, C.; Dash, S.; Balart, L.A.; Wu, T. MiR-122 in hepatitis B virus and hepatitis C virus dual infection. World J. Hepatol. 2015, 7, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wu, J.; Zhai, A.; Qian, J.; Wang, X.; Qaria, M.A.; Zhang, Q.; Li, Y.; Fang, Y.; Kao, W.; et al. HBV triggers APOBEC2 expression through miR-122 regulation and affects the proliferation of liver cancer cells. Int. J. Oncol. 2019, 55, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Han, C.; Zhang, J.; Lu, N.; Dash, S.; Feitelson, M.; Lim, K.; Wu, T. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology 2013, 58, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Xiao, X.; Jiang, Y.; Luo, K.; Tian, Y.; Peng, M.; Zhang, M.; Xu, Y.; Gong, G. HBx Down-Regulated Gld2 Plays a Critical Role in HBV-Related Dysregulation of miR-122. PLoS ONE 2014, 9, e92998. [Google Scholar] [CrossRef]
- Ding, J.; Ding, X.; Ning, J.; Yi, F.; Chen, J.; Zhao, D.; Zheng, J.; Liang, Z.; Hu, Z.; Du, Q. Circulating microRNA-122 as a potential biomarker for liver injury. Mol. Med. Rep. 2012, 5, 1428–1432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Fang, S.; Wang, M.; Xiong, A.; Zheng, C.; Wang, J.; Yin, C. Diagnostic value of circulating miRNA-122 for hepatitis B virus and/or hepatitis C virus-associated chronic viral hepatitis. Biosci. Rep. 2019, 39, BSR20190900. [Google Scholar] [CrossRef]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Chang, J.; Guo, J.-T.; Jiang, D.; Guo, H.; Taylor, J.M.; Block, T.M. Liver-Specific MicroRNA miR-122 Enhances the Replication of Hepatitis C Virus in Nonhepatic Cells. J. Virol. 2008, 82, 8215–8223. [Google Scholar] [CrossRef]
- Narbus, C.M.; Israelow, B.; Sourisseau, M.; Michta, M.L.; Hopcraft, S.E.; Zeiner, G.M.; Evans, M.J. HepG2 Cells Expressing MicroRNA miR-122 Support the Entire Hepatitis C Virus Life Cycle. J. Virol. 2011, 85, 12087–12092. [Google Scholar] [CrossRef]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Zheng, J.; Lambrecht, R.W.; Bonkovsky, H.L. Reciprocal Effects of Micro-RNA-122 on Expression of Heme Oxygenase-1 and Hepatitis C Virus Genes in Human Hepatocytes. Gastroenterology 2007, 133, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef]
- Wang, X.; Peng, J.; Wang, J.; Li, M.; Wu, D.; Wu, S.; Liao, J.; Dou, J. Hepatitis C virus core impacts expression of miR122 and miR204 involved in carcinogenic progression via regulation of TGFBRAP1 and HOTTIP expression. OncoTargets Ther. 2018, 11, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Bihrer, V.; Friedrich-Rust, M.; Kronenberger, B.; Forestier, N.; Haupenthal, J.; Shi, Y.; Peveling-Oberhag, J.; Radeke, H.; Sarrazin, C.; Herrmann, E.; et al. Serum miR-122 as a Biomarker of Necroinflammation in Patients With Chronic Hepatitis C Virus Infection. Am. J. Gastroenterol. 2011, 106, 1663–1669. [Google Scholar] [CrossRef]
- Tambyah, P.A.; Ching, C.S.; Sepramaniam, S.; Ali, J.M.; Armugam, A.; Jeyaseelan, K. microRNA expression in blood of dengue patients. Ann. Clin. Biochem. 2016, 53, 466–476. [Google Scholar] [CrossRef]
- Lee, T.-C.; Lin, Y.-L.; Liao, J.-T.; Su, C.-M.; Lin, C.-C.; Lin, W.-P.; Liao, C.-L. Utilizing liver-specific microRNA-122 to modulate replication of dengue virus replicon. Biochem. Biophys. Res. Commun. 2010, 396, 596–601. [Google Scholar] [CrossRef]
- Diallo, I.; Ho, J.; Laffont, B.; Laugier, J.; Benmoussa, A.; Lambert, M.; Husseini, Z.; Soule, G.; Kozak, R.; Kobinger, G.; et al. Altered microRNA Transcriptome in Cultured Human Liver Cells upon Infection with Ebola Virus. Int. J. Mol. Sci. 2021, 22, 3792. [Google Scholar] [CrossRef]
- Madara, J.J.; Han, Z.; Ruthel, G.; Freedman, B.D.; Harty, R.N. The multifunctional Ebola virus VP40 matrix protein is a promising therapeutic target. Futur. Virol. 2015, 10, 537–546. [Google Scholar] [CrossRef]
- Wang, S.; Liu, P.; Yang, P.; Zheng, J.; Zhao, D. Peripheral blood microRNAs expression is associated with infant respiratory syncytial virus infection. Oncotarget 2017, 8, 96627–96635. [Google Scholar] [CrossRef]
- Ioannidis, I.; McNally, B.; Willette, M.; Peeples, M.E.; Chaussabel, D.; Durbin, J.E.; Ramilo, O.; Mejias, A.; Flaño, E. Plasticity and Virus Specificity of the Airway Epithelial Cell Immune Response during Respiratory Virus Infection. J. Virol. 2012, 86, 5422–5436. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.A.; Haynes, L.M.; Jones, L.P.; Tripp, R.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of Toll-Like Receptor 4 in Innate Immunity to Respiratory Syncytial Virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Shepherd, B.; Crowe, J.E., Jr. Transforming Growth Factor Beta Is a Major Regulator of Human Neonatal Immune Responses following Respiratory Syncytial Virus Infection. J. Virol. 2010, 84, 12895–12902. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Salice, P.; Bosis, S.; Ghiglia, S.; Tremolati, E.; Tagliabue, C.; Gualtieri, L.; Barbier, P.; Galeone, C.; Marchisio, P.; et al. Altered cardiac rhythm in infants with bronchiolitis and respiratory syncytial virus infection. BMC Infect. Dis. 2010, 10, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.A.E.-G.; Zayed, K.M. Impact of Acute Bronchiolitis on Cardiac Functions and Serum microRNA-122 and 499. Am. J. Infect. Dis. 2016, 12, 11–19. [Google Scholar] [CrossRef]
- Collison, A.M.; Sokulsky, L.A.; Kepreotes, E.; de Siqueira, A.P.; Morten, M.; Edwards, M.R.; Walton, R.P.; Bartlett, N.W.; Yang, M.; Nguyen, T.H.; et al. miR-122 promotes virus-induced lung disease by targeting SOCS1. JCI Insight 2021, 6, e127933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, G.; Jiang, Y. The Emerging Roles of miR-125b in Cancers. Cancer Manag. Res. 2020, 12, 1079–1088. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.; et al. Modulation of miR-155 and miR-125b Levels following Lipopolysaccharide/TNF-α Stimulation and Their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef]
- Murphy, A.J.; Guyre, P.M.; Pioli, P.A. Estradiol Suppresses NF-κB Activation through Coordinated Regulation of let-7a and miR-125b in Primary Human Macrophages. J. Immunol. 2010, 184, 5029–5037. [Google Scholar] [CrossRef]
- Busch, S.; Auth, E.; Scholl, F.; Huenecke, S.; Koehl, U.; Suess, B.; Steinhilber, D. 5-Lipoxygenase Is a Direct Target of miR-19a-3p and miR-125b-5p. J. Immunol. 2015, 194, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; So, A.Y.-L.; Sinha, N.; Gibson, W.S.J.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b Potentiates Macrophage Activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, J.-P.; Li, B.; Zeng, C.; You, K.; Chen, M.-X.; Yuan, Y.; Zhuang, S.-M. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene 2013, 32, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Zhang, X.; Ma, Z.; Lin, Y.; Lu, M. MicroRNA-125b-5p mediates post-transcriptional regulation of hepatitis B virus replication via the LIN28B/let-7 axis. RNA Biol. 2017, 14, 1389–1398. [Google Scholar] [CrossRef]
- Li, F.; Zhou, P.; Deng, W.; Wang, J.; Mao, R.; Zhang, Y.; Li, J.; Yu, J.; Yang, F.; Huang, Y.; et al. Serum microRNA-125b correlates with hepatitis B viral replication and liver necroinflammation. Clin. Microbiol. Infect. 2016, 22, 384.e1–384.e10. [Google Scholar] [CrossRef]
- Chen, S.; Chen, H.; Gao, S.; Qiu, S.; Zhou, H.; Yu, M.; Tu, J. Differential expression of plasma microRNA-125b in hepatitis B virus-related liver diseases and diagnostic potential for hepatitis B virus-induced hepatocellular carcinoma. Hepatol. Res. 2017, 47, 312–320. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, J.; He, Y.; Zhan, X.; Zhao, R.; Huang, Y.; Xu, H.; Zhu, Z.; Liu, Q. miR-125b inhibits hepatitis B virus expression in vitro through targeting of the SCNN1A gene. Arch. Virol. 2014, 159, 3335–3343. [Google Scholar] [CrossRef]
- Peng, C.; Wang, H.; Zhang, W.-J.; Jie, S.-H.; Tong, Q.-X.; Lu, M.-J.; Yang, D.-L. Inhibitory effect of miR-125b on hepatitis C virus core protein-induced TLR2/MyD88 signaling in THP-1 cells. World J. Gastroenterol. 2016, 22, 4354–4361. [Google Scholar] [CrossRef]
- Dai, C.-Y.; Tsai, Y.-S.; Chou, W.-W.; Liu, T.; Huang, C.-F.; Wang, S.-C.; Tsai, P.-C.; Yeh, M.-L.; Hsieh, M.-Y.; Huang, C.-I.; et al. The IL-6/STAT3 pathway upregulates microRNA-125b expression in hepatitis C virus infection. Oncotarget 2018, 9, 11291–11302. [Google Scholar] [CrossRef] [PubMed]
- Shwetha, S.; Sharma, G.; Raheja, H.; Goel, A.; Aggarwal, R.; Das, S. Interaction of miR-125b-5p with Human antigen R mRNA: Mechanism of controlling HCV replication. Virus Res. 2018, 258, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sultana, C.; Rosca, A.; Ruta, S. Correlation Between miR-125b Expression and Liver Fibrosis in Patients with Chronic Hepatitis C. Hepat. Mon. 2019, 19, e84615. [Google Scholar] [CrossRef]
- Liu, W.; Hu, J.; Zhou, K.; Chen, F.; Wang, Z.; Liao, B.; Dai, Z.; Cao, Y.; Fan, J.; Zhou, J. Serum exosomal miR-125b is a novel prognostic marker for hepatocellular carcinoma. OncoTargets Ther. 2017, 10, 3843–3851. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/hepeviridae (accessed on 19 July 2022).
- Harms, D.; Choi, M.; Allers, K.; Wang, B.; Pietsch, H.; Papp, C.-P.; Hanisch, L.; Kurreck, J.; Hofmann, J.; Bock, C.-T. Specific circulating microRNAs during hepatitis E infection can serve as indicator for chronic hepatitis E. Sci. Rep. 2020, 10, 5337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, F.; Yang, D.; Peng, T.; Lu, G. Identification of miRNA-mRNA Crosstalk in Respiratory Syncytial Virus- (RSV-) Associated Pediatric Pneumonia through Integrated miRNAome and Transcriptome Analysis. Mediat. Inflamm. 2020, 2020, 8919534. [Google Scholar] [CrossRef]
- Makkoch, J.; Poomipak, W.; Saengchoowong, S.; Khongnomnan, K.; Praianantathavorn, K.; Jinato, T.; Poovorawan, Y.; Payungporn, S. Human microRNAs profiling in response to influenza A viruses (subtypes pH1N1, H3N2, and H5N1). Exp. Biol. Med. 2016, 241, 409–420. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Wu, J.; Deng, C.; Tan, J.; Liu, H.; Zhong, L. Lung adenocarcinoma patients have higher risk of SARS-CoV-2 infection. Aging 2021, 13, 1620–1632. [Google Scholar] [CrossRef]
- Chaudhuri, E.; Dash, S.; Balasubramaniam, M.; Padron, A.; Holland, J.; Sowd, G.A.; Villalta, F.; Engelman, A.N.; Pandhare, J.; Dash, C. The HIV-1 capsid-binding host factor CPSF6 is post-transcriptionally regulated by the cellular microRNA miR-125b. J. Biol. Chem. 2020, 295, 5081–5094. [Google Scholar] [CrossRef]
- Mantri, C.K.; Dash, J.P.; Mantri, J.V.; Dash, C.C.V. Cocaine Enhances HIV-1 Replication in CD4+ T Cells by Down-Regulating MiR-125b. PLoS ONE 2012, 7, e51387. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae (accessed on 19 July 2022).
- Ghosh, D.; Basu, A. Japanese Encephalitis—A Pathological and Clinical Perspective. PLOS Negl. Trop. Dis. 2009, 3, e437. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-W.; Tsai, K.-N.; Chen, Y.-S.; Chang, R.-Y. Differential miRNA Expression Profiling Reveals Correlation of miR125b-5p with Persistent Infection of Japanese Encephalitis Virus. Int. J. Mol. Sci. 2021, 22, 4218. [Google Scholar] [CrossRef] [PubMed]
- Mziaut, H.; Henniger, G.; Ganss, K.; Hempel, S.; Wolk, S.; McChord, J.; Chowdhury, K.; Ravassard, P.; Knoch, K.-P.; Krautz, C.; et al. MiR-132 controls pancreatic beta cell proliferation and survival through Pten/Akt/Foxo3 signaling. Mol. Metab. 2020, 31, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, A.; Liu, X.; Meisgen, F.; Grünler, J.; Botusan, I.R.; Narayanan, S.; Erikci, E.; Blomqvist, L.; Du, L.; et al. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J. Clin. Investig. 2015, 125, 3008–3026. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tong, Z.; Chen, K.; Hu, X.; Jin, H.; Hou, M. The Role of miRNA-132 against Apoptosis and Oxidative Stress in Heart Failure. BioMed Res. Int. 2018, 2018, 3452748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strum, J.C.; Johnson, J.H.; Ward, J.; Xie, H.; Feild, J.; Hester, A.; Alford, A.; Waters, K.M. MicroRNA 132 Regulates Nutritional Stress-Induced Chemokine Production through Repression of SirT1. Mol. Endocrinol. 2009, 23, 1876–1884. [Google Scholar] [CrossRef]
- Wei, X.; Tan, C.; Tang, C.; Ren, G.; Xiang, T.; Qiu, Z.; Liu, R.; Wu, Z. Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell. Signal. 2013, 25, 1037–1043. [Google Scholar] [CrossRef]
- Liu, B.; Yang, X.-F.; Liang, X.-P.; Wang, L.; Shao, M.-M.; Han, W.-X.; Wu, Y.-H. Expressions of MiR-132 in patients with chronic hepatitis B, posthepatitic cirrhosis and hepatitis B virus-related hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8431–8437. [Google Scholar]
- Zhang, F.; Lin, X.; Yang, X.; Lu, G.; Zhang, Q.; Zhang, C. MicroRNA-132-3p suppresses type I IFN response through targeting IRF1 to facilitate H1N1 influenza A virus infection. Biosci. Rep. 2019, 39, BSR20192769. [Google Scholar] [CrossRef]
- Hsu, A.C.-Y.; Parsons, K.; Moheimani, F.; Knight, D.A.; Hansbro, P.M.; Fujita, T.; Wark, P.A. Impaired Antiviral Stress Granule and IFN-β Enhanceosome Formation Enhances Susceptibility to Influenza Infection in Chronic Obstructive Pulmonary Disease Epithelium. Am. J. Respir. Cell Mol. Biol. 2016, 55, 117–127. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsdna-viruses/w/herpesviridae (accessed on 19 July 2022).
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.; González, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell. Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Pepose, J.S. Herpes simplex keratitis: Role of viral infection versus immune response. Surv. Ophthalmol. 1991, 35, 345–352. [Google Scholar] [CrossRef]
- Mulik, S.; Xu, J.; Reddy, P.B.; Rajasagi, N.K.; Gimenez, F.; Sharma, S.; Lu, P.Y.; Rouse, B.T. Role of miR-132 in Angiogenesis after Ocular Infection with Herpes Simplex Virus. Am. J. Pathol. 2012, 181, 525–534. [Google Scholar] [CrossRef]
- Taheri, F.; Ebrahimi, S.O.; Shareef, S.; Reiisi, S. Regulatory and immunomodulatory role of miR-34a in T cell immunity. Life Sci. 2020, 262, 118209. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, H.; Li, J.; Xue, R.; Liu, H.; Zhu, Z.; Pan, C.; Lin, Y.; Hu, A.; Gou, P.; et al. Elevated HMGB1 expression induced by hepatitis B virus X protein promotes epithelial-mesenchymal transition and angiogenesis through STAT3/miR-34a/NF-kappaB in primary liver cancer. Am. J. Cancer Res. 2021, 11, 479–494. [Google Scholar]
- FeiLi, X.; Wu, S.; Ye, W.; Tu, J.; Lou, L. MicroRNA-34a-5p inhibits liver fibrosis by regulating TGF-β1/Smad3 pathway in hepatic stellate cells. Cell Biol. Int. 2018, 42, 1370–1376. [Google Scholar] [CrossRef]
- Rana, M.A.; Ijaz, B.; Daud, M.; Tariq, S.; Nadeem, T.; Husnain, T. Interplay of Wnt β-catenin pathway and miRNAs in HBV pathogenesis leading to HCC. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 373–386. [Google Scholar] [CrossRef]
- Yang, P.; Li, Q.-J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Rossi, A.D.; Higa, L.M.; Herlinger, A.L.; Ribeiro-Alves, M.; de Menezes, M.T.; Giannini, A.L.M.; Cardoso, C.C.; Da Poian, A.T.; Tanuri, A.; Aguiar, R.S. Differential Expression of Human MicroRNAs During Dengue Virus Infection in THP-1 Monocytes. Front. Cell. Infect. Microbiol. 2021, 11, 714088. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Jeng, S.; McWeeney, S.K.; Hirsch, A.J. A MicroRNA Screen Identifies the Wnt Signaling Pathway as a Regulator of the Interferon Response during Flavivirus Infection. J. Virol. 2017, 91, e02388–e02404. [Google Scholar] [CrossRef]
- Fan, N.; Wang, J. MicroRNA 34a contributes to virus-mediated apoptosis through binding to its target gene Bax in influenza A virus infection. Biomed. Pharmacother. 2016, 83, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Othumpangat, S.; Beezhold, D.H.; Umbright, C.M.; Noti, J.D. Influenza Virus-Induced Novel miRNAs Regulate the STAT Pathway. Viruses 2021, 13, 967. [Google Scholar] [CrossRef]
- Centa, A.; Fonseca, A.S.; Ferreira, S.G.D.S.; Azevedo, M.L.V.; de Paula, C.B.V.; Nagashima, S.; Machado-Souza, C.; Miggiolaro, A.F.R.D.S.; Baena, C.P.; de Noronha, L.; et al. Deregulated miRNA expression is associated with endothelial dysfunction in post-mortem lung biopsies of COVID-19 patients. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L405–L412. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Dabrowski, M.; Jakiela, B.; Matalon, S.; Harrod, K.S.; Sanak, M.; Collawn, J.F. SARS-CoV-2 may regulate cellular responses through depletion of specific host miRNAs. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L444–L455. [Google Scholar] [CrossRef] [PubMed]
- Demirci, M.D.S.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. PeerJ 2020, 8, e9369. [Google Scholar] [CrossRef]
- Verdonck, K.; González, E.; Van Dooren, S.; Vandamme, A.-M.; Vanham, G.; Gotuzzo, E. Human T-lymphotropic virus 1: Recent knowledge about an ancient infection. Lancet Infect. Dis. 2007, 7, 266–281. [Google Scholar] [CrossRef]
- Sharma, V.K.; Raimondi, V.; Ruggero, K.; Pise-Masison, C.A.; Cavallari, I.; Silic-Benussi, M.; Ciminale, V.; D’Agostino, D.M. Expression of miR-34a in T-Cells Infected by Human T-Lymphotropic Virus. Front. Microbiol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef]
- Jenike, A.E.; Halushka, M.K. miR-21: A non-specific biomarker of all maladies. Biomark. Res. 2021, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M. A Tale of Two States: Normal and Transformed, With and Without Rigidity Sensing. Annu. Rev. Cell Dev. Biol. 2019, 35, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.; Zhang, M.; Dong, X.; Zheng, L.; Li, G.; Han, X.; Yao, Z.; Han, T.; Hong, W. Silencing lncRNA Lfar1 alleviates the classical activation and pyoptosis of macrophage in hepatic fibrosis. Cell Death Dis. 2020, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Yun, Y. HBx Protein of Hepatitis B Virus Activates Jak1-STAT Signaling. J. Biol. Chem. 1998, 273, 25510–25515. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Kang-Park, S.; Do, S.-I.; Lee, Y.I. The Hepatitis B Virus-X Protein Activates a Phosphatidylinositol 3-Kinase-dependent Survival Signaling Cascade. J. Biol. Chem. 2001, 276, 16969–16977. [Google Scholar] [CrossRef]
- Wu, H.-C.; Huang, C.-L.; Wang, H.-W.; Hsu, W.-F.; Tsai, T.-Y.; Chen, S.-H.; Peng, C.-Y. Serum miR-21 correlates with the histological stage of chronic hepatitis B-associated liver fibrosis. Int. J. Clin. Exp. Pathol. 2019, 12, 3819–3829. [Google Scholar]
- Li, W.; Yu, X.; Chen, X.; Wang, Z.; Yin, M.; Zhao, Z.; Zhu, C. HBV induces liver fibrosis via the TGF-β1/miR-21-5p pathway. Exp. Ther. Med. 2021, 21, 169. [Google Scholar] [CrossRef]
- Wei, J.; Feng, L.; Li, Z.; Xu, G.; Fan, X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. 2013, 67, 387–392. [Google Scholar] [CrossRef]
- Fu, R.-Q.; Hu, D.-P.; Hu, Y.-B.; Hong, L.; Sun, Q.-F.; Ding, J.-G. miR-21 promotes α-SMA and collagen I expression in hepatic stellate cells via the Smad7 signaling pathway. Mol. Med. Rep. 2017, 16, 4327–4333. [Google Scholar] [CrossRef]
- Qiu, X.; Dong, S.; Qiao, F.; Lu, S.; Song, Y.; Lao, Y.; Li, Y.; Zeng, T.; Hu, J.; Zhang, L.; et al. HBx-mediated miR-21 upregulation represses tumor-suppressor function of PDCD4 in hepatocellular carcinoma. Oncogene 2013, 32, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Damania, P.; Sen, B.; Dar, S.B.; Kumar, S.; Kumari, A.; Gupta, E.; Sarin, S.K.; Venugopal, S.K. Hepatitis B Virus Induces Cell Proliferation via HBx-Induced microRNA-21 in Hepatocellular Carcinoma by Targeting Programmed Cell Death Protein4 (PDCD4) and Phosphatase and Tensin Homologue (PTEN). PLoS ONE 2014, 9, e91745. [Google Scholar] [CrossRef]
- Li, C.H.; Xu, F.; Chow, S.; Feng, L.; Yin, D.; Ng, T.B.; Chen, Y. Hepatitis B virus X protein promotes hepatocellular carcinoma transformation through interleukin-6 activation of microRNA-21 expression. Eur. J. Cancer 2014, 50, 2560–2569. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Chow, S.C.; Yin, D.L.; Ng, T.B.; Chen, Y.C. Functional characterisation of hepatitis B viral X protein/microRNA-21 interaction in HBV-associated hepatocellular carcinoma. Hong Kong Med. J. 2016, 22, 37–42. [Google Scholar]
- Bandopadhyay, M.; Banerjee, A.; Sarkar, N.; Panigrahi, R.; Datta, S.; Pal, A.; Singh, S.P.; Biswas, A.; Chakrabarti, S.; Chakravarty, R. Tumor suppressor micro RNA miR-145 and onco micro RNAs miR-21 and miR-222 expressions are differentially modulated by Hepatitis B virus X protein in malignant hepatocytes. BMC Cancer 2014, 14, 721. [Google Scholar] [CrossRef]
- Yin, D.; Wang, Y.; Sai, W.; Zhang, L.; Miao, Y.; Cao, L.; Zhai, X.; Feng, X.; Yang, L. HBx-induced miR-21 suppresses cell apoptosis in hepatocellular carcinoma by targeting interleukin-12. Oncol. Rep. 2016, 36, 2305–2312. [Google Scholar] [CrossRef]
- Otania, T.; Nakamuraa, S.; Tokia, M.; Motodaa, R.; Kurimotob, M.; Oritaa, K. Identification of IFN-γ-Producing Cells in IL-12/IL-18-Treated Mice. Cell. Immunol. 1999, 198, 111–119. [Google Scholar] [CrossRef]
- Zeh, H.J.; Hurd, S.; Storkus, W.J.; Lotze, M.T. Interleukin-12 Promotes the Proliferation and Cytolytic Maturation of Immune Effectors: Implications for the immunotherapy of cancer. J. Immunother. 1993, 14, 155–161. [Google Scholar] [CrossRef]
- Clément, S.; Sobolewski, C.; Gomes, D.; Rojas, A.; Goossens, N.; Conzelmann, S.; Calo, N.; Negro, F.; Foti, M. Activation of the oncogenic miR-21-5p promotes HCV replication and steatosis induced by the viral core 3a protein. Liver Int. 2019, 39, 1226–1236. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-Induced miR-21 Contributes to Evasion of Host Immune System by Targeting MyD88 and IRAK1. PLOS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef]
- Marquez, R.T.; Bandyopadhyay, S.; Wendlandt, E.B.; Keck, K.; Hoffer, B.A.; Icardi, M.S.; Christensen, R.N.; Schmidt, W.N.; McCaffrey, A.P. Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab. Investig. 2010, 90, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Kanokudom, S.; Vilaivan, T.; Wikan, N.; Thepparit, C.; Smith, D.R.; Assavalapsakul, W. miR-21 promotes dengue virus serotype 2 replication in HepG2 cells. Antivir. Res. 2017, 142, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Duy, J.; Koehler, J.; Honko, A.; Schoepp, R.; Wauquier, N.; Gonzalez, J.-P.; Pitt, M.L.; Mucker, E.; Johnson, J.; O’Hearn, A.; et al. Circulating microRNA profiles of Ebola virus infection. Sci. Rep. 2016, 6, 24496. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Feng, P.; Gu, T. MicroRNA-21-3p modulates FGF2 to facilitate influenza A virus H5N1 replication by refraining type I interferon response. Biosci. Rep. 2020, 40, BSR20200158. [Google Scholar] [CrossRef]
- Xia, B.; Lu, J.; Wang, R.; Yang, Z.; Zhou, X.; Huang, P. miR-21-3p Regulates Influenza A Virus Replication by Targeting Histone Deacetylase-8. Front. Cell. Infect. Microbiol. 2018, 8, 175. [Google Scholar] [CrossRef]
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master Regulators of Inflammatory Signaling. Trends Mol. Med. 2018, 24, 66–84. [Google Scholar] [CrossRef]
- Lam, W.-Y.; Yeung, A.C.-M.; Ngai, K.L.-K.; Li, M.-S.; To, K.-F.; Tsui, S.K.-W.; Chan, P.K.-S. Effect of avian influenza A H5N1 infection on the expression of microRNA-141 in human respiratory epithelial cells. BMC Microbiol. 2013, 13, 104. [Google Scholar] [CrossRef]
- Tan, K.S.; Choi, H.; Jiang, X.; Yin, L.; Seet, J.E.; Patzel, V.; Engelward, B.P.; Chow, V.T. Micro-RNAs in regenerating lungs: An integrative systems biology analysis of murine influenza pneumonia. BMC Genom. 2014, 15, 587. [Google Scholar] [CrossRef]
- Zhong, Z.; Dong, Z.; Yang, L.; Gong, Z. miR-21 induces cell cycle at S phase and modulates cell proliferation by down-regulating hMSH2 in lung cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 1781–1788. [Google Scholar] [CrossRef]
- Vaporidi, K.; Vergadi, E.; Kaniaris, E.; Hatziapostolou, M.; Lagoudaki, E.; Georgopoulos, D.; Zapol, W.M.; Bloch, K.D.; Iliopoulos, D. Pulmonary microRNA profiling in a mouse model of ventilator-induced lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L199–L207. [Google Scholar] [CrossRef]
- Nersisyan, S.; Engibaryan, N.; Gorbonos, A.; Kirdey, K.; Makhonin, A.; Tonevitsky, A. Potential role of cellular miRNAs in coronavirus-host interplay. PeerJ 2020, 8, e9994. [Google Scholar] [CrossRef] [PubMed]
- Jafarinejad-Farsangi, S.; Jazi, M.M.; Rostamzadeh, F.; Hadizadeh, M. High affinity of host human microRNAs to SARS-CoV-2 genome: An in silico analysis. Non-Coding RNA Res. 2020, 5, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Corsello, T.; Kudlicki, A.S.; Komaravelli, N.; Casola, A. Respiratory Syncytial Virus Infection Changes Cargo Composition of Exosome Released from Airway Epithelial Cells. Sci. Rep. 2018, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; Vergara, H.S.J.; Mohapatra, S.; Mohapatra, S. Respiratory Syncytial Virus NS1 Protein Colocalizes with Mitochondrial Antiviral Signaling Protein MAVS following Infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef]
- Kim, R.Y.; Horvat, J.C.; Pinkerton, J.W.; Starkey, M.R.; Essilfie, A.T.; Mayall, J.R.; Nair, P.M.; Hansbro, N.G.; Jones, B.; Haw, T.J.; et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase–mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 2017, 139, 519–532. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, P.; Chen, M.; Xu, Y.; Zhao, D. miRNAs and Leukotrienes in Respiratory Syncytial Virus Infection. Front. Pediatr. 2021, 9, 602195. [Google Scholar] [CrossRef]
- Ruiz-De-León, M.J.; Jiménez-Sousa, M.A.; Moreno, S.; García, M.; Gutiérrez-Rivas, M.; León, A.; Montero-Alonso, M.; González-García, J.; Resino, S.; Rallón, N.; et al. Lower expression of plasma-derived exosome miR-21 levels in HIV-1 elite controllers with decreasing CD4 T cell count. J. Microbiol. Immunol. Infect. 2019, 52, 667–671. [Google Scholar] [CrossRef]
- Cojo, M.S.-D.; López-Huertas, M.R.; Díez-Fuertes, F.; Rodríguez-Mora, S.; Bermejo, M.; López-Campos, G.; Mateos, E.; Jiménez-Tormo, L.; Gómez-Esquer, F.; Díaz-Gil, G.; et al. Changes in the cellular microRNA profile by the intracellular expression of HIV-1 Tat regulator: A potential mechanism for resistance to apoptosis and impaired proliferation in HIV-1 infected CD4+ T cells. PLoS ONE 2017, 12, e0185677. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, T.; Wang, R.; Zhang, H.; Huang, X.; Yin, J.; Zhang, L.; Xu, X.; Wu, H. Plasma IP-10 Is Associated with Rapid Disease Progression in Early HIV-1 Infection. Viral Immunol. 2012, 25, 333–337. [Google Scholar] [CrossRef]
- Liu, P.P.; Mason, J.W. Advances in the Understanding of Myocarditis. Circulation 2001, 104, 1076–1082. [Google Scholar] [CrossRef]
- He, F.; Yao, H.; Xiao, Z.; Han, J.; Zou, J.; Liu, Z. Inhibition of IL-2 inducible T-cell kinase alleviates T-cell activation and murine myocardial inflammation associated with CVB3 infection. Mol. Immunol. 2014, 59, 30–38. [Google Scholar] [CrossRef]
- He, F.; Xiao, Z.; Yao, H.; Li, S.; Feng, M.; Wang, W.; Liu, Z.; Liu, Z.; Wu, J. The protective role of microRNA-21 against coxsackievirus B3 infection through targeting the MAP2K3/P38 MAPK signaling pathway. J. Transl. Med. 2019, 17, 335. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhang, H.M.; Qiu, Y.; Hanson, P.J.; Hemida, M.; Wei, W.; Hoodless, P.A.; Chu, F.; Yang, D. Coxsackievirus-Induced miR-21 Disrupts Cardiomyocyte Interactions via the Downregulation of Intercalated Disk Components. PLOS Pathog. 2014, 10, e1004070. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/negative-sense-rna-viruses/w/rhabdoviridae/805/genus-vesiculovirus (accessed on 19 July 2022).
- Pandey, N.; Rastogi, M.; Singh, S.K. Chandipura virus dysregulates the expression of hsa-miR-21-5p to activate NF-κB in human microglial cells. J. Biomed. Sci. 2021, 28, 52. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liang, H.; Deng, T.; Zhu, K.; Zhang, S.; Wang, N.; Jiang, X.; Wang, X.; Liu, R.; Zen, K.; et al. The identification of novel targets of miR-16 and characterization of their biological functions in cancer cells. Mol. Cancer 2013, 12, 92. [Google Scholar] [CrossRef]
- Yan, L.; Liang, M.; Hou, X.; Zhang, Y.; Zhang, H.; Guo, Z.; Jinyu, J.; Feng, Z.; Mei, Z. The role of microRNA-16 in the pathogenesis of autoimmune diseases: A comprehensive review. Biomed. Pharmacother. 2019, 112, 108583. [Google Scholar] [CrossRef]
- Wu, G.; Yu, F.; Xiao, Z.; Xu, K.; Xu, J.; Tang, W.; Wang, J.; Song, E. Hepatitis B virus X protein downregulates expression of the miR-16 family in malignant hepatocytes in vitro. Br. J. Cancer 2011, 105, 146–153. [Google Scholar] [CrossRef]
- Zhu, B.; Wei, X.-X.; Wang, T.-B.; Zhou, Y.-C.; Liu, A.-M.; Zhang, G.-W. Increased miR-16 expression induced by hepatitis C virus infection promotes liver fibrosis through downregulation of hepatocyte growth factor and Smad7. Arch. Virol. 2015, 160, 2043–2050. [Google Scholar] [CrossRef]
- Wang, H.; Tian, Z.; Xu, Y.; Wang, Q.; Ding, S.-W.; Li, Y. Altering Intracellular Localization of the RNA Interference Factors by Influenza A Virus Non-structural Protein 1. Front. Microbiol. 2020, 11, 590904. [Google Scholar] [CrossRef]
- Sun, X.; Sit, A.; Feinberg, M.W. Role of miR-181 family in regulating vascular inflammation and immunity. Trends Cardiovasc. Med. 2014, 24, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Henao-Mejia, J.; Harman, C.C.D.; Flavell, R.A. miR-181 and Metabolic Regulation in the Immune System. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Zhang, F.-R.; Shang, J.-Y.; Liu, Y.-Y.; Lv, X.-F.; Yuan, J.-N.; Zhang, T.-T.; Li, K.; Lin, X.-C.; Liu, X.; et al. Renal inhibition of miR-181a ameliorates 5-fluorouracil-induced mesangial cell apoptosis and nephrotoxicity. Cell Death Dis. 2018, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhou, G.; Li, G.; Chen, B.; Dong, P.; Zheng, J. Serum miR-181b Is Correlated with Hepatitis B Virus Replication and Disease Progression in Chronic Hepatitis B Patients. Dig. Dis. Sci. 2015, 60, 2346–2352. [Google Scholar] [CrossRef]
- Wang, B.; Li, W.; Guo, K.; Xiao, Y.; Wang, Y.; Fan, J. miR-181b Promotes hepatic stellate cells proliferation by targeting p27 and is elevated in the serum of cirrhosis patients. Biochem. Biophys. Res. Commun. 2012, 421, 4–8. [Google Scholar] [CrossRef]
- Zheng, J.; Wu, C.; Xu, Z.; Xia, P.; Dong, P.; Chen, B.; Yu, F. Hepatic stellate cell is activated by microRNA-181b via PTEN/Akt pathway. Mol. Cell. Biochem. 2015, 398, 1–9. [Google Scholar] [CrossRef]
- Zou, C.; Li, Y.; Cao, Y.; Zhang, J.; Jiang, J.; Sheng, Y.; Wang, S.; Huang, A.; Tang, H. Up-regulated MicroRNA-181a induces carcinogenesis in Hepatitis B virus-related hepatocellular carcinoma by targeting E2F5. BMC Cancer 2014, 14, 97. [Google Scholar] [CrossRef]
- Tian, Y.; Xiao, X.; Gong, X.; Peng, F.; Xu, Y.; Jiang, Y.; Gong, G. HBx promotes cell proliferation by disturbing the cross-talk between miR-181a and PTEN. Sci. Rep. 2017, 7, 40089. [Google Scholar] [CrossRef]
- Zou, C.; Chen, J.; Chen, K.; Wang, S.; Cao, Y.; Zhang, J.; Sheng, Y.; Huang, A.; Tang, H. Functional analysis of miR-181a and Fas involved in hepatitis B virus-related hepatocellular carcinoma pathogenesis. Exp. Cell Res. 2015, 331, 352–361. [Google Scholar] [CrossRef]
- Elhelw, D.S.; Mekky, R.Y.; El-Ekiaby, N.; Ahmed, R.; Eldin, M.A.M.; El-Sayed, M.; AbouElKhair, M.; Salah, A.; Zekri, A.R.; Esmat, G.; et al. Predictive prognostic role of miR-181a with discrepancy in the liver and serum of genotype 4 hepatitis C virus patients. Biomed. Rep. 2014, 2, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Boštjančič, E.; Bandelj, E.; Luzar, B.; Poljak, M.; Glavač, D. Hepatic expression of miR-122, miR-126, miR-136 and miR-181a and their correlation to histopathological and clinical characteristics of patients with hepatitis C. J. Viral Hepat. 2015, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Zhou, Y.; Ying, R.S.; Shi, L.; Cheng, Y.Q.; Ren, J.P.; Griffin, J.W.; Jia, Z.S.; Li, C.F.; Moorman, J.P.; et al. Hepatitis C virus-induced reduction in miR-181a impairs CD4+T-cell responses through overexpression of DUSP6. Hepatology 2015, 61, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Shrivastava, S.; Chowdhury, J.B.; Ray, R.; Ray, R.B. Transcriptional Suppression of miR-181c by Hepatitis C Virus Enhances Homeobox A1 Expression. J. Virol. 2014, 88, 7929–7940. [Google Scholar] [CrossRef]
- Patra, T.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C Virus Mediated Inhibition of miR-181c Activates ATM Signaling and Promotes Hepatocyte Growth. Hepatology 2020, 71, 780–793. [Google Scholar] [CrossRef]
- Lim, J.; Byun, J.; Guk, K.; Hwang, S.G.; Bae, P.K.; Jung, J.; Kang, T.; Lim, E.-K. Highly Sensitive in Vitro Diagnostic System of Pandemic Influenza A (H1N1) Virus Infection with Specific MicroRNA as a Biomarker. ACS Omega 2019, 4, 14560–14568. [Google Scholar] [CrossRef]
- Peng, F.; He, J.; Loo, J.F.C.; Yao, J.; Shi, L.; Liu, C.; Zhao, C.; Xie, W.; Shao, Y.; Kong, S.K.; et al. Identification of microRNAs in Throat Swab as the Biomarkers for Diagnosis of Influenza. Int. J. Med. Sci. 2016, 13, 77–84. [Google Scholar] [CrossRef]
- Atherton, L.J.; Jorquera, P.A.; Bakre, A.A.; Tripp, R.A. Determining Immune and miRNA Biomarkers Related to Respiratory Syncytial Virus (RSV) Vaccine Types. Front. Immunol. 2019, 10, 2323. [Google Scholar] [CrossRef]
- Su, J.-L.; Chen, P.-S.; Johansson, G.; Kuo, M.-L. Function and Regulation of Let-7 Family microRNAs. MicroRNA 2012, 1, 34–39. [Google Scholar] [CrossRef]
- Thammaiah, C.K.; Jayaram, S. Role of let-7 family microRNA in breast cancer. Non-Coding RNA Res. 2016, 1, 77–82. [Google Scholar] [CrossRef]
- Jiang, S. Recent findings regarding let-7 in immunity. Cancer Lett. 2018, 434, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Letafati, A.; Najafi, S.; Mottahedi, M.; Karimzadeh, M.; Shahini, A.; Garousi, S.; Abbasi-Kolli, M.; Nahand, J.S.; Zadeh, S.S.T.; Hamblin, M.R.; et al. MicroRNA let-7 and viral infections: Focus on mechanisms of action. Cell. Mol. Biol. Lett. 2022, 27, 1–47. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, Y.; Toh, S.T.; Sung, W.-K.; Tan, P.; Chow, P.; Chung, A.Y.; Jooi, L.L.; Lee, C.G.L. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription. J. Hepatol. 2010, 53, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Huang, P.; Ju, X.; Li, Z.; Wang, Y. Lin28B over-expression mediates the repression of let-7 by hepatitis B virus X protein in hepatoma cells. Int. J. Clin. Exp. Med. 2015, 8, 15108–15116. [Google Scholar]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Chen, J.; Liu, J.; Luo, Z.; Jiang, W.; Huang, J.; Qiu, Z.; Yue, W.; Wu, L. Expression of microRNA let-7a positively correlates with hepatitis B virus replication in hepatocellular carcinoma tissues. Exp. Biol. Med. 2017, 242, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Otsuka, M.; Ohno, M.; Kishikawa, T.; Yoshikawa, T.; Koike, K. Mutual antagonism between hepatitis B viral mRNA and host microRNA let-7. Sci. Rep. 2016, 6, 23237. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lowey, B.; Sodroski, C.; Krishnamurthy, S.; Alao, H.; Cha, H.; Chiu, S.; El-Diwany, R.; Ghany, M.G.; Liang, T.J. Cellular microRNA networks regulate host dependency of hepatitis C virus infection. Nat. Commun. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Cheng, J.-C.; Yeh, Y.-J.; Tseng, C.-P.; Hsu, S.-D.; Chang, Y.-L.; Sakamoto, N.; Huang, H.-D. Let-7b is a novel regulator of hepatitis C virus replication. Cell. Mol. Life Sci. 2012, 69, 2621–2633. [Google Scholar] [CrossRef]
- Yeh, Y.-J.; Tseng, C.-P.; Hsu, S.-D.; Huang, H.-Y.; Lai, M.M.C.; Huang, H.-D.; Cheng, J.-C. Dual Effects of Let-7b in the Early Stage of Hepatitis C Virus Infection. J. Virol. 2021, 95, e01800–e01820. [Google Scholar] [CrossRef]
- Cheng, M.; Si, Y.; Niu, Y.; Liu, X.; Li, X.; Zhao, J.; Jin, Q.; Yang, W. High-Throughput Profiling of Alpha Interferon- and Interleukin-28B-Regulated MicroRNAs and Identification of let-7s with Anti-Hepatitis C Virus Activity by Targeting IGF2BP. J. Virol. 2013, 87, 9707–9718. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Aizawa, N.; Enomoto, H.; Nishiguchi, S.; Toyoda, H.; Kumada, T.; Iio, E.; Ito, K.; Ogawa, S.; Isogawa, M.; et al. Circulating let-7 Levels in Serum Correlate with the Severity of Hepatic Fibrosis in Chronic Hepatitis C. Open Forum Infect. Dis. 2018, 5, ofy268. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; De Giorgi, V.; Schechterly, C.; Wang, R.Y.; Farci, P.; Tanaka, Y.; Alter, H.J. Circulating let-7 levels in plasma and extracellular vesicles correlate with hepatic fibrosis progression in chronic hepatitis C. Hepatology 2016, 64, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Cueto, M.; Medina-Martínez, I.; del Angel, R.; Berumen, J.; Gutiérrez-Escolano, A.L.; Yocupicio-Monroy, M. Let-7c overexpression inhibits dengue virus replication in human hepatoma Huh-7 cells. Virus Res. 2015, 196, 105–112. [Google Scholar] [CrossRef]
- Zhang, S.; Gu, D.; Ouyang, X.; Xie, W. Proinflammatory effects of the hemagglutinin protein of the avian influenza A (H7N9) virus and microRNA-mediated homeostasis response in THP-1 cells. Mol. Med. Rep. 2015, 12, 6241–6246. [Google Scholar] [CrossRef]
- Zhu, Z.; Qi, Y.; Ge, A.; Zhu, Y.; Xu, K.; Ji, H.; Shi, Z.; Cui, L.; Zhou, M. Comprehensive Characterization of Serum MicroRNA Profile in Response to the Emerging Avian Influenza A (H7N9) Virus Infection in Humans. Viruses 2014, 6, 1525–1539. [Google Scholar] [CrossRef]
- Ma, Y.-J.; Yang, J.; Fan, X.-L.; Zhao, H.-B.; Hu, W.; Li, Z.-P.; Yu, G.; Ding, X.-R.; Wang, J.-Z.; Bo, X.-C.; et al. Cellular microRNA let-7c inhibits M1 protein expression of the H1N1 influenza A virus in infected human lung epithelial cells. J. Cell. Mol. Med. 2012, 16, 2539–2546. [Google Scholar] [CrossRef]
- Bakre, A.; Mitchell, P.; Coleman, J.K.; Jones, L.P.; Saavedra, G.; Teng, M.; Tompkins, S.; Tripp, R.A. Respiratory syncytial virus modifies microRNAs regulating host genes that affect virus replication. J. Gen. Virol. 2012, 93, 2346–2356. [Google Scholar] [CrossRef]
- Thornburg, N.; Hayward, S.L.; Crowe, J.E. Respiratory Syncytial Virus Regulates Human MicroRNAs by Using Mechanisms Involving Beta Interferon and NF-κB. mBio 2012, 3, e00220–e00232. [Google Scholar] [CrossRef]
- Bakre, A.A.; Harcourt, J.L.; Haynes, L.M.; Anderson, L.J.; Tripp, R.A. The Central Conserved Region (CCR) of Respiratory Syncytial Virus (RSV) G Protein Modulates Host miRNA Expression and Alters the Cellular Response to Infection. Vaccines 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Han, J.; Chen, S.; Xie, R.; Yang, J.; Zhou, T.; Zhang, Q.; Xia, R. MicroLet-7b Regulates Neutrophil Function and Dampens Neutrophilic Inflammation by Suppressing the Canonical TLR4/NF-κB Pathway. Front. Immunol. 2021, 12, 653344. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, Y.; Luo, D.; Zhuang, Z.; Jin, H.; Zhou, H.; Li, X.; Lin, H.; Zheng, X.; Zhang, J.; et al. Therapeutic potential of C1632 by inhibition of SARS-CoV-2 replication and viral-induced inflammation through upregulating let-7. Signal Transduct. Target. Ther. 2021, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Suzuki, K.; Seddiki, N.; Kaplan, W.; Cowley, M.J.; Hood, C.L.; Clancy, J.L.; Murray, D.D.; Méndez, C.; Gelgor, L.; et al. Differential Regulation of the Let-7 Family of MicroRNAs in CD4+ T Cells Alters IL-10 Expression. J. Immunol. 2012, 188, 6238–6246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, Y.; Zhang, S.; Luo, H.; Zhang, H. HIV-1 Infection-Induced Suppression of the Let-7i/IL-2 Axis Contributes to CD4+ T Cell Death. Sci. Rep. 2016, 6, 25341. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Akbar, I.; Kumari, B.; Vrati, S.; Basu, A.; Banerjee, A. Japanese Encephalitis Virus-induced let-7a/b interacted with the NOTCH—TLR 7 pathway in microglia and facilitated neuronal death via caspase activation. J. Neurochem. 2019, 149, 518–534. [Google Scholar] [CrossRef]
- Bin, S.; Xin, L.; Lin, Z.; Jinhua, Z.; Rui, G.; Xiang, Z. Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced cartilage cell ferroptosis. Exp. Mol. Pathol. 2021, 118, 104570. [Google Scholar] [CrossRef]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. Serum MiRNA panel as potential biomarkers for chronic hepatitis B with persistently normal alanine aminotransferase. Clin. Chim. Acta 2015, 451, 232–239. [Google Scholar] [CrossRef]
- Horii, R.; Honda, M.; Shirasaki, T.; Shimakami, T.; Shimizu, R.; Yamanaka, S.; Murai, K.; Kawaguchi, K.; Arai, K.; Yamashita, T.; et al. MicroRNA-10a Impairs Liver Metabolism in Hepatitis C Virus-Related Cirrhosis Through Deregulation of the Circadian Clock Gene Brain and Muscle Aryl Hydrocarbon Receptor Nuclear Translocator-Like 1. Hepatol. Commun. 2019, 3, 1687–1703. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Lin, L.; Wu, S.; Guo, Z.; Wang, T.; Qin, Y.; Wang, R.; Zhong, X.; Wu, X.; Wang, Y.; et al. MiR-10a* up-regulates coxsackievirus B3 biosynthesis by targeting the 3D-coding sequence. Nucleic Acids Res. 2013, 41, 3760–3771. [Google Scholar] [CrossRef] [PubMed]
- Le Pendu, J.; Abrantes, J.; Bertagnoli, S.; Guitton, J.-S.; Le Gall-Reculé, G.; Lopes, A.M.; Marchandeau, S.; Alda, F.; Almeida, T.; Célio, A.P.; et al. Proposal for a unified classification system and nomenclature of lagoviruses. J. Gen. Virol. 2017, 98, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, J.; Van Der Loo, W.; Le Pendu, J.; Esteves, P.J. Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): A review. Vet. Res. 2012, 43, 12. [Google Scholar] [CrossRef] [PubMed]
- Hukowska-Szematowicz, B.; Maciejak-Jastrzębska, A.; Blatkiewicz, M.; Maciak, K.; Góra, M.; Janiszewska, J.; Burzyńska, B. Changes in MicroRNA Expression during Rabbit Hemorrhagic Disease Virus (RHDV) Infection. Viruses 2020, 12, 965. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brahmakshatriya, V.; Lupiani, B.; Reddy, S.M.; Soibam, B.; Benham, A.L.; Gunaratne, P.; Liu, H.-C.; Trakooljul, N.; Ing, N.; et al. Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers. BMC Genom. 2012, 13, 278. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsdna-viruses/w/herpesviridae/1611/genus-mardivirus (accessed on 19 July 2022).
- Burnside, J.; Morgan, R. Emerging roles of chicken and viral microRNAs in avian disease. BMC Proc. 2011, 5, S2. [Google Scholar] [CrossRef]
- Burnside, J.; Morgan, R.W. Genomics and Marek’s disease virus. Cytogenet. Genome Res. 2007, 117, 376–387. [Google Scholar] [CrossRef]
- Yao, Y.; Zhao, Y.; Smith, L.P.; Lawrie, C.; Saunders, N.; Watson, M.; Nair, V. Differential expression of microRNAs in Marek’s disease virus-transformed T-lymphoma cell lines. J. Gen. Virol. 2009, 90, 1551–1559. [Google Scholar] [CrossRef]
- Skovgaard, K.; Cirera, S.; Vasby, D.; Podolska, A.; Breum, S.; Dürrwald, R.; Schlegel, M.; Heegaard, P.M. Expression of innate immune genes, proteins and microRNAs in lung tissue of pigs infected experimentally with influenza virus (H1N2). Innate Immun. 2013, 19, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Song, Y.; He, L.; Wan, X.; Lai, L.; Dai, F.; Liu, Y.; Wang, Q. MicroRNA-223 Promotes Type I Interferon Production in Antiviral Innate Immunity by Targeting Forkhead Box Protein O3 (FOXO3). J. Biol. Chem. 2016, 291, 14706–14716. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brahmakshatriya, V.; Zhu, H.; Lupiani, B.; Reddy, S.M.; Yoon, B.-J.; Gunaratne, P.H.; Kim, J.H.; Chen, R.; Wang, J.; et al. Identification of differentially expressed miRNAs in chicken lung and trachea with avian influenza virus infection by a deep sequencing approach. BMC Genom. 2009, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xue, M.; Wang, C.; Wang, J.; Chen, P. Bursopentine as a Novel Immunoadjuvant Enhances both Humoral and Cell-Mediated Immune Responses to Inactivated H9N2 Avian Influenza Virus in Chickens. Clin. Vaccine Immunol. 2011, 18, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A Morbillivirus that Caused Fatal Fisease in Horses and Humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Marsh, G.A.; Jenkins, K.A.; Gantier, M.P.; Tizard, M.L.; Middleton, D.; Lowenthal, J.W.; Haining, J.; Izzard, L.; Gough, T.J.; et al. Promotion of Hendra Virus Replication by MicroRNA 146a. J. Virol. 2013, 87, 3782–3791. [Google Scholar] [CrossRef]
- Cowled, C.; Foo, C.-H.; Deffrasnes, C.; Rootes, C.L.; Williams, D.T.; Middleton, D.; Wang, L.-F.; Bean, A.G.D.; Stewart, C.R. Circulating microRNA profiles of Hendra virus infection in horses. Sci. Rep. 2017, 7, 7431. [Google Scholar] [CrossRef]
- Witter, R.L.S.; Schat, K. Diseases of Poultry. In Marek’s Disease; Agricultural Research Service US Department of Agriculture: Urbana, IL, USA, 2003; pp. 407–465. [Google Scholar]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/picornaviridae/707/genus-aphthovirus (accessed on 19 July 2022).
- Mason, P.W.; Grubman, M.J.; Baxt, B. Molecular basis of pathogenesis of FMDV. Virus Res. 2003, 91, 9–32. [Google Scholar] [CrossRef]
- Mason, P.W.; Chinsangaram, J.; Moraes, M.P.; Mayr, G.A.; Grubman, M.J. Engineering better vaccines for foot-and-mouth disease. Dev. Biol. 2003, 114, 79–88. [Google Scholar]
- Stenfeldt, C.; Arzt, J.; Smoliga, G.; LaRocco, M.; Gutkoska, J.; Lawrence, P. Proof-of-concept study: Profile of circulating microRNAs in Bovine serum harvested during acute and persistent FMDV infection. Virol. J. 2017, 14, 71. [Google Scholar] [CrossRef]
- Quinn, S.R.; O’Neill, L. A trio of microRNAs that control Toll-like receptor signalling. Int. Immunol. 2011, 23, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/arteriviridae/1590/genus-betaarterivirus (accessed on 19 July 2022).
- Chand, R.J.; Trible, B.R.; Rowland, R.R. Pathogenesis of porcine reproductive and respiratory syndrome virus. Curr. Opin. Virol. 2012, 2, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.A.; Yoo, D.; Liu, H.-C. Characterization of the microRNAome in Porcine Reproductive and Respiratory Syndrome Virus Infected Macrophages. PLoS ONE 2013, 8, e82054. [Google Scholar] [CrossRef]
- Ye, D.; Shen, Z.; Zhou, S. Function of microRNA-145 and mechanisms underlying its role in malignant tumor diagnosis and treatment. Cancer Manag. Res. 2019, 11, 969–979. [Google Scholar] [CrossRef]
- He, T.; Feng, G.; Chen, H.; Wang, L.; Wang, Y. Identification of host encoded microRNAs interacting with novel swine-origin influenza A (H1N1) virus and swine influenza virus. Bioinformation 2009, 4, 112–118. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/negative-sense-rna-viruses/w/rhabdoviridae/795/genus-lyssavirus (accessed on 19 July 2022).
- Zhao, P.; Zhao, L.; Zhang, K.; Feng, H.; Wang, H.; Wang, T.; Xu, T.; Feng, N.; Wang, C.; Gao, Y.; et al. Infection with street strain rabies virus induces modulation of the microRNA profile of the mouse brain. Virol. J. 2012, 9, 159. [Google Scholar] [CrossRef]
- Booth, V.; Keizer, D.; Kamphuis, M.B.; Clark-Lewis, A.I.; Sykes, B.D. The CXCR3 Binding Chemokine IP-10/CXCL10: Structure and Receptor Interactions. Biochemistry 2002, 41, 10418–10425. [Google Scholar] [CrossRef]
- Shi, N.; Zhang, X.Y.; Dong, C.; Hou, J.L.; Zhang, M.L.; Guan, Z.H.; Li, Z.Y.; Duan, M. Alterations in microRNA expression profile in rabies virus-infected mouse neurons. Acta Virol. 2014, 58, 120–127. [Google Scholar] [CrossRef]
- Liu, T.; Xu, Z.; Ou, D.; Liu, J.; Zhang, J. The miR-15a/16 gene cluster in human cancer: A systematic review. J. Cell. Physiol. 2019, 234, 5496–5506. [Google Scholar] [CrossRef]
- Indrieri, A.; Carrella, S.; Carotenuto, P.; Banfi, S.; Franco, B. The Pervasive Role of the miR-181 Family in Development, Neurodegeneration, and Cancer. Int. J. Mol. Sci. 2020, 21, 2092. [Google Scholar] [CrossRef]
- Peng, X.; Gao, Q.S.; Zhou, L.; Chen, Z.H.; Lu, S.; Huang, H.J.; Zhan, C.Y.; Xiang, M. microRNAs in avian influenza virus H9N2-infected and non-infected chicken embryo fibroblasts. Genet. Mol. Res. 2015, 14, 9081–9091. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-K.; Zhang, Q.; Gao, L.; Li, N.; Chen, X.-X.; Feng, W.-H. Increasing Expression of MicroRNA 181 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication and Has Implications for Controlling Virus Infection. J. Virol. 2013, 87, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Luo, J.; Zhang, H.; Chang, S.; Song, J. MiRNA expression signatures induced by Marek’s disease virus infection in chickens. Genomics 2012, 99, 152–159. [Google Scholar] [CrossRef]
- Payne, L.N.; Nair, V. The long view: 40 years of avian leukosis research. Avian Pathol. 2012, 41, 11–19. [Google Scholar] [CrossRef]
- Ji, J.; Shang, H.; Zhang, H.; Li, H.; Ma, J.; Bi, Y.; Xie, Q. Temporal changes of microRNA gga-let-7b and gga-let-7i expression in chickens challenged with subgroup J avian leukosis virus. Vet. Res. Commun. 2017, 41, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ji, J.; Xie, Q.; Shang, H.; Zhang, H.; Xin, X.; Chen, F.; Sun, B.; Xue, C.; Ma, J.; et al. Aberrant expression of liver microRNA in chickens infected with subgroup avian leukosis virus. Virus Res. 2012, 169, 268–271. [Google Scholar] [CrossRef] [PubMed]




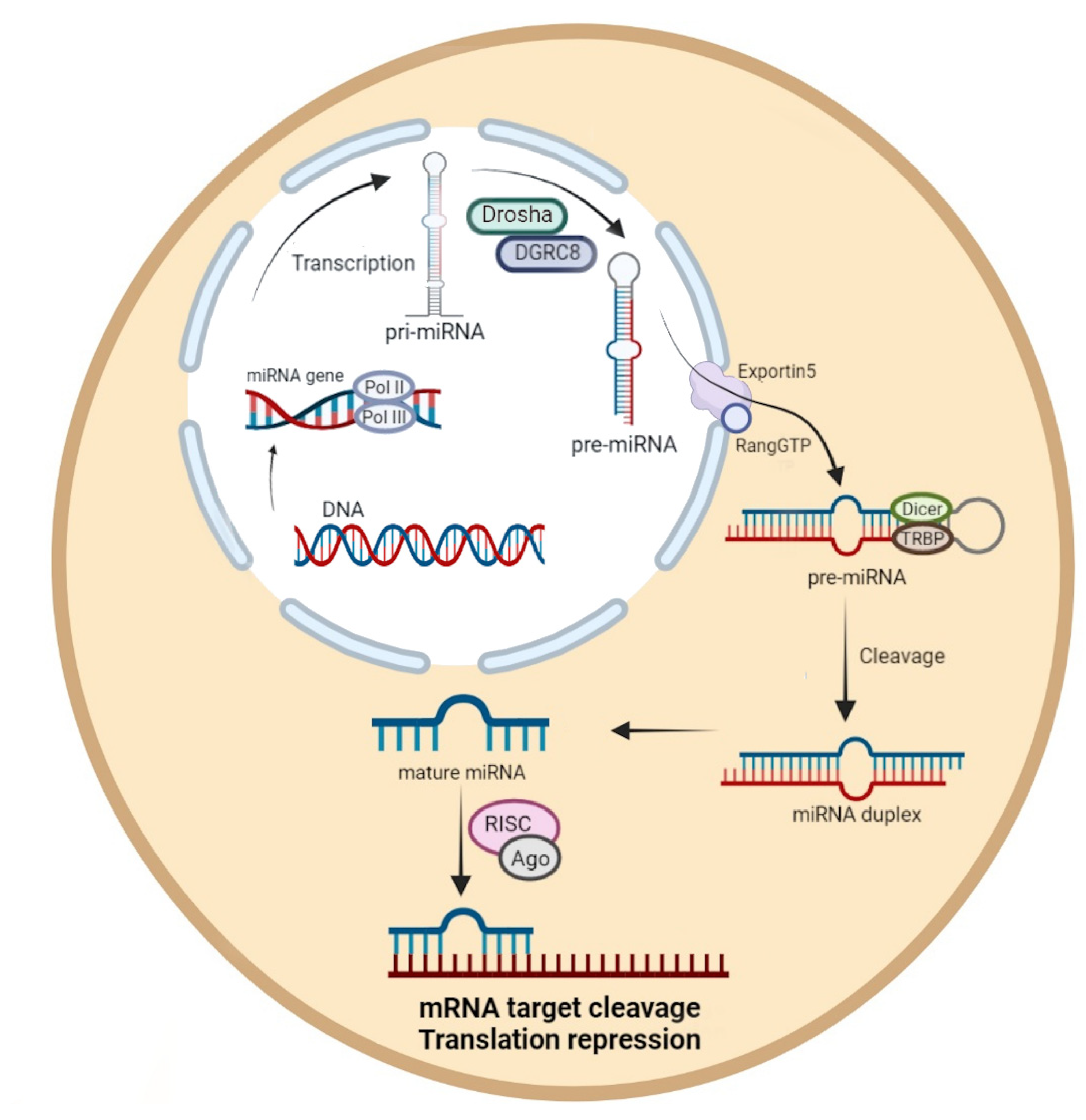{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Virus/Disease | Target Genes | Biological Function/Modified Pathways | Reference |
|---|---|---|---|---|
| miR-155 | HBV/HB | SOCS1 | Enhances the phosphorylation of STAT1 and STAT3; enhances JAK/STAT signaling pathway; suppresses HBV infection in hepatocytes; regulates IFN-γ production; inhibits Akt/mTOR pathway | [23,26,29] |
| C/EBP-β | Modulates HBV replication | [24] | ||
| BCL-6, SHIP-1, SOCS-1 | Induces inflammatory cytokine production | [28] | ||
| HCV/HC | TNF-α | Pro-inflammatory response | [33] | |
| Tim3/T-bet | Increases IFN-γ production; regulates Tim-3/T-bet/STAT-5 signaling and cytokine expression in NK cells; immune injury during chronic viral disease | [36] | ||
| ISG15, TLR3 | Innate and adaptive immune response | [34] | ||
| APC | Wnt pathway; apoptosis; cell proliferation | [39] | ||
| DENV/dengue | Bach1 | Replication of DEVN; enhances anti-viral IFN responses | [43] | |
| SOCS1 | Regulates cytokine signal transduction | [41] | ||
| WNV/ West Nile fever | IL-13, BDNF, CCR9 | Cell survival | [45] | |
| IL-1β, IL-12, IL-6, IL-15, GM-CSF | Anti-viral response | [47] | ||
| IAV/influenza | S1PR1 | Inflammatory response; activates S1PR1/NF-κB/pro-inflammatory cytokine pathway | [51] | |
| IFN type I | Anti-viral response | [53] | ||
| RSV/upper respiratory tract infections | IFN type I | Anti-viral response | [53] | |
| TNF-α, IL-1β, IL-6, IL-8 | Pro-inflammatory response | [59] | ||
| SHIP1, Kif1 | Antigen presentation | [61,62] | ||
| SOCS1 | Enhances activation of STAT1 and up-regulation of ISGs gene | [63] | ||
| SARS-CoV-2/ COVID-19 | STAT1, STAT3, TGFB1, SMAD3, IRF1, AKT1, MYB, BCL6, TP6, HIF1A, FOXP3, JUNB, NFKB1 | Immune response; apoptosis | [71] | |
| SOCS1, IL-6, IL-1β, CSF1R CD274, TLF6, TNF | Regulates the host immune response; miR-155-5p–IL-6/TNF/IL-1β axis | [75] | ||
| IL-1α, G-CSF, IL-9, MIP-2, IL-12-P70 VEGF, IP-10, IFN-γ, MCP-1, MIG, MIP-1α, M−CSF, TNF-α, MIP-1β | Alleviates inflammation and lung cytokine storm | [73] | ||
| HAdV/respiratory infections (colds) | IFN-β | Anti-viral response | [80,81] | |
| SOCS1 | Enhances type I IFN Anti-viral response | [82] | ||
| SHIP1 | Enhances IFN type I signaling | [20] | ||
| HRV/COPD | HRV1B | Inhibits viral replication | [84] | |
| miR-223 | HCV/CHC | NF-κB | Chronic liver inflammation | [94] |
| DENV/dengue | STMN1 | DENV replication | [95] | |
| IAV/influenza | TNF-α | Pro-inflammatory response | [97,101] | |
| PI3K, IGF1R, GPCR, PP2A, PKA and Ca2+ channel | Represses the activity of CREB; T-cell development and cell survival | [96] | ||
| IL1RN, MDA5, STAT1 | Cell death; apoptosis | [100] | ||
| SARS-CoV/SARS | NF-κB | Activated CCR1, the inflammatory chemokine receptor for CCL3 and CCL5 enhances lung fibrosis | [105] | |
| SARS-CoV-2/ COVID-19 | TRAF6, FOXO1, TLR4, STMN1, PI3K/AKT, CXCL2, CCL3, IL-6, IFN-I, IL-1β, Caspase-1 and mainly NLRP3, IKKα, NF-κB | Regulates inflammatory processes; anti-oxidant and anti-viral role | [108] | |
| HIV-1/AIDS | RhoB Sp3, LIF | Inhibits HIV-1 production in resting primary CD4+ T-cells Activates the AKT–NF-κB pathway Viral replication | [110] [112] [112] | |
| miR-146a | HBV/CHB | NF-κB | Production of pro-inflammatory cytokines (TNF-α, IL-6, IL-8, IL-12, and IL-18) | [118] |
| STAT1 | Viral persistence Decreases cytotoxicity of T-cells | [117] | ||
| CFH | Regulation of the complement alternative pathway | [121] | ||
| XIAP | Regulation of HBV replication; regulation of XIAP-MDM2/p53 pathway | [118] | ||
| ZEB2 | Regulation of HBV transcription and replication | [119] | ||
| TRAF6, IRAK1 | Regulation of FEN1 by NF-κB activity; promotes HBV DNA replication | [120,124] | ||
| HCV/HC | SOCS1 | STAT3 inhibition; increases IL-23, IL-10, and TGF-β expression | [125] | |
| DENV/dengue | TRAF6 LC3 | Inhibits IFN-β and IL-28A/B; increases DENV2 replication Autophagy; viral replication | [129] [130] | |
| IAV/influenza | TRAF6 | Production of type I IFN; anti-viral response | [133] | |
| IRAK1 | IL-7, VEGF and, JAK-STAT signaling pathway | [134] | ||
| RSV/upper respiratory tract infections | TRAF6, IRAK1 | Viral replication | [137,138] | |
| SARS-CoV-2/ COVID-19 | TRAF6, IRAK1, IRAK2 | NF-κB pro-inflammatory signaling pathway | [139] | |
| STAT1 | Regulates JAK/STAT pathway | [139] | ||
| miR-122 | HBV/HB | cyclin G1 | Replication of HBV | [149] |
| HO-1 | Replication of HBV; oxidative stress | [153,154,155] | ||
| IFN type 1 | Replication of HBV; anti-viral response | [156] | ||
| SOCS3 | JAK/STAT pathway signaling; cytokine signaling | [157] | ||
| HCV/HC | viral 5′-NCR | Replication of HCV | [166] | |
| Bach1 | HO-1 gene regulation; HCV replication | [170] | ||
| IFN | Anti-viral response | [171] | ||
| TGFBRAP1 | Promotes HCC progression induced by HCV | [172] | ||
| DENV/dengue | CYP7A1, IGFR1, SFR, RAC1, RHOA, cyclin G1 | Immune response | [131] | |
| ZEBOV/RESTV/ Ebola | viral vp40 gene | Regulation of virus replication | [176,177] | |
| RSV/upper respiratory tract infections | Wnt | Inflammatory and immune response | [178] | |
| IL1R1 | NF-κB activation, inflammatory response | [179,180] | ||
| TLR4 | Innate immune response: NF-κB activation; cytokine secretion; inflammatory response | [180] | ||
| iNOS | Anti-viral response | [184] | ||
| HRV/COPD | CXCL2 | Chemotaxis | [185] | |
| SOCS1 | Regulates cytokine signal transduction | [185] | ||
| miR-125b | HBV/HB | LIN28B | Regulates let-7 and stimulates HBV replication | [194] |
| SCNN1A | Inhibits HBV core protein expression | [197] | ||
| HCV/HC | TLR2 | TLR2/MyD88 signaling pathway; phosphorylation of NF-κBp65, ERK, and P38 | [198] | |
| HuR | Viral replication | [200] | ||
| RSV/upper respiratory tract infections | NF-κB, MAPK | Immune response | [205] | |
| IAV/influenza | MAPK | Cell proliferation; apoptosis | [206] | |
| SARS-CoV-2/ COVID-19 | ACE2 | Activation of immune system | [207] | |
| HIV-1/AIDS | CPSF6 | Regulation of HIV-1 nuclear entry and viral replication | [208] | |
| JEV/Japanese encephalitis | STAT3, Map2k7, Triap1 | Reduces genome replication and virus titers | [212] | |
| miR-132 | HBV/HB | Akt | Development of HCC induced by HBV | [217] |
| IAV/influenza | IRF1 | Regulation of IFN-α and IFN-β production | [219] | |
| MAPK3 | Regulates innate immune signaling pathways | [134] | ||
| HSV/orofacial and genital disease | Ras | Inflammation | [224] | |
| miR-34a | HBV/HB | Wnt1 | Regulation of Wnt/β-catenin pathway; contributes to HCC induced by HBV | [229] |
| HMGB1 | Innate immune response; apoptosis | [227] | ||
| CCL22 | Chemotactic for monocytes, dendritic cells, natural killer cells, and T lymphocytes | [230] | ||
| Smad4 | Regulation of TGF-β/Smad3 pathway activity; liver fibrosis | [228] | ||
| DENV/dengue WNV/West Nile fever | Wnt | Activates type I IFN response; regulation of WNT/β-catenin signaling; anti-viral state | [233] | |
| IAV/influenza | Bax | Apoptosis | [234] | |
| STAT3 | Anti-inflammatory function Inhibits NF-κB gene reporters | [235] | ||
| SARS-CoV-2/ COVID-19 | Bcl-2, Bax, KLF4 IL-6R | Apoptosis Inflammation | [236] | |
| HTLV-1/leukemia | SIRT1 Bax | Apoptosis | [240] | |
| miR-21 | HBV/CHB | PTEN | PTEN/Akt signaling; activation of HSCs; apoptosis | [250,251] |
| Smad7 | Promotes α-SMA and collagen I expression in HSCs; liver fibrosis | [250,251] | ||
| TGF-β1 | Positive feedback loop; liver fibrosis | [249] | ||
| PDCD4 | Apoptosis | [252] | ||
| IL-12 | Regulates proliferation of NK and T-cells | [257] | ||
| HCV/HC | PTEN | Apoptosis | [260] | |
| MyD88 IRAK1 | Production of IFN type I; anti-viral response | [261] | ||
| Smad7 | Fibrosis | [262] | ||
| IAV/influenza | CCL1, CCL17, CCL19, IL-22, C2orf28 | Inflammation; apoptosis | [268] | |
| TGF-β | Fibrosis | [271] | ||
| Ad/respiratory infection | p53 TGF-β | Apoptosis | [80] | |
| SARS-CoV-2/ COVID-19 | ORF1ab ORF3a S protein gene | Regulation of viral entry | [273] | |
| CCL20 | Chemotactic response TGF-β and Akt signaling | [139] | ||
| IRAK1 | Participates in NF-κB pro-inflammatory signaling pathway | [139,273] | ||
| CXCL-10 | Pro-inflammatory response; chemotaxis | [273] | ||
| HIV-1/AIDS | PTEN PDCD4 CDKN1B | Apoptosis | [279] | |
| IP-10 | Inflammation | [280] | ||
| CVB3 | YOD1 | Metabolism of proteins; protein ubiquitination | [284] | |
| CHPV | PTEN | Regulation of phosphorylation of AKT and subunit p65 of NF-ĸB; production of pro-inflammatory cytokines (IL-6, TNF-α) | [286] | |
| miR-16 | HBV/HB | CCND1 | Cell proliferation | [290] |
| c-Myc | Cellular metabolism and proliferation | [290] | ||
| Bcl-2 | Apoptosis | [290] | ||
| HCV/HC | HGF | Hepatocyte proliferation | [291] | |
| Smad7 | Fibrosis | [291] | ||
| Human CoV (MERS, SARS, SARS-CoV-2) | polyprotein 1ab coding region | Regulation of viral replication | [272] | |
| SARS-CoV-2/ COVID-19 | MK2 CCND1 | Cell proliferation | [273] | |
| RSV/upper respiratory tract infections | TLR4 RIG-1 | Activation of NF-κB; pro-inflammatory | [58] | |
| miR-181a | HBV/HB | PTEN | Cell proliferation; apoptosis; development of hepatocellular carcinoma | [300] |
| CLDN1 | Blocks HCV entry into cells | [306] | ||
| E2F5 | Development of HCC induced by HBV | [301] | ||
| Fas | Apoptosis and regulation of HCC cell proliferation | [302] | ||
| HCV/CHC | DUSP6 | Regulation of T-cell response | [305] | |
| miR-181b | HBV/HB | p27 PTEN | Regulation of hepatic stellate cell proliferation; pro-fibrotic role | [298,299] |
| miR-181c | HCV/HC | HOXA1 | Cell growth regulator; activates STAT3 and STAT5 | [306] |
| ATM | Apoptosis | [307] | ||
| CDK-2 cyclin-A | Cell cycle | [307] | ||
| IAV/influenza | Bcl-2 | Apoptosis | [206] | |
| IL-2, TNF-α | Immune response | [206] | ||
| let-7a | HBV/HB | STAT3 | Immune suppression | [315,317] |
| HCV/HC | CHUK, IKBKE, XPNPEP1 | Regulation of HCV assembly or secretion | [320] | |
| JEV/Japanese encephalitis | cPARP | Apoptosis | [337] | |
| let-7b | HCV/HC | IFN type I SOCS1 IKKα | Immune response; anti-viral response; stimulation of JAK/STAT signaling pathway | [322] |
| GF2BP1 | Regulation of HCV replication | [323] | ||
| SARS-CoV-2/ COVID-19 | RELB, IL-6, KHSRP, EHMT2, KDM2B, CMYC, LIN28B | Immune response; repair function | [71] | |
| TLR4 | Regulation of inflammation via NF-κB | [333] | ||
| IL-6, IL-8, TNF-α, IL-10 | Regulation of pro- and anti-inflammatory cytokine production | [333] | ||
| ORF1ab (virus gene) | Regulation of viral replication | [238] | ||
| HIV-1/AIDS | IL-10 | Anti-viral response | [335] | |
| JEV/Japanese encephalitis | cPARP | Apoptosis | [337] | |
| let-7c | DENV/dengue | Bach1 | Regulation of HO-1; regulation of viral replication; oxidative stress; anti-inflammatory response | [326] |
| let-7f | RSV/upper respiratory tract infections | CCL7 SOCS3 | Anti-viral cytokine response | [330] |
| let-7i | HIV-1/AIDS | IL-2 | Th-cell death | [336] |
| miR-10a | HCV/HC | RORA | Immune response | [340] |
| SREBP1 FANS SREBP2 PGC1α | Lipid homeostasis; cholesterol biosynthesis; fatty acid synthesis; regulation of fatty acid metabolism | [340] | ||
| Bmal1 | Liver damage | [340] | ||
| RSV/upper respiratory tract infections | NF-κB MAPK | Immune response | [341,342,343] |
| MicroRNA | Virus/Disease | Target Genes | Biological Function/Modified Pathways | Reference |
|---|---|---|---|---|
| miR-155 | Lagovirus europaeus/RHDV/ RHD | JUN, IGFR1, KRAS, TNF-α, TGF-β, IL-1β, IL-6, IFN | Immune response | [347] |
| AIV/influenza | MX1 | Anti-viral response | [348] | |
| JNK pathway | Apoptosis | [348] | ||
| miR-223 | VSV/vesicular stomatitis | FOXO3 | Regulation of IFN type I production | [354] |
| miR-146a | H1N2/influenza | IRAK1 STAT2 TLR2 | Innate immune response; regulation of IFN type I production | [353] |
| HeV | RIG-I | Innate immune response | [359] | |
| RFN11 | Regulation of viral replication | [359] | ||
| VSV/vesicular stomatitis | IRAK1, IRAK2 | Regulation of IFN type I production | [116] | |
| FMDV/FMD | TRAF6 IRAK1 | Regulation of NF-κB activation; innate immune response | [365] | |
| PRRSV/PRRS | C1QTNF3 MAFB | Regulation of immune response | [369] | |
| miR-145 | AIV/influenza | HA | Regulation of pathogenicity of influenza virus and immunosuppression | [371] |
| RABV/rabies | MYC, STAM, STAT4, SOCS7 | JAK/STAT signaling pathway | [373] | |
| IL17RB TGFBR2 | Receptor for the pro-inflammatory cytokines IL17B and IL17E, controlling the growth and/or differentiation of hematopoietic cells Regulation of cell cycle arrest in epithelial and hematopoietic cells; control of mesenchymal cell proliferation and differentiation; wound healing; extracellular matrix production; immunosuppression; and carcinogenesis | [373] | ||
| MYC, SMAD3, INHBB, TGFBR2, SMAD5 | TGF-beta signaling pathway | [373] | ||
| ARF6, CFL2, LAT, ARPC5, CRKL | Fc gamma R-mediated phagocytosis | [373] | ||
| miR-21 | H1N2/influenza | CXXL10 | Chemoattractant for monocytes/macrophages, T-cells, NK cells, and dendritic cells; may also promote T-cell adhesion to endothelial cells | [353,355] |
| miR-16 miR-15a | Lagovirus europaeus/RHDV/RHD | HGF Bcl-2 | Cell proliferation during the liver regeneration process Apoptosis | [347] |
| AIV/influenza | CXCL10 | Chemoattractant for monocytes/macrophages, T-cells, NK cells, and dendritic cells; may also promote T-cell adhesion to endothelial cells | [353] | |
| TLR7 | Activation of innate immunity | [353] | ||
| miR-181a | PRRSV/PRRS | ORF7 (virus gene) | Inhibits viral gene expression and virus protein production | [379] |
| miR-181b | FMDV/FMD | RASSF1A NF-κB | Regulates cellular proliferation and inflammatory response | [365] |
| AC9 | Immunomodulatory effect | [365] | ||
| IFNα | Anti-viral response | [365] | ||
| miR-181c | PRRSV/PRRS | ORF7 (virus gene) | Inhibits viral gene expression and virus protein production | [379] |
| let-7g | FMDV/FMD | LOX | Cell proliferation | [365] |
| caspase-3 | Apoptosis | [365] | ||
| let-7i | MDV/MD | ATF2 | Proliferation, apoptosis | [380] |
| miR-122 | Lagovirus europaeus/RHDV/RHD | HFG, c-Met STAT1, STAT3 | Hepatic homeostasis Apoptosis | [347] |
| AIV/influenza | MX1, IL-8, IRF-7, TNFRS19 | Immune response | [348] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostrycharz, E.; Hukowska-Szematowicz, B. Micro-Players of Great Significance—Host microRNA Signature in Viral Infections in Humans and Animals. Int. J. Mol. Sci. 2022, 23, 10536. https://doi.org/10.3390/ijms231810536
Ostrycharz E, Hukowska-Szematowicz B. Micro-Players of Great Significance—Host microRNA Signature in Viral Infections in Humans and Animals. International Journal of Molecular Sciences. 2022; 23(18):10536. https://doi.org/10.3390/ijms231810536
Chicago/Turabian StyleOstrycharz, Ewa, and Beata Hukowska-Szematowicz. 2022. "Micro-Players of Great Significance—Host microRNA Signature in Viral Infections in Humans and Animals" International Journal of Molecular Sciences 23, no. 18: 10536. https://doi.org/10.3390/ijms231810536