

Updated Confirmatory Diagnosis for Mucopolysaccharidoses in Taiwanese Infants and the Application of Gene Variants
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
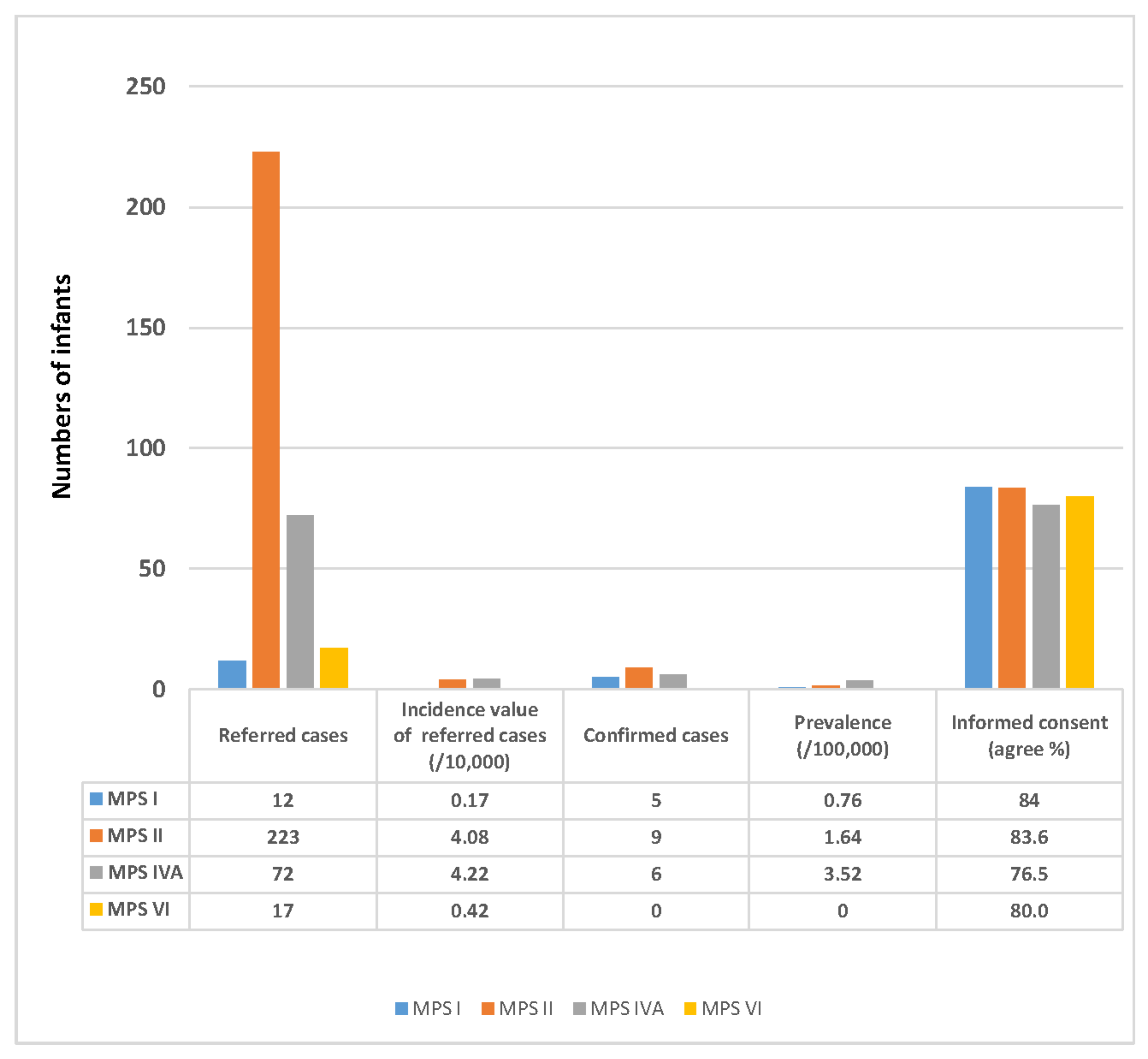
2.1. The Number of Referred Infants from the NBS Program and Prevalence of MPS
2.2. The ACMG Classification of Gene Variants from Different MPS Types in Taiwan
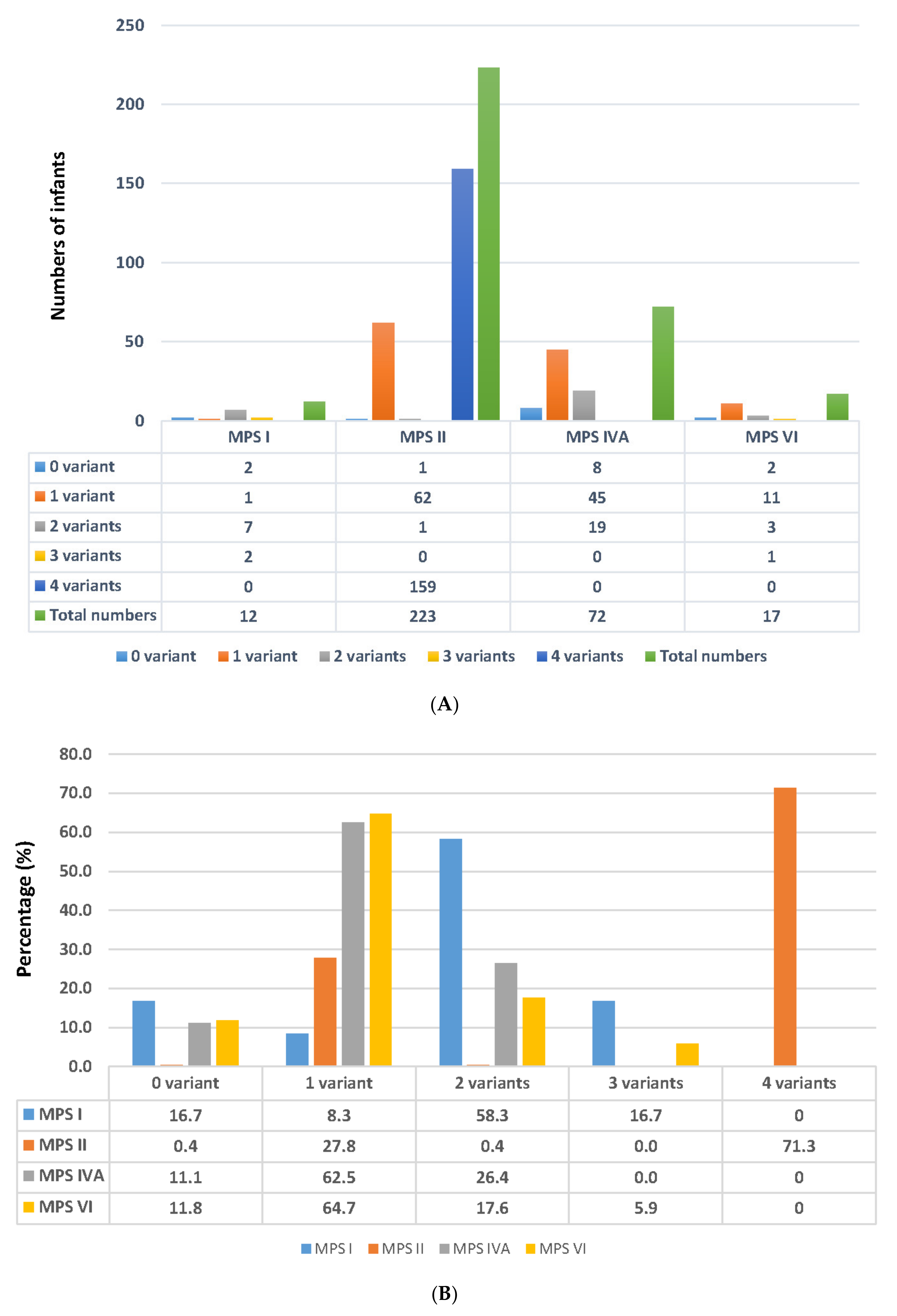
2.3. Number of Variants Detected in the Suspected Infants Referred from the NBS Program
2.3.1. Results Found in MPS I
2.3.2. Results Found in MPS II
2.3.3. Results Found in MPS IVA
2.3.4. Results Found in MPS VI
2.4. Variations of MPS Genes According to ACMG Classification
2.4.1. The ACMG Classification of Variants Found in Infants with Confirmed and Suspected MPS I
2.4.2. The ACMG Classification of Variants Found in Infants with Confirmed and Suspected MPS II
2.4.3. The ACMG Classification of Variants Found in Infants with Confirmed or Suspected MPS IVA
2.4.4. The ACMG Classification of Variants Found in Infants with Suspected MPS VI
2.5. Expression of Enzyme Activity by In Vitro COS-7 Cell Transfection Assay
2.6. Quantification of Urinary GAG-Derived Disaccharides by Tandem Mass Spectrometry Assay Was Crucial to Confirm the Diagnosis of MPS
3. Discussion
4. Materials and Methods
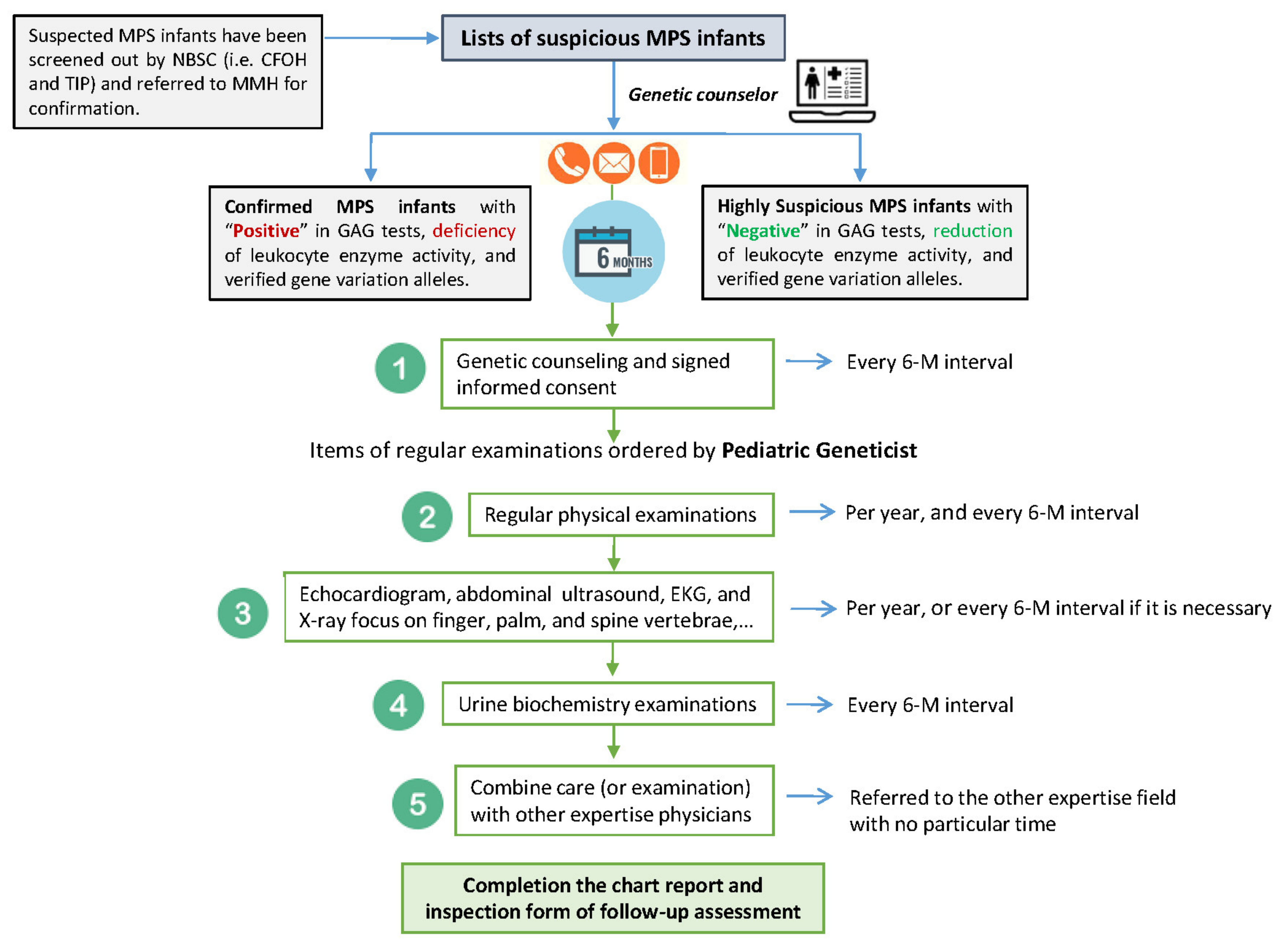
4.1. Suspected MPS Infants Referred to MMH for Confirmation
4.2. Quantification of GAGs; the DMB/Cre Ratio
4.3. GAG-Derived Disaccharide Quantification by LC-MS/MS-Based Assay
4.4. Leukocyte Enzyme Activity Assay
4.5. Molecular DNA Analysis
4.6. RNA Sequencing Analysis
4.7. Expression of Enzyme Activity by In Vitro COS-7 Cell Transfection Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neufeld, E.F. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Applegarth, D.A.; Dimmick, J.E.; Hall, J.G. Organelle Diseases: Clinical Features, Diagnosis, Pathogenesis and Management, Chapman & Hall Medical; Chapman & Hall: London, UK, 1997; pp. 45–97. [Google Scholar]
- Wraith, J. Mucopolysaccharidoses. Curr. Paediatr. 1996, 6, 74–79. [Google Scholar] [CrossRef]
- Muenzer, J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Federica Deodato, F.; Parin, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 120. [Google Scholar] [CrossRef]
- Tomatsu, S.; Alméciga-Díaz, C.J.; Montaño, A.M.; Yabe, H.; Tanaka, A.; Dung, V.C.; Giugliani, R.; Kubaski, F.; Mason, R.W.; Mason, E.; et al. Therapies for the bone in mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Noberto, G.; Jones, S.A.; Kyosen, O.K.; Martins, A.M.; Phornphutkul, C.; et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: A retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Fang, Y.Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.J.; Liao, H.C.; Gelb, M.H.; Chuang, C.K.; Liu, M.Y.; Chen, H.J.; Kao, S.M.; Lin, H.Y.; Huang, Y.H.; Kumar, A.B.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for Mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef]
- Chuang, C.K.; Lee, C.L.; Tu, R.Y.; Lo, Y.T.; Sisca, F.; Chang, Y.H.; Liu, M.Y.; Liu, H.Y.; Chen, H.J.; Kao, S.M.; et al. Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis. Diagnostics 2021, 11, 1583. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lee, C.L.; Chang, C.Y.; Chiu, P.C.; Chien, Y.H.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; Lin, S.J.; Lin, J.L.; et al. Survival and diagnostic age of 175 Taiwanese patients with mucopolysaccharidoses (1985–2019). Orphanet J. Rare Dis. 2020, 15, 314. [Google Scholar] [CrossRef]
- Harrison, S.M.; Leslie GBiesecker, L.G.; Rehm, H.L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 2019, 103, e93. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar]
- Harrison, S.M.; Heidi, L.; Rehm, H.L. Is ‘likely pathogenic’ really 90% likely? Reclassification data in ClinVar. Genome Med. 2019, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Tu, R.Y.; Chern, S.R.; Lo, Y.T.; Fran, S.; Wei, F.J.; Huang, S.F.; Tsai, S.Y.; Chang, Y.H.; Lee, C.L.; et al. Identification and functional characterization of IDS gene mutations underlying Taiwanese Hunter Syndrome (mucopolysaccharidosis type II). Int. J. Mol. Sci. 2020, 21, 114. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Tsai, C.C.; Liu, H.L.; Lin, S.P. A modified liquid chromatography/tandem mass spectrometry method for predominant disaccharide units of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Orphanet J. Rare Dis. 2014, 9, 135. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Tu, R.Y.; Chen, T.L.; Lin, S.P.; Chuang, C.K. Normalization of glycosaminoglycan-derived disaccharides detected by tandem mass spectrometry assay for the diagnosis of mucopolysaccharidosis. Sci. Rep. 2019, 9, 10755. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, S.P.; Chung, S.F. Diagnostic Screening for Mucopolysaccharidoses by the Dimethylmethylene Blue Method and Two Dimensional Electrophoresis. Chin. Med. J. 2001, 64, 15–22. [Google Scholar]
- Auray-Blais, C.; Bhérer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. [Google Scholar] [CrossRef]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef]
- Zhang, H.; Young, S.P.; Auray-Blais, C.; Orchard, P.J.; Tolar, J.; Millington, D.S. Analysis of glycosaminoglycans in cerebrospinal fluid from patients with mucopolysaccharidoses by isotope-dilution ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2011, 57, 1005–1012. [Google Scholar] [CrossRef]
- Wood, T.; Bodamer, O.A.; Burin, M.G.; D’Almeida, V.; Fietz, M.; Giugliani, R.; Hawley, S.M.; Hendriksz, C.J.; Hwu, W.L.; Ketteridge, D.; et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol. Genet. Metab. 2012, 106, 73–82. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Gutierrez, M.A.; Peña, O.M.; Firescu, G.G.T.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pathogenic Variants | |||||
|---|---|---|---|---|---|
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| IDUA (MPS I) | c.2T>C | p.M1T | IDS (MPS II) | c.137A>C | p.D46A |
| c.265C>T | p.R89W | c.253G>A | p.A85T | ||
| c.532G>A | p.E178K | c.254C>T | p.A85V | ||
| c.590G>T | p.G197V | c.311 A>T | p.D104V | ||
| c.606C>G | p.Y202Ter | c.589C>T | p.P197S | ||
| c.617C>T | p.S206L | c.797C>G | p.P266R | ||
| c.911delT | p.V304GfsTer13 | c.805G>A | p.D269N | ||
| c.1037T>G | p.L346R | c.890G>A | p.R297H | ||
| c.1395delC | p.G466AfsTer59 | c.998C>T | p.S333L | ||
| c.1422_1423dupCT | p.Y475SfsTer51 | c.1025A>G | p.H342R | ||
| c.1861C>T | p.R621Ter | c.1400C>T | p.P467L | ||
| c.1898C>T | p.S633L | ||||
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| GALNS (MPS IVA) | c.131G>T | p.G44V | ARSB (MPS VI) | c.478C>T | p.R160Ter |
| c.139G>A | p.G47R | c.943C>T | p.R315Ter | ||
| c.169C>T | p.P57S | c.1197C>G | p.F399L | ||
| c.265G>T | p.G89Ter | c.1350G>T | p.W450C | ||
| c.281G>A | p.R94H | c.1394C>G | p.S465Ter | ||
| c.319G>A | p.A107T | ||||
| c.374C>T | p.P125L | ||||
| c.857C>T | p.T286M | ||||
| c.953T>G | p.M318R | ||||
| c.1009delC | p.H337TfsTer19 | ||||
| c.1019G>A | p.G340D | ||||
| c.1483-2A>G |
| Likely Pathogenic | |||||
|---|---|---|---|---|---|
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| IDUA (MPS I) | c.300-3C>G | IDS (MPS II) | c.142C>T | p.R48C | |
| c.590-2A>C | c.659T>C | p.F220S | |||
| c.1139A>G | p.Q380R | c.851C>T | p.P284L | ||
| c.1359_1384del26bp | p.S453RfsTer47 | c.880-2A>T | |||
| c.1634delAinsGGG | p.E545GfsTer16 | c.1106C>G | |||
| c.1874A>C | p.Y625S | c.1188delA | p.Q396HfsTer44 | ||
| c.1478G>A | p.R493H | ||||
| c.1513T>C | p.F505L | ||||
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| GALNS (MPS IVA) | c.106_111delCTGCTC | p. L36_Lu37del | ARSB (MPS VI) | c.215T>C | p.L72P |
| c.190_191delinsAT | p.A64I | c.245T>C | p.L82P | ||
| c.226A>C | p.Asn76His | c.395T>C | p.L132P | ||
| c.430G>A | p.Gly144Ser | c.424A>G | p.M142V | ||
| c.265G>C | p.Gly89Arg | c.716A>G | p.Q239R | ||
| c.641T>C | p.Leu214Pro | c.905G>A | p.G302E | ||
| c.425A>G | p.H142R | c.908G>A | p.G303E | ||
| c.638C>T | p.A213V | c.1033C>T | p.R345W | ||
| c.704C>A | p.T235K | c.1277A>G | p.N426S | ||
| c.706C>T | p.H236Y | ||||
| c.782T>C | p.I261T | ||||
| c.971C>A | p.A324E | ||||
| c.985C>A | p.H329N | ||||
| c.1108C>T | p.P370S | ||||
| c.1127G>A | p.R376Q | ||||
| c.1493C>T | p.P498L | ||||
| c.1496C>T | p.P499L | ||||
| c.1498G>T | p.G500C |
| Uncertain Significance | |||||
|---|---|---|---|---|---|
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| IDUA (MPS I) | c.76G>A | p.A26T | IDS (MPS II) | c.103+34_56dup | |
| c.95T>G | p.V32G | c.301C>T | p.R101C | ||
| c.179A>C | p.Q60P | c.817C>T | p.R273W | ||
| c.343G>A | p.D115N | c.1006+5G>C | |||
| c.355G>T | p.D119Y | c.1122C>T | p.Gly374= | ||
| c.590-7G>C | |||||
| c.1079T>G | p.F360C | Gene | cDNA change | Amino acid change | |
| c.1091C>T | p.T364M | ARSB (MPS VI) | c.43C>G | p.P15A | |
| c.1093C>G | p.L365V | c.113_121del9 | p.G38_G40del3 | ||
| c.1195_1197delGAG | p.E399del | ||||
| c.1463G>C | p.R488P | ||||
| c.1816G>T | p.V606L | ||||
| c.1828+5G>A | |||||
| Gene | cDNA change | Amino acid change | |||
| GALNS (MPS IVA) | c.887C>T | p.A296V | |||
| c.1567T>G | p.Ter523Eext*92 | ||||
| c.*3C>G |
| Benign | |||||
|---|---|---|---|---|---|
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| IDUA | c.1081G>A | p.A361T | IDS | c.684A>G | p.Pro228= |
| c.1180+184T>C | |||||
| c.1499C>T | p.T500I | ||||
| Gene | cDNA change | Amino acid change | Gene | cDNA change | Amino acid change |
| GALNS | c.121-210C>T | ARSB | c.313-26T>C | ||
| c.345C>T | p.Gly115= | c.1072G>A | p.V358 M | ||
| c.692C>G | p.Ala231Gly | c.1143-27A>C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, C.-K.; Tu, Y.-R.; Lee, C.-L.; Lo, Y.-T.; Chang, Y.-H.; Liu, M.-Y.; Liu, H.-Y.; Chen, H.-J.; Kao, S.-M.; Wang, L.-Y.; et al. Updated Confirmatory Diagnosis for Mucopolysaccharidoses in Taiwanese Infants and the Application of Gene Variants. Int. J. Mol. Sci. 2022, 23, 9979. https://doi.org/10.3390/ijms23179979
Chuang C-K, Tu Y-R, Lee C-L, Lo Y-T, Chang Y-H, Liu M-Y, Liu H-Y, Chen H-J, Kao S-M, Wang L-Y, et al. Updated Confirmatory Diagnosis for Mucopolysaccharidoses in Taiwanese Infants and the Application of Gene Variants. International Journal of Molecular Sciences. 2022; 23(17):9979. https://doi.org/10.3390/ijms23179979
Chicago/Turabian StyleChuang, Chih-Kuang, Yuan-Rong Tu, Chung-Lin Lee, Yun-Ting Lo, Ya-Hui Chang, Mei-Ying Liu, Hsin-Yun Liu, Hsiao-Jan Chen, Shu-Min Kao, Li-Yun Wang, and et al. 2022. "Updated Confirmatory Diagnosis for Mucopolysaccharidoses in Taiwanese Infants and the Application of Gene Variants" International Journal of Molecular Sciences 23, no. 17: 9979. https://doi.org/10.3390/ijms23179979