
Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions


Abstract
:1. Introduction
1.1. Peptides as Anticancer Agents
1.2. Focus of the Review
2. Sam Domains
2.1. The Chameleon Domain
2.2. Structural Properties
2.2.1. Sam Fold
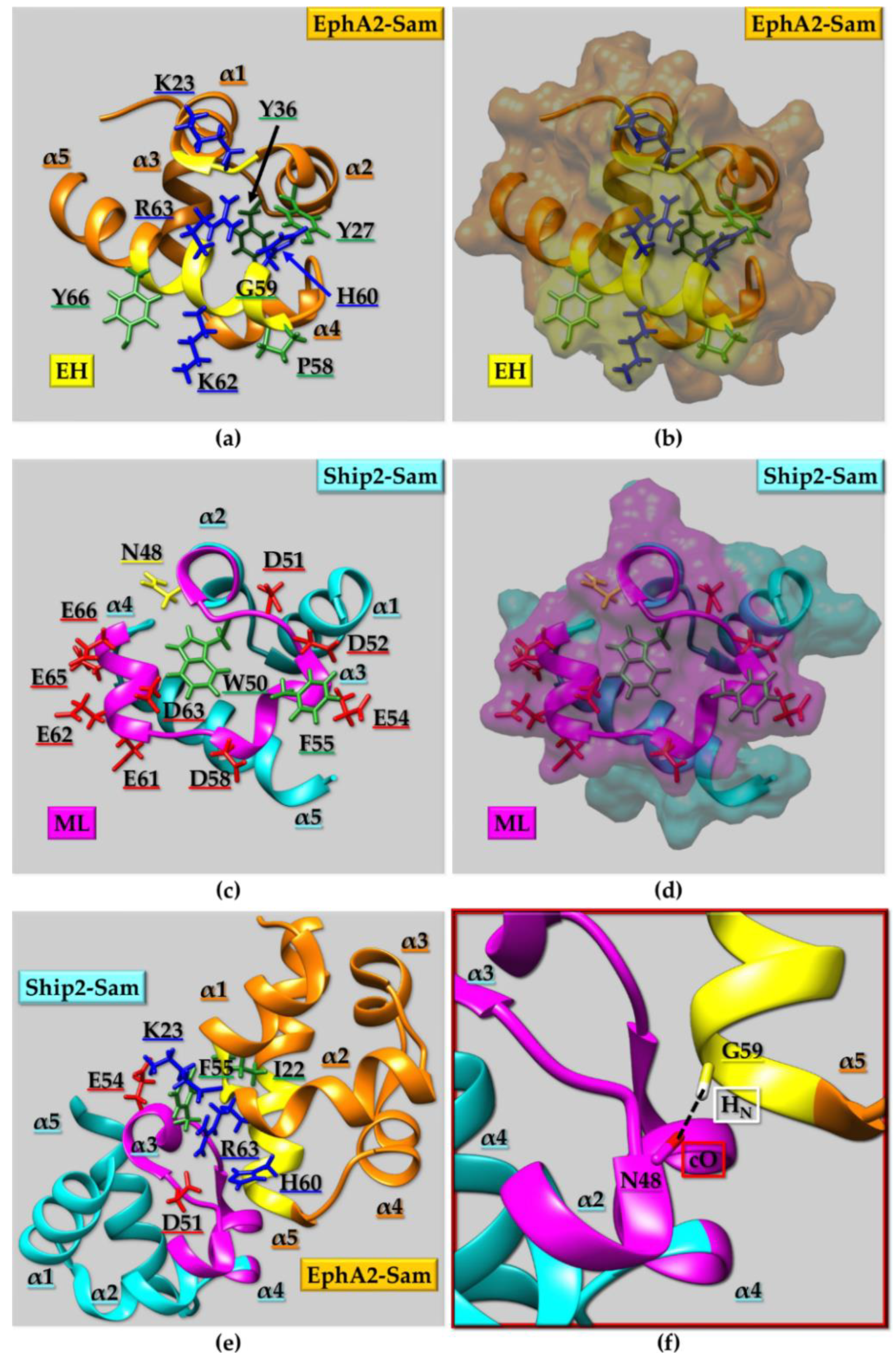
2.2.2. Sam Domains Self-Association and the Mid-Loop/End-Helix (ML/EH) Model
3. The EphA2 Receptor and Its Controversial Role in Cancer
3.1. Possible Therapeutic Approaches
4. EphA2-Sam and Its Interaction Network
4.1. Ship2 (SH2-Containing 5′-Inositol Phosphatase)
4.1.1. Ship2-Sam and Its Complex with EphA2-Sam: Structural and Functional Insights
4.2. The Adaptor Protein Odin
4.2.1. Odin-Sam1 and Its Complex with EphA2-Sam: Structural and Functional Insights
5. Peptides Targeting EphA2-Sam and Its Interactome
5.1. Protein Dissection Approaches
5.1.1. Mid-Loop Peptides
5.1.2. End-Helix Peptides
5.2. KRI3 Analogues
5.3. Helical Peptides
5.3.1. Helical Linear Peptides
5.3.2. Stapled Peptides
5.4. Designing Anticancer Peptides through In Silico Methods
5.4.1. Virtual Screening Strategies to Identify Linear Peptides Targeting EphA2-Sam
5.4.2. Cyclic Peptide Libraries against EphA2-Sam
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 29 July 2022).
- International Agency for Research on Cancer—World Health Organization. Estimated Numbers from 2020 to 2040, Males and Females, Age [0–85+]. Available online: https://gco.iarc.fr/tomorrow/en/dataviz/trends (accessed on 29 July 2022).
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.A.; Simal-Gandara, J.; Eun, J.B.; Shim, J.H.; Abd El-Aty, A.M. Bioactivities, Applications, Safety, and Health Benefits of Bioactive Peptides From Food and By-Products: A Review. Front. Nutr. 2021, 8, 815640. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Babajani, A.; Sarrami Forooshani, R.; Yazdani, M.; Rezaei-Tavirani, M. Clinical Applications and Anticancer Effects of Antimicrobial Peptides: From Bench to Bedside. Front. Oncol. 2022, 12, 819563. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, M.; Zhang, F.; Wang, Y.; Ouyang, J.; Luo, X.; Yang, H.; Zhang, D.; Chen, Y.; Yu, H.; et al. Design of a Sea Snake Antimicrobial Peptide Derivative with Therapeutic Potential against Drug-Resistant Bacterial Infection. ACS Infect. Dis. 2020, 6, 2451–2467. [Google Scholar] [CrossRef]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Lyu, Y.; Chen, T.; Shang, L.; Yang, Y.; Li, Z.; Zhu, J.; Shan, A. Design of Trp-Rich Dodecapeptides with Broad-Spectrum Antimicrobial Potency and Membrane-Disruptive Mechanism. J. Med. Chem. 2019, 62, 6941–6957. [Google Scholar] [CrossRef]
- Scheenstra, M.R.; van den Belt, M.; Tjeerdsma-van Bokhoven, J.L.M.; Schneider, V.A.F.; Ordonez, S.R.; van Dijk, A.; Veldhuizen, E.J.A.; Haagsman, H.P. Cathelicidins PMAP-36, LL-37 and CATH-2 are similar peptides with different modes of action. Sci. Rep. 2019, 9, 4780. [Google Scholar] [CrossRef]
- Bommineni, Y.R.; Pham, G.H.; Sunkara, L.T.; Achanta, M.; Zhang, G. Immune regulatory activities of fowlicidin-1, a cathelicidin host defense peptide. Mol. Immunol. 2014, 59, 55–63. [Google Scholar] [CrossRef]
- Wei, L.; Gao, J.; Zhang, S.; Wu, S.; Xie, Z.; Ling, G.; Kuang, Y.Q.; Yang, Y.; Yu, H.; Wang, Y. Identification and Characterization of the First Cathelicidin from Sea Snakes with Potent Antimicrobial and Anti-inflammatory Activity and Special Mechanism. J. Biol. Chem. 2015, 290, 16633–16652. [Google Scholar] [CrossRef] [PubMed]
- Bellavita, R.; Casciaro, B.; Di Maro, S.; Brancaccio, D.; Carotenuto, A.; Falanga, A.; Cappiello, F.; Buommino, E.; Galdiero, S.; Novellino, E.; et al. First-in-Class Cyclic Temporin L Analogue: Design, Synthesis, and Antimicrobial Assessment. J. Med. Chem. 2021, 64, 11675–11694. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, A.; Avitabile, C.; Cirillo, A.; Moretta, A.; Merlino, A.; Paduano, L.; Duilio, A.; Romanelli, A. The antimicrobial peptide Temporin L impairs E. coli cell division by interacting with FtsZ and the divisome complex. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129606. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Epand, R.F.; Rosenfeld, Y.; Peleg, A.; Barra, D.; Epand, R.M.; Shai, Y. Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J. Biol. Chem. 2008, 283, 22907–22917. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.C.; Mangoni, M.L.; Rufo, A.; Luzi, C.; Barra, D.; Zhao, H.; Kinnunen, P.K.; Bozzi, A.; Di Giulio, A.; Simmaco, M. Temporin L: Antimicrobial, haemolytic and cytotoxic activities, and effects on membrane permeabilization in lipid vesicles. Biochem. J. 2002, 368, 91–100. [Google Scholar] [CrossRef]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef]
- Calderone, R.; Sun, N.; Gay-Andrieu, F.; Groutas, W.; Weerawarna, P.; Prasad, S.; Alex, D.; Li, D. Antifungal drug discovery: The process and outcomes. Future Microbiol. 2014, 9, 791–805. [Google Scholar] [CrossRef]
- Strom, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Sharma, K.K.; Maurya, I.K.; Khan, S.I.; Jacob, M.R.; Kumar, V.; Tikoo, K.; Jain, R. Discovery of a Membrane-Active, Ring-Modified Histidine Containing Ultrashort Amphiphilic Peptide That Exhibits Potent Inhibition of Cryptococcus neoformans. J. Med. Chem. 2017, 60, 6607–6621. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.A.; Edgerton, M.; Levine, M.J. Salivary histatin 5: Dependence of sequence, chain length, and helical conformation for candidacidal activity. J. Biol. Chem. 1990, 265, 3898–3905. [Google Scholar] [CrossRef]
- Lawyer, C.; Pai, S.; Watabe, M.; Borgia, P.; Mashimo, T.; Eagleton, L.; Watabe, K. Antimicrobial activity of a 13 amino acid tryptophan-rich peptide derived from a putative porcine precursor protein of a novel family of antibacterial peptides. FEBS Lett. 1996, 390, 95–98. [Google Scholar] [CrossRef]
- Almaaytah, A.; Qaoud, M.T.; Khalil Mohammed, G.; Abualhaijaa, A.; Knappe, D.; Hoffmann, R.; Al-Balas, Q. Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine. Pharmaceuticals 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Almaaytah, A.; Darwish, R.M. The Design of Alapropoginine, a Novel Conjugated Ultrashort Antimicrobial Peptide with Potent Synergistic Antimicrobial Activity in Combination with Conventional Antibiotics. Antibiotics 2021, 10, 712. [Google Scholar] [CrossRef]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267–2280. [Google Scholar] [CrossRef]
- Shai, Y.; Makovitzky, A.; Avrahami, D. Host defense peptides and lipopeptides: Modes of action and potential candidates for the treatment of bacterial and fungal infections. Curr. Protein. Pept. Sci. 2006, 7, 479–486. [Google Scholar] [CrossRef]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef]
- Peng, J.; Lu, Q.; Liu, X.; Deng, Y.; Shang, T.; Yuan, L.; Zhang, H.; Zeng, Q. Antibacterial effect of synthetic ultra-short lipopeptide on Streptococcus agalactiae and its active on bacterial mastitis in mice. Biochem. Biophys. Res. Commun. 2022, 601, 153–159. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed]
- Philippe, G.J.B.; Craik, D.J.; Henriques, S.T. Converting peptides into drugs targeting intracellular protein-protein interactions. Drug Discov. Today 2021, 26, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.; Jones, S.; Langel, U. Cell-penetrating peptides in protein mimicry and cancer therapeutics. Adv. Drug Deliv. Rev. 2022, 180, 114044. [Google Scholar] [CrossRef] [PubMed]
- Berillo, D.; Yeskendir, A.; Zharkinbekov, Z.; Raziyeva, K.; Saparov, A. Peptide-Based Drug Delivery Systems. Medicina 2021, 57, 1209. [Google Scholar] [CrossRef] [PubMed]
- Thundimadathil, J. Cancer treatment using peptides: Current therapies and future prospects. J. Amino Acids 2012, 2012, 967347. [Google Scholar] [CrossRef]
- Abd-Aziz, N.; Poh, C.L. Development of Peptide-Based Vaccines for Cancer. J. Oncol. 2022, 2022, 9749363. [Google Scholar] [CrossRef]
- Wang, T.; Liu, X.; Ng, Y.Y.; Tarleton, K.; Tran, A.; Tran, T.; Xue, W.Y.; Youssef, P.; Yuan, P.; Zhang, D.; et al. Milk-Derived Proteins and Peptides in Head and Neck Carcinoma Treatment. Biomolecules 2022, 12, 290. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, H.; Fakhri, S.; Aschner, M.; Cheang, W.S. Therapeutic potential of marine peptides in cervical and ovarian cancers. Mol. Cell Biochem. 2022, 477, 605–619. [Google Scholar] [CrossRef]
- Wang, L.; Dong, C.; Li, X.; Han, W.; Su, X. Anticancer potential of bioactive peptides from animal sources (Review). Oncol. Rep. 2017, 38, 637–651. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, N.; Wang, W.; Xu, Z.; Wu, Y.; Luo, X. An electrochemical biosensor for alpha-fetoprotein detection in human serum based on peptides containing isomer D-Amino acids with enhanced stability and antifouling property. Biosens. Bioelectron. 2021, 190, 113466. [Google Scholar] [CrossRef]
- Xie, J.; Yang, C.; Liu, Q.; Li, J.; Liang, R.; Shen, C.; Zhang, Y.; Wang, K.; Liu, L.; Shezad, K.; et al. Encapsulation of Hydrophilic and Hydrophobic Peptides into Hollow Mesoporous Silica Nanoparticles for Enhancement of Antitumor Immune Response. Small 2017, 13, 1701741. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Mahidhara, G.; Roy, K.; Sasidharan, S.; Krishnakumar, S.; Prasad, N.; Sehgal, R.; Kanwar, R.K. Fe-bLf nanoformulation targets survivin to kill colon cancer stem cells and maintains absorption of iron, calcium and zinc. Nanomedicine 2015, 10, 35–55. [Google Scholar] [CrossRef]
- Qiao, Z.Y.; Hou, C.Y.; Zhang, D.; Liu, Y.; Lin, Y.X.; An, H.W.; Li, X.J.; Wang, H. Self-assembly of cytotoxic peptide conjugated poly(beta-amino ester)s for synergistic cancer chemotherapy. J. Mater. Chem. B 2015, 3, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef] [PubMed]
- Ahangarzadeh, S.; Kanafi, M.M.; Hosseinzadeh, S.; Mokhtarzadeh, A.; Barati, M.; Ranjbari, J.; Tayebi, L. Bicyclic peptides: Types, synthesis and applications. Drug Discov. Today 2019, 24, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.M.; Elnagdy, S.; et al. Cyclic Dipeptides: The Biological and Structural Landscape with Special Focus on the Anti-Cancer Proline-Based Scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, Y.; Zang, T. DRACP: A novel method for identification of anticancer peptides. BMC Bioinform. 2020, 21, 559. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.Y.; Tseng, Y.J.; Kao, H.J.; Chen, C.H.; Yang, H.H.; Weng, S.L. Identification of subtypes of anticancer peptides based on sequential features and physicochemical properties. Sci. Rep. 2021, 11, 13594. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef]
- Novel Drug Approvals for 2019. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019 (accessed on 20 August 2022).
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Novel Drug Approvals for 2020. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020 (accessed on 20 August 2022).
- Novel Drug Approvals for 2021. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2021 (accessed on 20 August 2022).
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Tirzepatide: First Approval. Drugs 2022, 82, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Novel Drug Approvals for 2022. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2022 (accessed on 20 August 2022).
- Vincenzi, M.; Mercurio, F.A.; Leone, M. Protein Interaction Domains: Structural Features and Drug Discovery Applications (Part 2). Curr. Med. Chem. 2021, 28, 854–892. [Google Scholar] [CrossRef]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. Sam Domains in Multiple Diseases. Curr. Med. Chem. 2020, 27, 450–476. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Leone, M. The Sam Domain of EphA2 Receptor and its Relevance to Cancer: A Novel Challenge for Drug Discovery? Curr. Med. Chem. 2016, 23, 4718–4734. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Kim, C.A.; Bowie, J.U. SAM domains: Uniform structure, diversity of function. Trends Biochem. Sci. 2003, 28, 625–628. [Google Scholar] [CrossRef]
- Denay, G.; Vachon, G.; Dumas, R.; Zubieta, C.; Parcy, F. Plant SAM-Domain Proteins Start to Reveal Their Roles. Trends Plant Sci. 2017, 22, 718–725. [Google Scholar] [CrossRef]
- Qiao, F.; Bowie, J.U. The many faces of SAM. Sci. STKE 2005, 2005, re7. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Cellitti, J.; Pellecchia, M. NMR studies of a heterotypic Sam-Sam domain association: The interaction between the lipid phosphatase Ship2 and the EphA2 receptor. Biochemistry 2008, 47, 12721–12728. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Cellitti, J.; Pellecchia, M. The Sam domain of the lipid phosphatase Ship2 adopts a common model to interact with Arap3-Sam and EphA2-Sam. BMC Struct. Biol. 2009, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Marasco, D.; Pirone, L.; Pedone, E.M.; Pellecchia, M.; Leone, M. Solution structure of the first Sam domain of Odin and binding studies with the EphA2 receptor. Biochemistry 2012, 51, 2136–2145. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Marasco, D.; Pirone, L.; Scognamiglio, P.L.; Pedone, E.M.; Pellecchia, M.; Leone, M. Heterotypic Sam-Sam association between Odin-Sam1 and Arap3-Sam: Binding affinity and structural insights. ChemBioChem 2013, 14, 100–106. [Google Scholar] [CrossRef]
- Knight, M.J.; Leettola, C.; Gingery, M.; Li, H.; Bowie, J.U. A human sterile alpha motif domain polymerizome. Protein Sci. 2011, 20, 1697–1706. [Google Scholar] [CrossRef]
- Meruelo, A.D.; Bowie, J.U. Identifying polymer-forming SAM domains. Proteins 2009, 74, 1–5. [Google Scholar] [CrossRef]
- Harada, B.T.; Knight, M.J.; Imai, S.; Qiao, F.; Ramachander, R.; Sawaya, M.R.; Gingery, M.; Sakane, F.; Bowie, J.U. Regulation of enzyme localization by polymerization: Polymer formation by the SAM domain of diacylglycerol kinase delta1. Structure 2008, 16, 380–387. [Google Scholar] [CrossRef]
- Stafford, R.L.; Hinde, E.; Knight, M.J.; Pennella, M.A.; Ear, J.; Digman, M.A.; Gratton, E.; Bowie, J.U. Tandem SAM domain structure of human Caskin1: A presynaptic, self-assembling scaffold for CASK. Structure 2011, 19, 1826–1836. [Google Scholar] [CrossRef]
- Inoue, H.; Baba, T.; Sato, S.; Ohtsuki, R.; Takemori, A.; Watanabe, T.; Tagaya, M.; Tani, K. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim. Biophys. Acta 2012, 1823, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Rufini, S.; Lena, A.M.; Cadot, B.; Mele, S.; Amelio, I.; Terrinoni, A.; Desideri, A.; Melino, G.; Candi, E. The sterile alpha-motif (SAM) domain of p63 binds in vitro monoasialoganglioside (GM1) micelles. Biochem. Pharmacol. 2011, 82, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Song, H.; Kim, C.A.; Sawaya, M.R.; Hunter, J.B.; Gingery, M.; Rebay, I.; Courey, A.J.; Bowie, J.U. Derepression by depolymerization; structural insights into the regulation of Yan by Mae. Cell 2004, 118, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.J.; Warner, N.; Maini, J.; Chan Tung, K.W.; Zakaria, H.; Pawson, T.; Donaldson, L.W. Saccharomyces cerevisiae Ste50 binds the MAPKKK Ste11 through a head-to-tail SAM domain interaction. J. Mol. Biol. 2006, 356, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fung, K.L.; Jin, D.Y.; Chung, S.S.; Ching, Y.P.; Ng, I.O.; Sze, K.H.; Ko, B.C.; Sun, H. Solution structures, dynamics, and lipid-binding of the sterile alpha-motif domain of the deleted in liver cancer 2. Proteins 2007, 67, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.J.; Donaldson, L.W. The NMR structure of the murine DLC2 SAM domain reveals a variant fold that is similar to a four-helix bundle. BMC Struct. Biol. 2007, 7, 34. [Google Scholar] [CrossRef]
- Johnson, P.E.; Donaldson, L.W. RNA recognition by the Vts1p SAM domain. Nat. Struct. Mol. Biol. 2006, 13, 177–178. [Google Scholar] [CrossRef]
- Ramachander, R.; Bowie, J.U. SAM domains can utilize similar surfaces for the formation of polymers and closed oligomers. J. Mol. Biol. 2004, 342, 1353–1358. [Google Scholar] [CrossRef]
- Thanos, C.D.; Goodwill, K.E.; Bowie, J.U. Oligomeric structure of the human EphB2 receptor SAM domain. Science 1999, 283, 833–836. [Google Scholar] [CrossRef]
- Kukuk, L.; Dingley, A.J.; Granzin, J.; Nagel-Steger, L.; Thiagarajan-Rosenkranz, P.; Ciupka, D.; Hanel, K.; Batra-Safferling, R.; Pacheco, V.; Stoldt, M.; et al. Structure of the SLy1 SAM homodimer reveals a new interface for SAM domain self-association. Sci. Rep. 2019, 9, 54. [Google Scholar] [CrossRef]
- Tran, H.H.; Kim, C.A.; Faham, S.; Siddall, M.C.; Bowie, J.U. Native interface of the SAM domain polymer of TEL. BMC Struct. Biol. 2002, 2, 5. [Google Scholar] [CrossRef]
- Zhang, J.; Graham, T.G.; Vivekanand, P.; Cote, L.; Cetera, M.; Rebay, I. Sterile alpha motif domain-mediated self-association plays an essential role in modulating the activity of the Drosophila ETS family transcriptional repressor Yan. Mol. Cell. Biol. 2010, 30, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Harada, B.; Song, H.; Whitelegge, J.; Courey, A.J.; Bowie, J.U. Mae inhibits Pointed-P2 transcriptional activity by blocking its MAPK docking site. EMBO J. 2006, 25, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Kurabi, A.; Brener, S.; Mobli, M.; Kwan, J.J.; Donaldson, L.W. A nuclear localization signal at the SAM-SAM domain interface of AIDA-1 suggests a requirement for domain uncoupling prior to nuclear import. J. Mol. Biol. 2009, 392, 1168–1177. [Google Scholar] [CrossRef]
- Knight, M.J.; Joubert, M.K.; Plotkowski, M.L.; Kropat, J.; Gingery, M.; Sakane, F.; Merchant, S.S.; Bowie, J.U. Zinc binding drives sheet formation by the SAM domain of diacylglycerol kinase delta. Biochemistry 2010, 49, 9667–9676. [Google Scholar] [CrossRef]
- Di Pietro, S.M.; Cascio, D.; Feliciano, D.; Bowie, J.U.; Payne, G.S. Regulation of clathrin adaptor function in endocytosis: Novel role for the SAM domain. EMBO J. 2010, 29, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Vincenzi, M.; Marasco, D.; De Luca, S.; Pedone, E.M.; Leone, M. Exploring the Ability of Cyclic Peptides to Target SAM Domains: A Computational and Experimental Study. ChemBioChem 2020, 21, 702–711. [Google Scholar] [CrossRef]
- Smirnova, E.; Kwan, J.J.; Siu, R.; Gao, X.; Zoidl, G.; Demeler, B.; Saridakis, V.; Donaldson, L.W. A new mode of SAM domain mediated oligomerization observed in the CASKIN2 neuronal scaffolding protein. Cell Commun. Signal. 2016, 14, 17. [Google Scholar] [CrossRef]
- Sporny, M.; Guez-Haddad, J.; Khazma, T.; Yaron, A.; Dessau, M.; Shkolnisky, Y.; Mim, C.; Isupov, M.N.; Zalk, R.; Hons, M.; et al. Structural basis for SARM1 inhibition and activation under energetic stress. eLife 2020, 9, e62021. [Google Scholar] [CrossRef]
- Rothe, B.; Leettola, C.N.; Leal-Esteban, L.; Cascio, D.; Fortier, S.; Isenschmid, M.; Bowie, J.U.; Constam, D.B. Crystal Structure of Bicc1 SAM Polymer and Mapping of Interactions between the Ciliopathy-Associated Proteins Bicc1, ANKS3, and ANKS6. Structure 2018, 26, 209–224.e6. [Google Scholar] [CrossRef]
- Sporny, M.; Guez-Haddad, J.; Lebendiker, M.; Ulisse, V.; Volf, A.; Mim, C.; Isupov, M.N.; Opatowsky, Y. Structural Evidence for an Octameric Ring Arrangement of SARM1. J. Mol. Biol. 2019, 431, 3591–3605. [Google Scholar] [CrossRef]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Perez White, B.E.; Getsios, S. Eph receptor and ephrin function in breast, gut, and skin epithelia. Cell. Adh. Migr. 2014, 8, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef]
- Ieguchi, K.; Maru, Y. Roles of EphA1/A2 and ephrin-A1 in cancer. Cancer Sci. 2019, 110, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Psilopatis, I.; Pergaris, A.; Vrettou, K.; Tsourouflis, G.; Theocharis, S. The EPH/Ephrin System in Gynecological Cancers: Focusing on the Roots of Carcinogenesis for Better Patient Management. Int. J. Mol. Sci. 2022, 23, 3249. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, R.A.; Hunter, T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol. Cell. Biol. 1990, 10, 6316–6324. [Google Scholar] [CrossRef]
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495. [Google Scholar] [CrossRef]
- Liang, L.Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef]
- Park, J.E.; Son, A.I.; Zhou, R. Roles of EphA2 in Development and Disease. Genes 2013, 4, 334–357. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Wang, B. Eph/ephrin signaling in epithelial development and homeostasis. Int. J. Biochem. Cell Biol. 2009, 41, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, A009159. [Google Scholar] [CrossRef] [PubMed]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell. Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M. Clinical relevance of Ephs and ephrins in cancer: Lessons from breast, colorectal, and lung cancer profiling. Semin. Cell Dev. Biol. 2012, 23, 102–108. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrinA1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Ireton, R.C.; Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 149–157. [Google Scholar] [CrossRef]
- Tandon, M.; Vemula, S.V.; Mittal, S.K. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin. Ther. Targets 2011, 15, 31–51. [Google Scholar] [CrossRef]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, D.; Li, G.; Ma, J.; Chen, P.; Yuan, W.; Hou, F.; Ge, J.; Zhong, M.; Tang, Y.; et al. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene 2014, 33, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef]
- Barquilla, A.; Lamberto, I.; Noberini, R.; Heynen-Genel, S.; Brill, L.M.; Pasquale, E.B. Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol. Biol. Cell 2016, 27, 2757–2770. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yamada, N.; Tanaka, T.; Hori, T.; Yokoyama, S.; Hayakawa, Y.; Yano, S.; Fukuoka, J.; Koizumi, K.; Saiki, I.; et al. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat. Commun. 2015, 6, 7679. [Google Scholar] [CrossRef] [PubMed]
- London, M.; Gallo, E. The EphA2 and cancer connection: Potential for immune-based interventions. Mol. Biol. Rep. 2020, 47, 8037–8048. [Google Scholar] [CrossRef] [PubMed]
- Fattet, L.; Jung, H.Y.; Matsumoto, M.W.; Aubol, B.E.; Kumar, A.; Adams, J.A.; Chen, A.C.; Sah, R.L.; Engler, A.J.; Pasquale, E.B.; et al. Matrix Rigidity Controls Epithelial-Mesenchymal Plasticity and Tumor Metastasis via a Mechanoresponsive EPHA2/LYN Complex. Dev. Cell 2020, 54, 302–316.e7. [Google Scholar] [CrossRef] [PubMed]
- Brannan, J.M.; Sen, B.; Saigal, B.; Prudkin, L.; Behrens, C.; Solis, L.; Dong, W.; Bekele, B.N.; Wistuba, I.; Johnson, F.M. EphA2 in the early pathogenesis and progression of non-small cell lung cancer. Cancer Prev. Res. 2009, 2, 1039–1049. [Google Scholar] [CrossRef]
- Amato, K.R.; Wang, S.; Tan, L.; Hastings, A.K.; Song, W.; Lovly, C.M.; Meador, C.B.; Ye, F.; Lu, P.; Balko, J.M.; et al. EPHA2 Blockade Overcomes Acquired Resistance to EGFR Kinase Inhibitors in Lung Cancer. Cancer Res. 2016, 76, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.B.; Stockhausen, M.T.; Poulsen, H.S. Cell adhesion and EGFR activation regulate EphA2 expression in cancer. Cell. Signal. 2010, 22, 636–644. [Google Scholar] [CrossRef]
- Zhang, C.; Smalley, I.; Emmons, M.F.; Sharma, R.; Izumi, V.; Messina, J.; Koomen, J.M.; Pasquale, E.B.; Forsyth, P.A.; Smalley, K.S.M. Noncanonical EphA2 Signaling Is a Driver of Tumor-Endothelial Cell Interactions and Metastatic Dissemination in BRAF InhibitorResistant Melanoma. J. Investig. Dermatol. 2021, 141, 840–851.e4. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Gehring, M.P.; Light, T.P.; Horne, C.R.; Matsumoto, M.W.; Hristova, K.; Pasquale, E.B. Regulation of the EphA2 receptor intracellular region by phosphomimetic negative charges in the kinase-SAM linker. Nat. Commun. 2021, 12, 7047. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Udayakumar, D.; Zhang, G.; Ji, Z.; Njauw, C.N.; Mroz, P.; Tsao, H. EphA2 is a critical oncogene in melanoma. Oncogene 2011, 30, 4921–4929. [Google Scholar] [CrossRef]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004, 23, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yuan, X.; Li, Z.; Tu, H.; Li, D.; Qing, J.; Wang, H.; Zhang, L. RNA interference targeting EphA2 inhibits proliferation, induces apoptosis, and cooperates with cytotoxic drugs in human glioma cells. Surg. Neurol. 2008, 70, 562–568; discussion 568–569. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Wang, S.; Hastings, A.K.; Youngblood, V.M.; Santapuram, P.R.; Chen, H.; Cates, J.M.; Colvin, D.C.; Ye, F.; Brantley-Sieders, D.M.; et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J. Clin. Investig. 2014, 124, 2037–2049. [Google Scholar] [CrossRef]
- Zhang, T.; Li, J.; Ma, X.; Yang, Y.; Sun, W.; Jin, W.; Wang, L.; He, Y.; Yang, F.; Yi, Z.; et al. Inhibition of HDACs-EphA2 Signaling Axis with WW437 Demonstrates Promising Preclinical Antitumor Activity in Breast Cancer. eBioMedicine 2018, 31, 276–286. [Google Scholar] [CrossRef]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Carles-Kinch, K.; Kilpatrick, K.E.; Stewart, J.C.; Kinch, M.S. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 2002, 62, 2840–2847. [Google Scholar]
- Landen, C.N., Jr.; Lu, C.; Han, L.Y.; Coffman, K.T.; Bruckheimer, E.; Halder, J.; Mangala, L.S.; Merritt, W.M.; Lin, Y.G.; Gao, C.; et al. Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J. Natl. Cancer Inst. 2006, 98, 1558–1570. [Google Scholar] [CrossRef]
- Jackson, D.; Gooya, J.; Mao, S.; Kinneer, K.; Xu, L.; Camara, M.; Fazenbaker, C.; Fleming, R.; Swamynathan, S.; Meyer, D.; et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008, 68, 9367–9374. [Google Scholar] [CrossRef]
- Lee, J.W.; Stone, R.L.; Lee, S.J.; Nam, E.J.; Roh, J.W.; Nick, A.M.; Han, H.D.; Shahzad, M.M.; Kim, H.S.; Mangala, L.S.; et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin. Cancer Res. 2010, 16, 2562–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, S.J.; Pasquale, E.B. Targeting the Eph System with Peptides and Peptide Conjugates. Curr. Drug Targets 2015, 16, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Jiang, J.; Wang, H.; He, Y.; Zhao, X.; Xue, Y.; Xu, H. Design, synthesis, and in vivo evaluation of GO-SWL-Ahx-K-SWL. Bioorg. Med. Chem. Lett. 2022, 70, 128802. [Google Scholar] [CrossRef] [PubMed]
- Koolpe, M.; Dail, M.; Pasquale, E.B. An ephrin mimetic peptide that selectively targets the EphA2 receptor. J. Biol. Chem. 2002, 277, 46974–46979. [Google Scholar] [CrossRef]
- Mitra, S.; Duggineni, S.; Koolpe, M.; Zhu, X.; Huang, Z.; Pasquale, E.B. Structure-activity relationship analysis of peptides targeting the EphA2 receptor. Biochemistry 2010, 49, 6687–6695. [Google Scholar] [CrossRef]
- Wang, S.; Placzek, W.J.; Stebbins, J.L.; Mitra, S.; Noberini, R.; Koolpe, M.; Zhang, Z.; Dahl, R.; Pasquale, E.B.; Pellecchia, M. Novel targeted system to deliver chemotherapeutic drugs to EphA2-expressing cancer cells. J. Med. Chem. 2012, 55, 2427–2436. [Google Scholar] [CrossRef]
- Wang, S.; Noberini, R.; Stebbins, J.L.; Das, S.; Zhang, Z.; Wu, B.; Mitra, S.; Billet, S.; Fernandez, A.; Bhowmick, N.A.; et al. Targeted delivery of paclitaxel to EphA2-expressing cancer cells. Clin. Cancer Res. 2013, 19, 128–137. [Google Scholar] [CrossRef]
- Salem, A.F.; Wang, S.; Billet, S.; Chen, J.F.; Udompholkul, P.; Gambini, L.; Baggio, C.; Tseng, H.R.; Posadas, E.M.; Bhowmick, N.A.; et al. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061. [Google Scholar] [CrossRef]
- Mudd, G.E.; Brown, A.; Chen, L.; van Rietschoten, K.; Watcham, S.; Teufel, D.P.; Pavan, S.; Lani, R.; Huxley, P.; Bennett, G.S. Identification and Optimization of EphA2-Selective Bicycles for the Delivery of Cytotoxic Payloads. J. Med. Chem. 2020, 63, 4107–4116. [Google Scholar] [CrossRef]
- Giorgio, C.; Hassan Mohamed, I.; Flammini, L.; Barocelli, E.; Incerti, M.; Lodola, A.; Tognolini, M. Lithocholic acid is an Eph-ephrin ligand interfering with Eph-kinase activation. PLoS ONE 2011, 6, e18128. [Google Scholar] [CrossRef]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino acid conjugates of lithocholic acid as antagonists of the EphA2 receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Russo, S.; Incerti, M.; Bugatti, A.; Vacondio, F.; Barocelli, E.; Mor, M.; Pala, D.; Hassan-Mohamed, I.; Gioiello, A.; et al. Biochemical characterization of EphA2 antagonists with improved physico-chemical properties by cell-based assays and surface plasmon resonance analysis. Biochem. Pharmacol. 2016, 99, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Jannu, A.K.; Puppala, E.R.; Gawali, B.; Syamprasad, N.P.; Alexander, A.; Marepally, S.; Chella, N.; Gangasani, J.K.; Naidu, V.G.M. Lithocholic acid-tryptophan conjugate (UniPR126) based mixed micelle as a nano carrier for specific delivery of niclosamide to prostate cancer via EphA2 receptor. Int. J. Pharm. 2021, 605, 120819. [Google Scholar] [CrossRef] [PubMed]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with Many (Re)purposes. ChemMedChem 2018, 13, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 is a competitive small molecule Eph-ephrin antagonist blocking in vitro angiogenesis at low micromolar concentrations. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Delle Monache, S.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef]
- Heinzlmeir, S.; Kudlinzki, D.; Sreeramulu, S.; Klaeger, S.; Gande, S.L.; Linhard, V.; Wilhelm, M.; Qiao, H.; Helm, D.; Ruprecht, B.; et al. Chemical Proteomics and Structural Biology Define EPHA2 Inhibition by Clinical Kinase Drugs. ACS Chem. Biol. 2016, 11, 3400–3411. [Google Scholar] [CrossRef]
- Mitri, Z.; Nanda, R.; Blackwell, K.; Costelloe, C.M.; Hood, I.; Wei, C.; Brewster, A.M.; Ibrahim, N.K.; Koenig, K.B.; Hortobagyi, G.N.; et al. TBCRC-010: Phase I/II Study of Dasatinib in Combination with Zoledronic Acid for the Treatment of Breast Cancer Bone Metastasis. Clin. Cancer Res. 2016, 22, 5706–5712. [Google Scholar] [CrossRef]
- Singh, D.R.; Cao, Q.; King, C.; Salotto, M.; Ahmed, F.; Zhou, X.Y.; Pasquale, E.B.; Hristova, K. Unliganded EphA3 dimerization promoted by the SAM domain. Biochem. J. 2015, 471, 101–109. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Light, T.P.; Gomez-Soler, M.; Wang, Z.; Karl, K.; Zapata-Mercado, E.; Gehring, M.P.; Lechtenberg, B.C.; Pogorelov, T.V.; Hristova, K.; Pasquale, E.B. A cancer mutation promotes EphA4 oligomerization and signaling by altering the conformation of the SAM domain. J. Biol. Chem. 2021, 297, 100876. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Ahmed, F.; Paul, M.D.; Gedam, M.; Pasquale, E.B.; Hristova, K. The SAM domain inhibits EphA2 interactions in the plasma membrane. Biochim. Biophys. Acta 2017, 1864, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hapiak, V.; Zheng, J.; Muller-Greven, J.; Bowman, D.; Lingerak, R.; Buck, M.; Wang, B.C.; Smith, A.W. A role of the SAM domain in EphA2 receptor activation. Sci. Rep. 2017, 7, 45084. [Google Scholar] [CrossRef] [PubMed]
- Kullander, K.; Mather, N.K.; Diella, F.; Dottori, M.; Boyd, A.W.; Klein, R. Kinase-dependent and kinase-independent functions of EphA4 receptors in major axon tract formation in vivo. Neuron 2001, 29, 73–84. [Google Scholar] [CrossRef]
- Singh, D.R.; Pasquale, E.B.; Hristova, K. A small peptide promotes EphA2 kinase-dependent signaling by stabilizing EphA2 dimers. Biochim. Biophys. Acta 2016, 1860, 1922–1928. [Google Scholar] [CrossRef]
- Lee, H.J.; Hota, P.K.; Chugha, P.; Guo, H.; Miao, H.; Zhang, L.; Kim, S.J.; Stetzik, L.; Wang, B.C.; Buck, M. NMR structure of a heterodimeric SAM:SAM complex: Characterization and manipulation of EphA2 binding reveal new cellular functions of SHIP2. Structure 2012, 20, 41–55. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, Y.; Li, J.; Chen, W.; Li, G.; Wan, J.; Liu, W.; Zhang, M. Specific Eph receptor-cytoplasmic effector signaling mediated by SAM-SAM domain interactions. eLife 2018, 7, e35677. [Google Scholar] [CrossRef]
- Borthakur, S.; Lee, H.; Kim, S.; Wang, B.C.; Buck, M. Binding and function of phosphotyrosines of the Ephrin A2 (EphA2) receptor using synthetic sterile alpha motif (SAM) domains. J. Biol. Chem. 2014, 289, 19694–19703. [Google Scholar] [CrossRef]
- Zhuang, G.; Hunter, S.; Hwang, Y.; Chen, J. Regulation of EphA2 receptor endocytosis by SHIP2 lipid phosphatase via phosphatidylinositol 3-Kinase-dependent Rac1 activation. J. Biol. Chem. 2007, 282, 2683–2694. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; Kim, Y.; Yoo, S.; Park, E.; Park, S. The SAM domains of Anks family proteins are critically involved in modulating the degradation of EphA receptors. Mol. Cell. Biol. 2010, 30, 1582–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elong Edimo, W.; Schurmans, S.; Roger, P.P.; Erneux, C. SHIP2 signaling in normal and pathological situations: Its impact on cell proliferation. Adv. Biol. Regul. 2014, 54, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Erneux, C.; Potter, B.V. SHIP2: Structure, Function and Inhibition. ChemBioChem 2017, 18, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Pedicone, C.; Meyer, S.T.; Chisholm, J.D.; Kerr, W.G. Targeting SHIP1 and SHIP2 in Cancer. Cancers 2021, 13, 890. [Google Scholar] [CrossRef]
- Csolle, M.P.; Ooms, L.M.; Papa, A.; Mitchell, C.A. PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9189. [Google Scholar] [CrossRef]
- Lazar, D.F.; Saltiel, A.R. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat. Rev. Drug Discov. 2006, 5, 333–342. [Google Scholar] [CrossRef]
- Li, Z.L.; Buck, M. Modified Potential Functions Result in Enhanced Predictions of a Protein Complex by All-Atom Molecular Dynamics Simulations, Confirming a Stepwise Association Process for Native Protein-Protein Interactions. J. Chem. Theory Comput. 2019, 15, 4318–4331. [Google Scholar] [CrossRef]
- Zhang, L.; Borthakur, S.; Buck, M. Dissociation of a Dynamic Protein Complex Studied by All-Atom Molecular Simulations. Biophys. J. 2016, 110, 877–886. [Google Scholar] [CrossRef]
- Li, Z.L.; Mattos, C.; Buck, M. Computational studies of the principle of dynamic-change-driven protein interactions. Structure 2022, 30, 909–919. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 29–32, 51–55. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Noh, H.; Mun, J.; Gu, C.; Sever, S.; Park, S. Anks1a regulates COPII-mediated anterograde transport of receptor tyrosine kinases critical for tumorigenesis. Nat. Commun. 2016, 7, 12799. [Google Scholar] [CrossRef] [PubMed]
- Emaduddin, M.; Edelmann, M.J.; Kessler, B.M.; Feller, S.M. Odin (ANKS1A) is a Src family kinase target in colorectal cancer cells. Cell Commun. Signal. 2008, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Chaerkady, R.; Kandasamy, K.; Gucek, M.; Cole, R.N.; Pandey, A. The interactome of a PTB domain-containing adapter protein, Odin, revealed by SILAC. J. Proteomics 2011, 74, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Sydorskyy, Y.; St-Germain, J.R.; Taylor, P.; Tsao, M.S.; Moran, M.F. Odin (ANKS1A) modulates EGF receptor recycling and stability. PLoS ONE 2013, 8, e64817. [Google Scholar] [CrossRef]
- Shin, J.; Gu, C.; Park, E.; Park, S. Identification of phosphotyrosine binding domain-containing proteins as novel downstream targets of the EphA8 signaling function. Mol. Cell. Biol. 2007, 27, 8113–8126. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Neira, J.L. Structural dissection of the C-terminal sterile alpha motif (SAM) of human p73. Arch. Biochem. Biophys. 2014, 558, 133–142. [Google Scholar] [CrossRef]
- Neira, J.L.; Diaz-Garcia, C.; Prieto, M.; Coutinho, A. The C-terminal SAM domain of p73 binds to the N terminus of MDM2. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 760–770. [Google Scholar] [CrossRef]
- Joshi, R.; Qin, L.; Cao, X.; Zhong, S.; Voss, C.; Min, W.; Li, S.S.C. DLC1 SAM domain-binding peptides inhibit cancer cell growth and migration by inactivating RhoA. J. Biol. Chem. 2020, 295, 645–656. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Scognamiglio, P.L.; Di Natale, C.; Marasco, D.; Pellecchia, M.; Leone, M. CD and NMR conformational studies of a peptide encompassing the Mid Loop interface of Ship2-Sam. Biopolymers 2014, 101, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Scognamiglio, P.L.; Marasco, D.; Pedone, E.M.; Saviano, M.; Leone, M. Peptide Fragments of Odin-Sam1: Conformational Analysis and Interaction Studies with EphA2-Sam. ChemBioChem 2015, 16, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Iannitti, R.; Marasco, D.; Pedone, E.M.; Palumbo, R.; Leone, M. The Sam-Sam interaction between Ship2 and the EphA2 receptor: Design and analysis of peptide inhibitors. Sci. Rep. 2017, 7, 17474. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Anna Mercurio, F.; Di Natale, C.; Palumbo, R.; Pirone, L.; La Manna, S.; Marasco, D.; Maria Pedone, E.; Leone, M. Targeting Ship2-Sam with peptide ligands: Novel insights from a multidisciplinary approach. Bioorg. Chem. 2022, 122, 105680. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Marasco, D.; Di Natale, C.; Pirone, L.; Costantini, S.; Pedone, E.M.; Leone, M. Targeting EphA2-Sam and Its Interactome: Design and Evaluation of Helical Peptides Enriched in Charged Residues. ChemBioChem 2016, 17, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.A.; Pirone, L.; Di Natale, C.; Marasco, D.; Pedone, E.M.; Leone, M. Sam domain-based stapled peptides: Structural analysis and interaction studies with the Sam domains from the EphA2 receptor and the lipid phosphatase Ship2. Bioorg. Chem. 2018, 80, 602–610. [Google Scholar] [CrossRef]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Marasco, D.; Calce, E.; Vincenzi, M.; Pedone, E.M.; De Luca, S.; Leone, M. Design and analysis of EphA2-SAM peptide ligands: A multi-disciplinary screening approach. Bioorg. Chem. 2019, 84, 434–443. [Google Scholar] [CrossRef]
- Leone, M.; Freeze, H.H.; Chan, C.S.; Pellecchia, M. The Nuclear Overhauser Effect in the lead identification process. Curr. Drug Discov. Technol. 2006, 3, 91–100. [Google Scholar] [CrossRef]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. About TFE: Old and New Findings. Curr. Protein Pept. Sci. 2019, 20, 425–451. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Feng, Q.; Yan, Q.; Hao, X.; Chen, Y. Alpha-helical cationic anticancer peptides: A promising candidate for novel anticancer drugs. Mini Rev. Med. Chem. 2015, 15, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef]
- Bouclier, C.; Simon, M.; Laconde, G.; Pellerano, M.; Diot, S.; Lantuejoul, S.; Busser, B.; Vanwonterghem, L.; Vollaire, J.; Josserand, V.; et al. Stapled peptide targeting the CDK4/Cyclin D interface combined with Abemaciclib inhibits KRAS mutant lung cancer growth. Theranostics 2020, 10, 2008–2028. [Google Scholar] [CrossRef]
- Yang, Q.; Qiu, X.; Zhang, X.; Yu, Y.; Li, N.; Wei, X.; Feng, G.; Li, Y.; Zhao, Y.; Wang, R. Optimization of Beclin 1-Targeting Stapled Peptides by Staple Scanning Leads to Enhanced Antiproliferative Potency in Cancer Cells. J. Med. Chem. 2021, 64, 13475–13486. [Google Scholar] [CrossRef]
- Forood, B.; Feliciano, E.J.; Nambiar, K.P. Stabilization of alpha-helical structures in short peptides via end capping. Proc. Natl. Acad. Sci. USA 1993, 90, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Munoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994, 1, 399–409. [Google Scholar] [CrossRef]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hashemi, Z.S.; Zarei, M.; Fath, M.K.; Ganji, M.; Farahani, M.S.; Afsharnouri, F.; Pourzardosht, N.; Khalesi, B.; Jahangiri, A.; Rahbar, M.R.; et al. In silico Approaches for the Design and Optimization of Interfering Peptides Against Protein-Protein Interactions. Front. Mol. Biosci. 2021, 8, 669431. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Target Disease or Application Field | Year of Approval |
|---|---|---|
| Scenesse® | Erythropoietic protoporphyria | 2019 [52,53] |
| 68Ga-DOTATOC | Positron Emission Tomography (PET) for localization of somatostatin receptor-positive NEuroendocrine Tumors (NETs) in adult and pediatric patients | 2019 [52,53] |
| Vyleesi® | Hypoactive sexual desire disorder in premenopausal women | 2019 [52,53,54] |
| 68Ga-PSMA-11 | PET imaging of Prostate-Specific Membrane Antigen (PSMA)-positive lesions in men affected by prostate cancer | 2020 [55,56] |
| Imcivree® | Chronic weight management in adults and pediatric patients (age ≥ 6 years old) with obesity | 2020 [54,55,56] |
| DetectnetTM | PET for localization of somatostatin receptor-positive NETs in adult patients | 2020 [55,56] |
| Sogroya® | Replacement therapy for growth hormone deficiency in adults | 2020 [56] |
| Voxzogo® | Achondroplasia in pediatric patients who are 5 years of ageand older with open epiphyses | 2021 [57,58] |
| KorsuvaTM | Moderate-to-severe pruritus linked to chronic kidney disease in adults undertaking hemodialysis | 2021 [57,58] |
| Bylvay® | Pruritus in patients (age ≥ 3 months old) affected by progressive familial intrahepatic cholestasis | 2021 [57,58] |
| Pylarify® | PET imaging of PSMA-positive tumors in men with prostate cancer | 2021 [57,58] |
| Empaveli® | Paroxysmal nocturnal hemoglobinuria in adults | 2021 [57,58] |
| Zegalogue® | Severe hypoglycemia in patients (age ≥ 6 years old) with diabetes | 2021 [57,58] |
| Pepaxto® | Relapsed or refractory multiple myeloma in adult patients | 2021 [57,58] |
| Lupkynis® | Lupus nephritis in adults | 2021 [54,57,58] |
| MounjaroTM | Improvement of glycemic control in adults with type 2 diabetes mellitus, used in combination with diet and exercise | 2022 [59,60] |
| Peptide Name | Peptide Sequence | |
|---|---|---|
| Ship2-Sam dissection | Shiptide | Ac-EGLVHNGWDDLEFLSDITEEDL-NH2 |
| Odin-Sam1 dissection | Pep1 | Ac-SKLLLNGFDDVHFLGSNVMEEQ-NH2 |
| Pep2 | Ac-SKLLLNGFDDVHFLGSNVMEEQ DLRDIGISDPQHRRKLLQAAR-NH2 | |
| Pep3 | Ac-DLRDIGISDPQHRRKLLQAAR-NH2 | |
| EphA2-Sam dissection | S13WT | Ac-KRIGVRLPGHQKRIAYSLLGLKDQV-NH2 |
| S13-SS |  | |
| KRI | Ac-GHQKRIAY-NH2 | |
| KRI2 | Ac-KRIAYKRIAY-NH2 | |
| KRI3 | Ac-KRIAYKRIAYKRIAY-NH2 | |
| KRI3 analogues | KRI3-YM | Ac-KRIAAKRIAAKRIAA-NH2 |
| KRI3-IM | Ac-KRKAYKRKAYKRKAY-NH2 | |
| KRI4 | Ac-KRIAYKRIAYKRIAYKRIAY-NH2 | |
| cKRI3 |  | |
| Helical peptides enriched in charged residues | S13H1 | Ac-DPETEEIAYKLAMLKAQ-NH2 |
| S13H4 | Ac-DPETKRIAYKLAMLKAQ-NH2 | |
| S13H5 | Ac-DPETEEIAKRLAMLAQK-NH2 | |
| S13H6 | Ac-DPETKRIAEELAMLAQK-NH2 | |
| S13H7 | Ac-DPETEEIAWILAMLAQK-NH2 | |
| Stapled peptides | A5ST |  |
| S13ST |  | |
| S13STshort |  | |
| Peptides from computational screening approaches | SML6 | Ac-VHNGWDDLEFfSDITEEDLEEA-NH2 |
| SML7 | Ac-VENGWDDLEFLSDITEEDLEEA-NH2 | |
| SML8 | Ac-VHNGWDDLEFWSDITEEDLEEA-NH2 | |
| SML9 | Ac-VHNyWDDLEFLSDITEEDLEEA-NH2 | |
| SML10 | Ac-VHNGWDDLEFQSDITEEDLEEA-NH2 | |
| SML11 | Ac-VHNGWDDLEFLSDITEEDLnEA-NH2 | |
| ShipH1 | Ac-NGWDDLEFLEDIwEEDL-NH2 | |
| ShipH2 | Ac-NGWDDnEFdSDITEEDL-NH2 | |
| CTRL | Ac-EGLVHNGWDDLEFLSDITEEDLEEA-NH2 | |
| C131 |  | |
| C4 |  | |
| A5 |  | |
| C1 |  | |
| C5 |  | |
| C6 |  | |
| B2 |  | |
| C7 |  | |
| C8 |  | |
| C9 |  |
| Peptide/Protein Complex | KD (µM) |
|---|---|
| Pep2/EphA2-Sam | 221.97 ± 0.01 (SPR) [188] |
| Pep3/EphA2-Sam | 523.11 ± 0.01 (SPR) [188] |
| KRI3/Ship2-Sam | 83 ± 8 (SPR) [189] |
| KRI3-IM/Ship2-Sam | 309 ± 4 (MST) [190] |
| cKRI3/Ship2-Sam | 140 ± 20 (SPR) [190] 73 ± 5 (MST) [190] |
| S13H4/Odin-Sam1 | 320 (SPR) [191] |
| S13H7/Odin-Sam1 | 62 ± 3 (SPR) [191] 68 ± 3 (MST) |
| S13ST/Ship2-Sam | 52.2 ± 0.7 (MST) [192] |
| ShipH1/EphA2-Sam | 72.4 ± 0.5 (MST) [193] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercurio, F.A.; Vincenzi, M.; Leone, M. Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions. Int. J. Mol. Sci. 2022, 23, 10397. https://doi.org/10.3390/ijms231810397
Mercurio FA, Vincenzi M, Leone M. Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions. International Journal of Molecular Sciences. 2022; 23(18):10397. https://doi.org/10.3390/ijms231810397
Chicago/Turabian StyleMercurio, Flavia Anna, Marian Vincenzi, and Marilisa Leone. 2022. "Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions" International Journal of Molecular Sciences 23, no. 18: 10397. https://doi.org/10.3390/ijms231810397