
Targeting Lineage-Specific Transcription Factors and Cytokines of the Th17/Treg Axis by Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone Attenuates TNBS-Induced Experimental Colitis
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. The Effects of Pyrrolo[3,4-d]pyridazinone Derivatives on Body Weight, Disease Activity Index, and Colon Index in Rats with TNBS-Induced Colitis
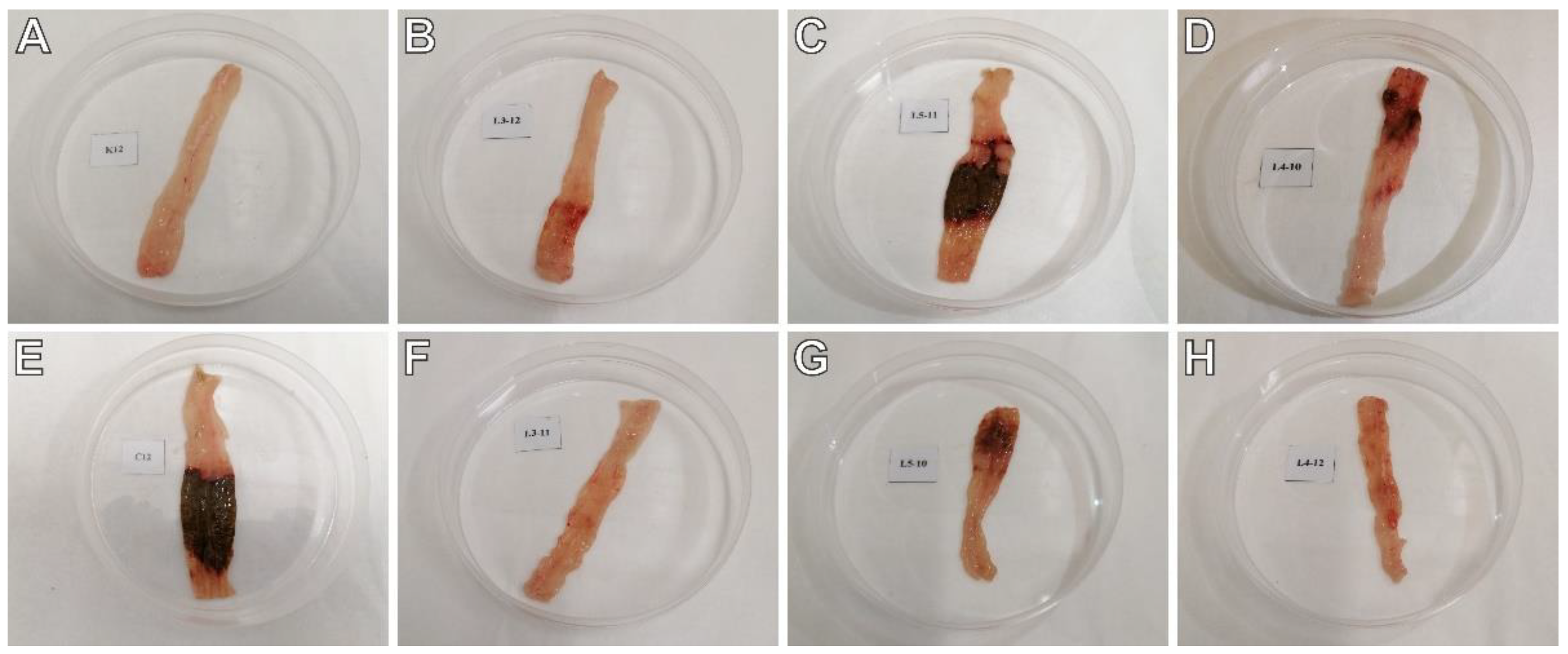
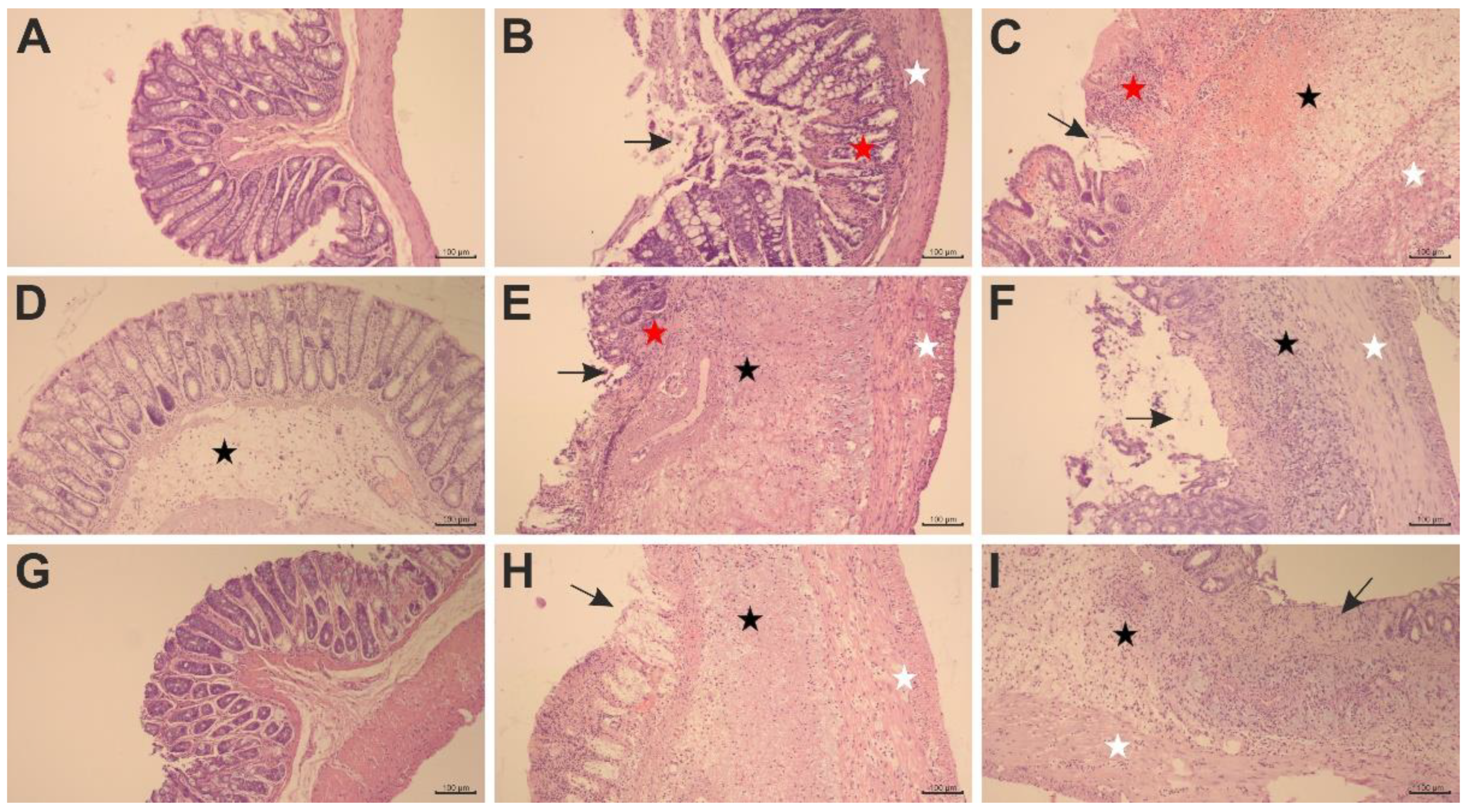
2.2. The Effects of Pyrrolo[3,4-d]pyridazinone Derivatives on TNBS-Induced Macro- and Microscopic Colon Tissue Damage in Rats
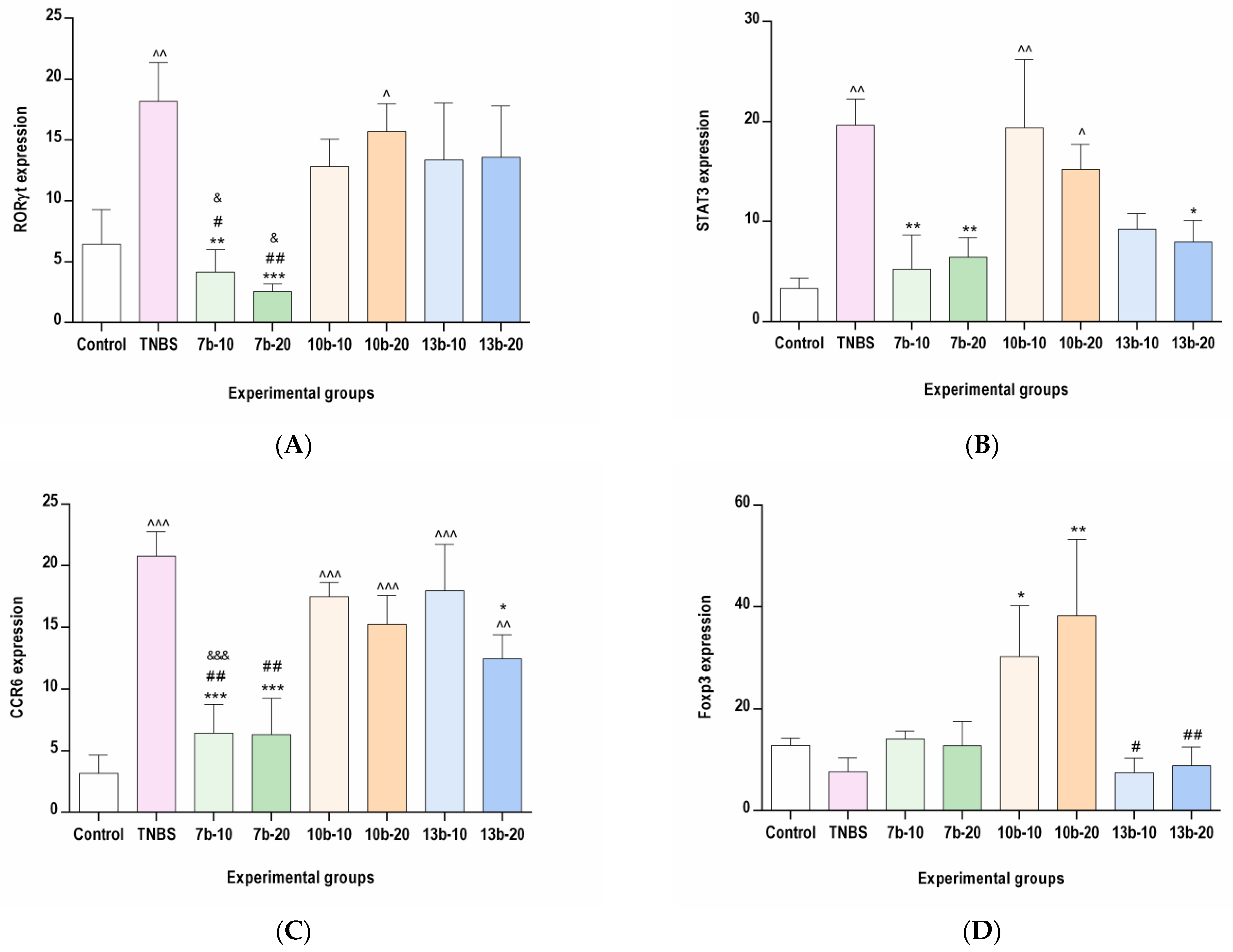
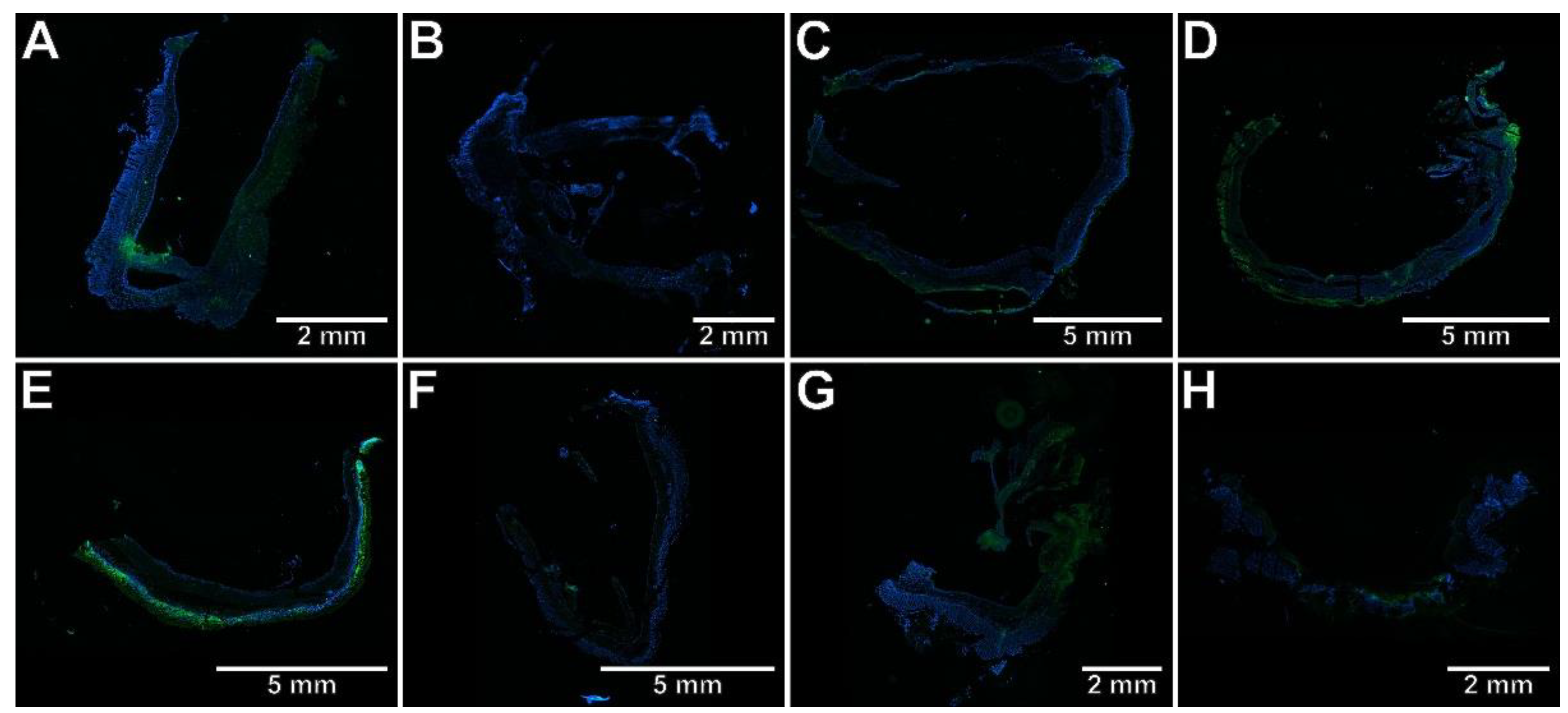
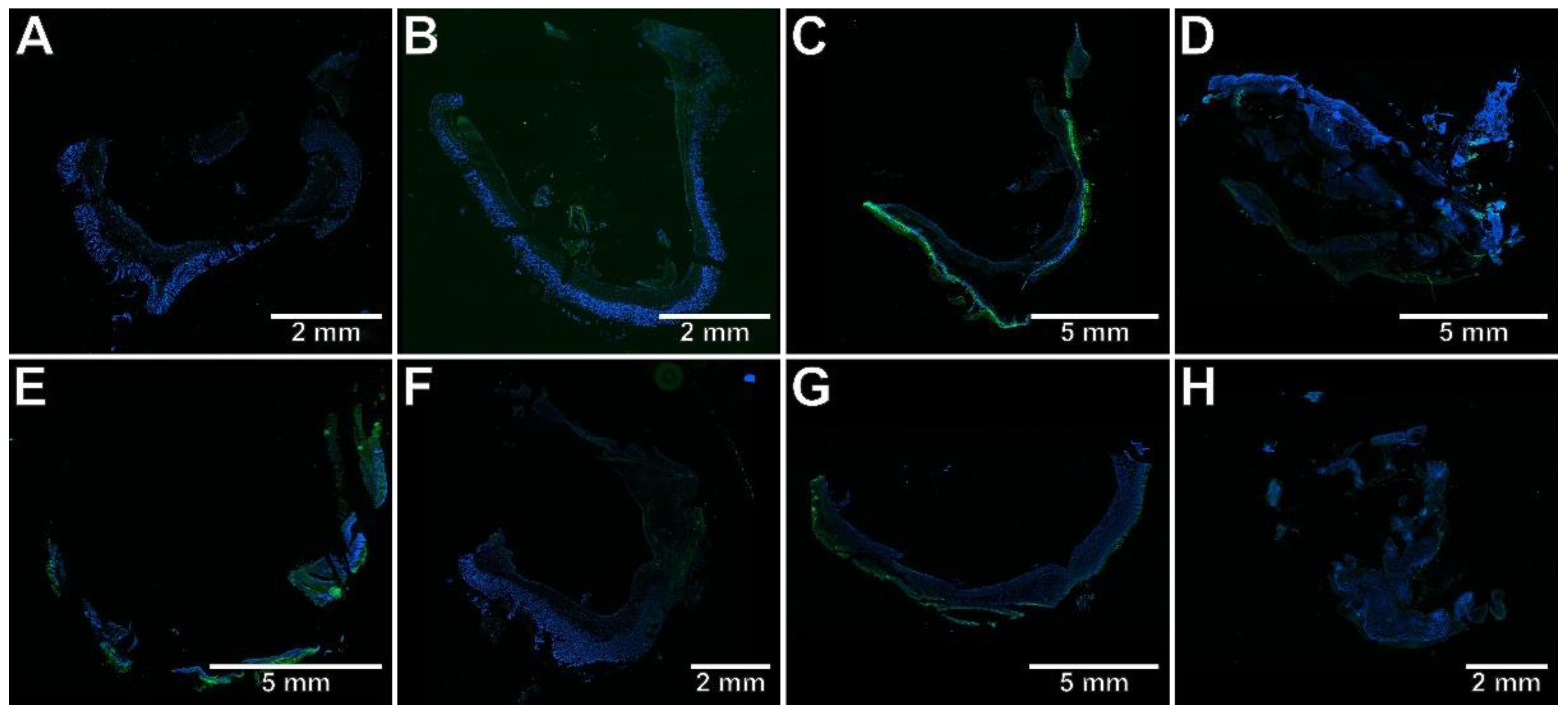
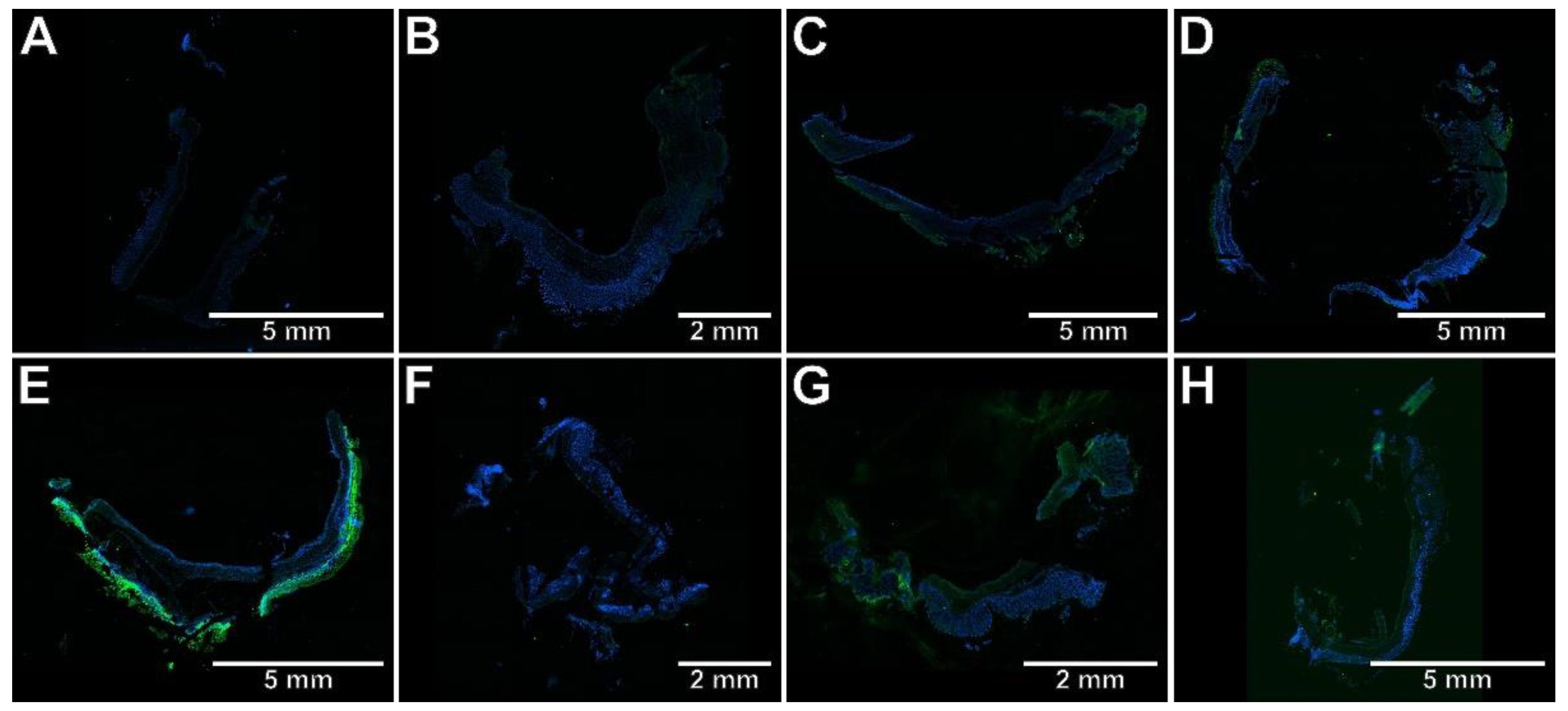
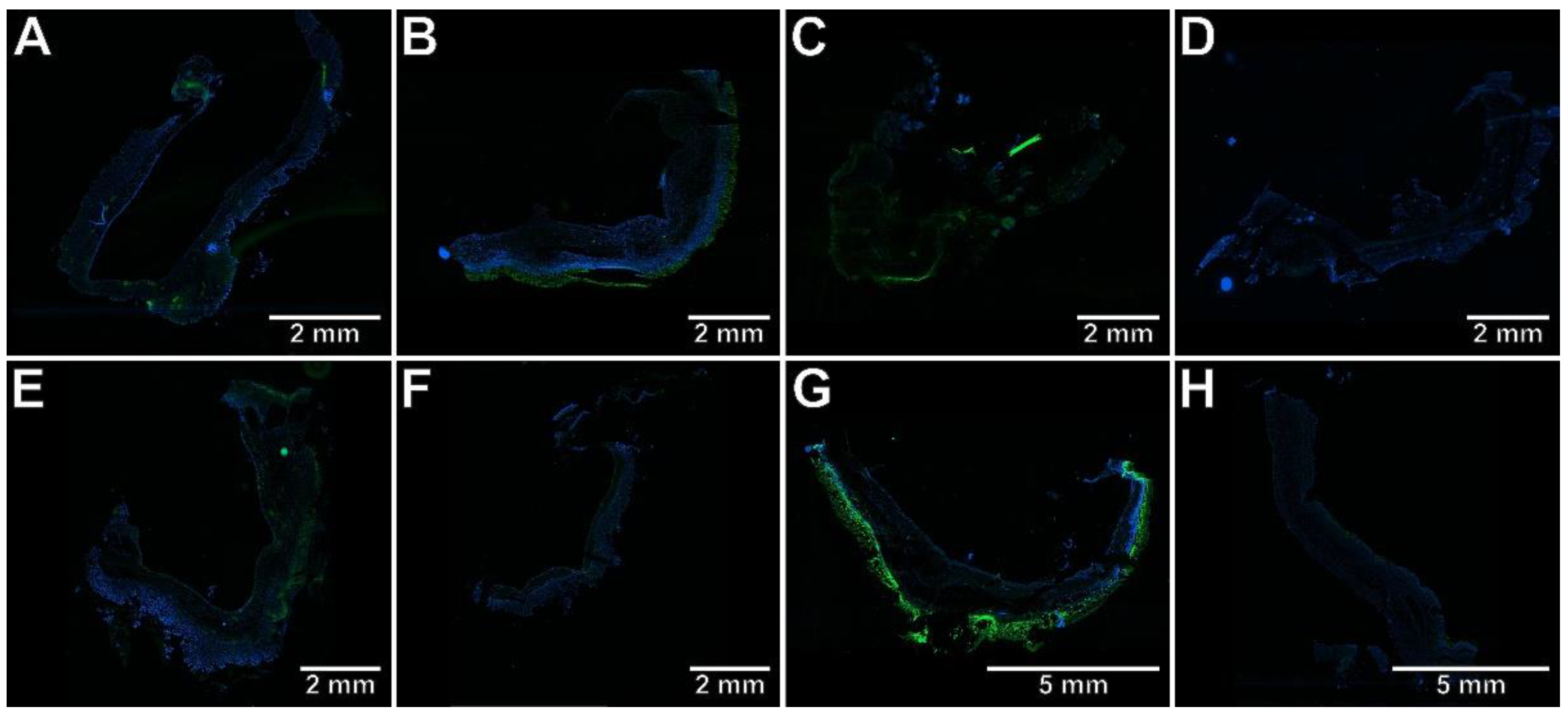
2.3. The Effects of Pyrrolo[3,4-d]pyridazinone Derivatives on the Colonic Expression of RORγt, STAT3, CCR6, and Foxp3 in Rats with TNBS-Induced Colitis
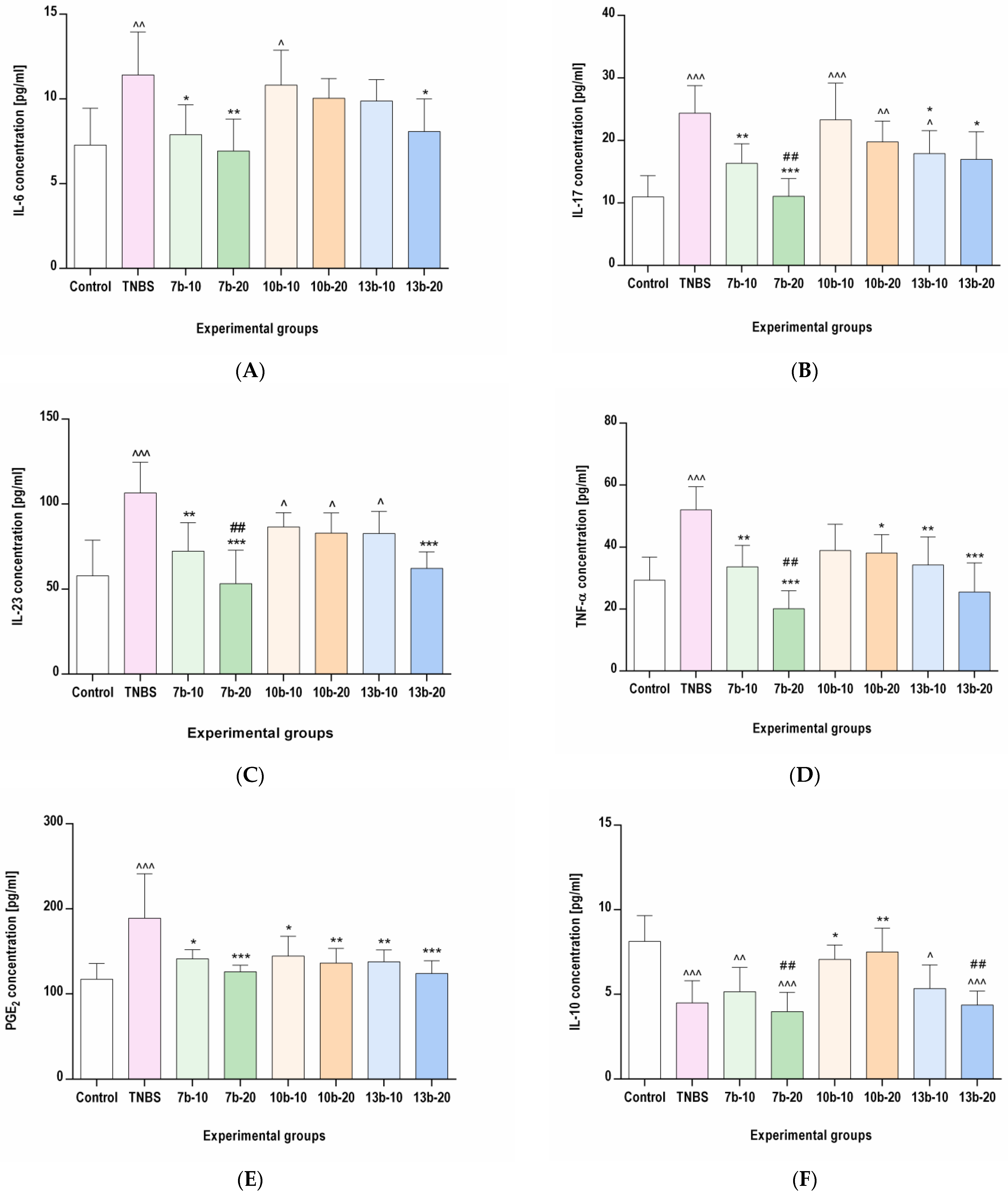
2.4. The Effects of Pyrrolo[3,4-d]pyridazinone Derivatives on the Colonic Levels of IL-6, IL-17, IL-23, TNF-α, PGE2, and IL-10 in Rats with TNBS-Induced Colitis
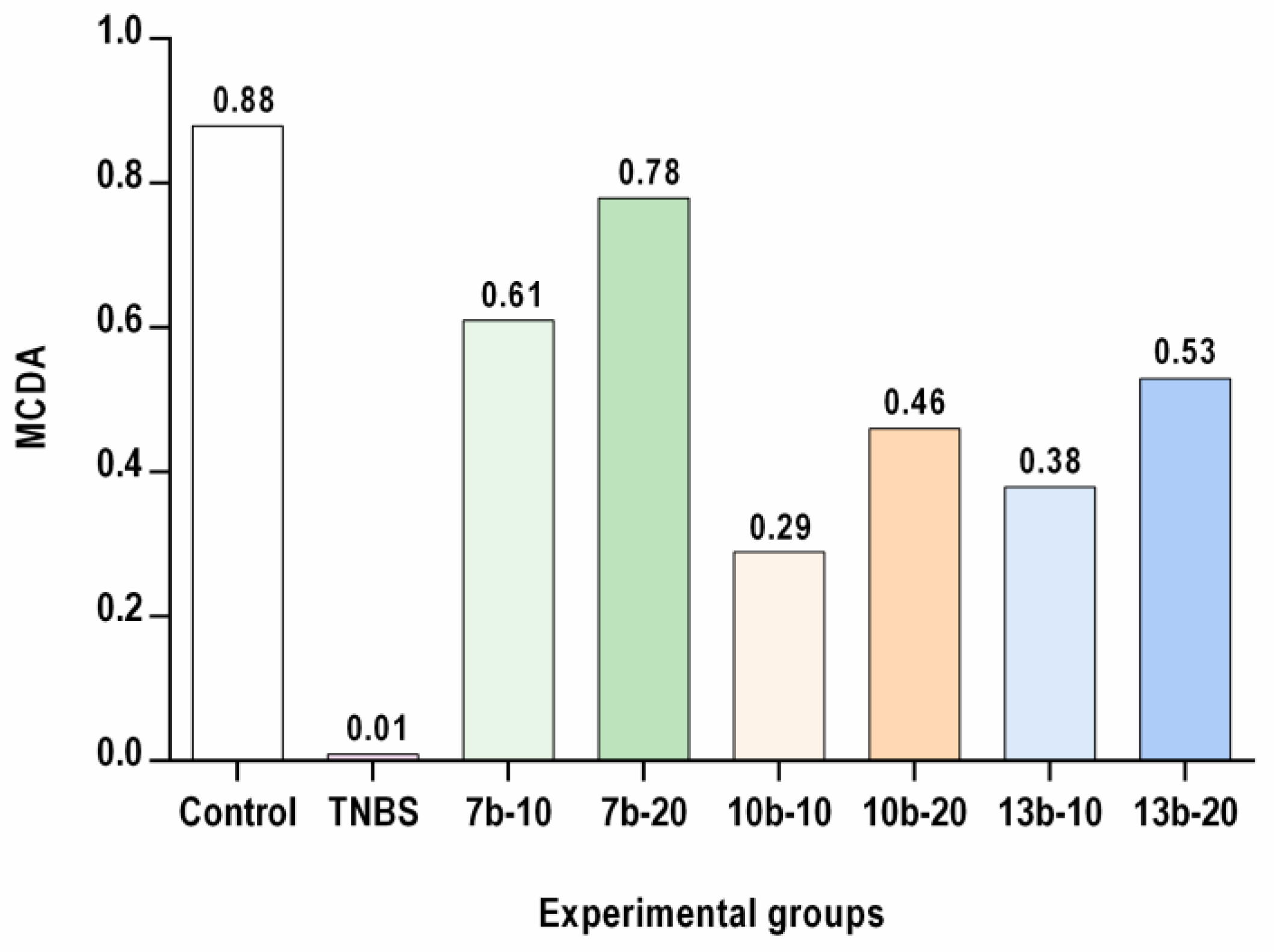
2.5. Multi-Criteria Decision Analysis (MCDA)
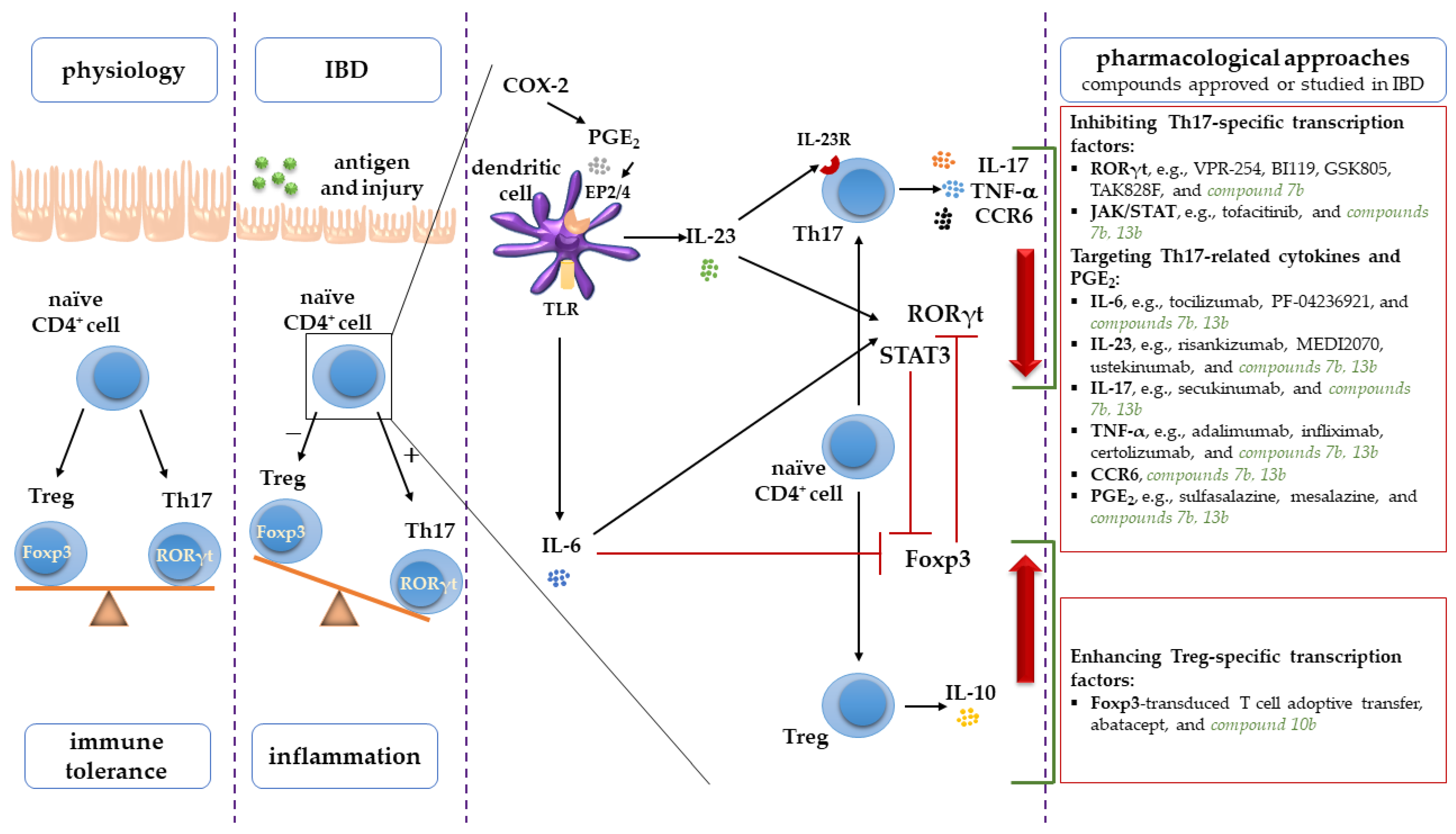
3. Discussion

4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Animals
4.3. Ethical Statement
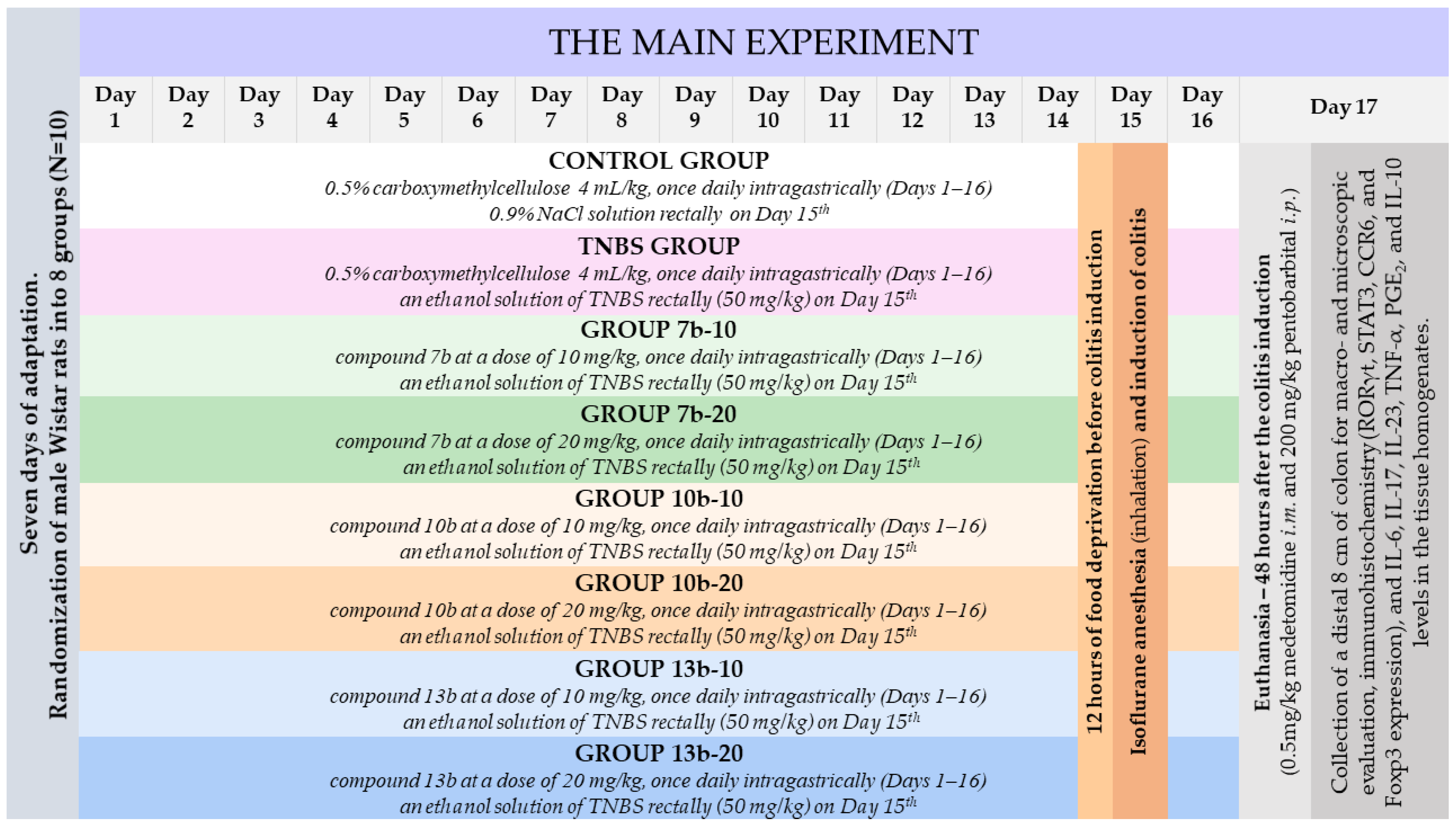
4.4. Experimental Design
- One group pretreated with 0.5% CMC solution (vehicle) intragastrically (i.g.) and receiving once normal saline per rectum (p.r.), i.e., the control group;
- One group pretreated with 0.5% CMC i.g. and receiving once TNBS solution p.r., i.e., the colitis (TNBS) group;
- Six groups pretreated i.g. with compound 7b or 10b or 13b at the doses of 10 or 20 mg/kg and receiving once TNBS solution p.r., i.e., the 7b-10, 7b-20, 10b-10, 10b-20, 13b-10, 13b-20 groups, respectively.
4.5. Induction of Experimental Colitis
4.6. Assessment of the Body Weight, Disease Activity Index, and Colon Index
4.7. Macro- and Microscopic Assessment of the Colon Tissues
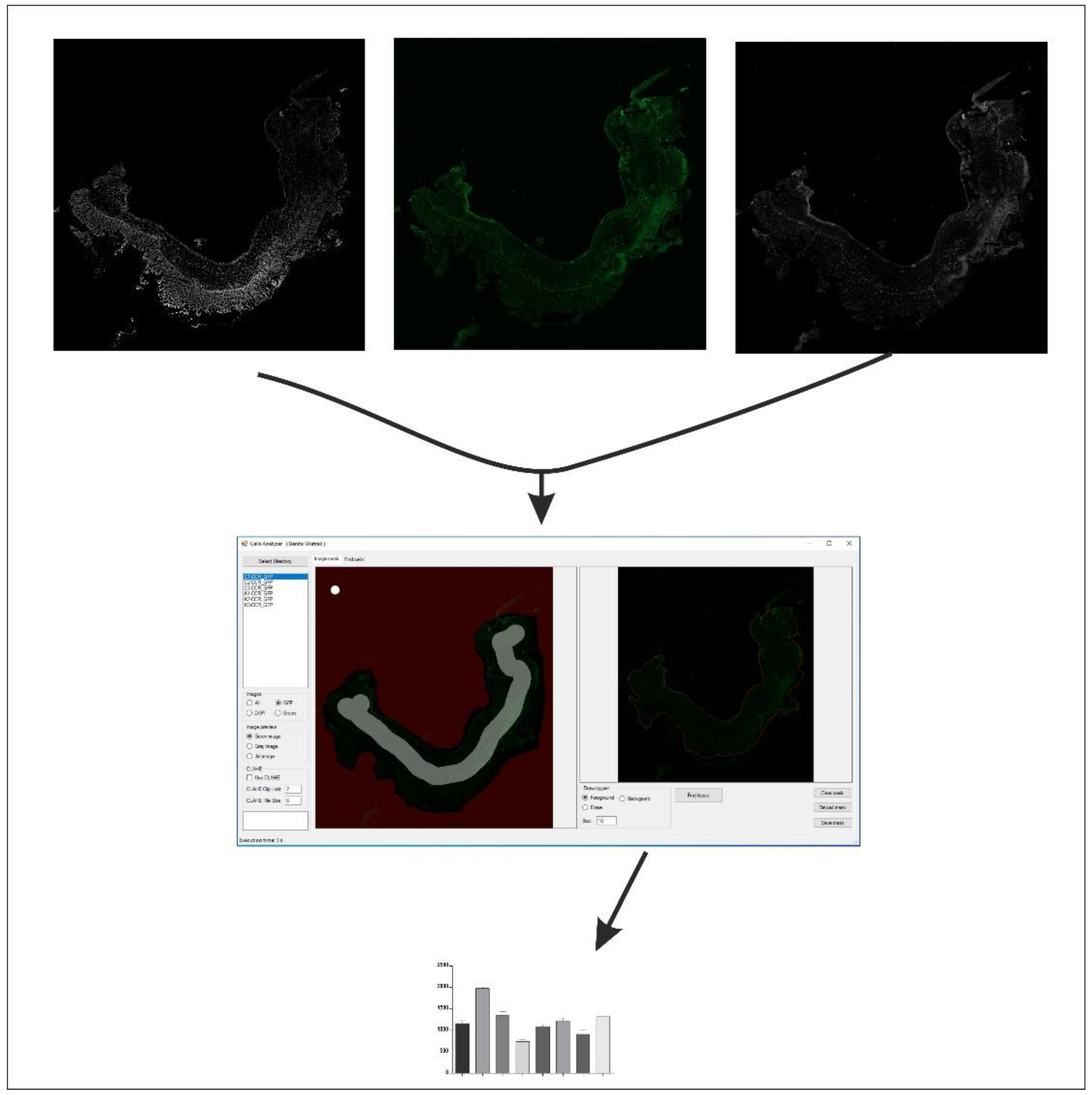
4.8. Immunohistochemical Assessment of RORγt, STAT3, CCR6, and Foxp3 Expression in the Colon Tissues
4.9. Assessment of IL-6, IL-17, IL-23, TNF-α, PGE2, and IL-10 Levels in the Colon Tissues
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A Review of the Diagnosis, Prevention, and Treatment Methods of Inflammatory Bowel Disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef]
- Gálvez, J. Role of Th17 Cells in the Pathogenesis of Human IBD. ISRN Inflamm. 2014, 2014, 928461. [Google Scholar] [CrossRef]
- Lönnfors, S.; Vermeire, S.; Greco, M.; Hommes, D.; Bell, C.; Avedano, L. IBD and Health-Related Quality of Life—Discovering the True Impact. J. Crohns Colitis 2014, 8, 1281–1286. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.; Gębczak, K.; Gębarowski, T.; Maniewska, J. Synthesis, COX-1/2 Inhibition and Antioxidant Activities of New Oxicam Analogues Designed as Potential Chemopreventive Agents. Acta Biochim. Pol. 2018, 65, 199–207. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef]
- GBD 2017 Inflammatory Bowel Disease Collaborators The Global, Regional, and National Burden of Inflammatory Bowel Disease in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [CrossRef]
- Yan, J.-B.; Luo, M.-M.; Chen, Z.-Y.; He, B.-H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef]
- Stockinger, B.; Omenetti, S. The Dichotomous Nature of T Helper 17 Cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Ueno, A.; Ghosh, A.; Hung, D.; Li, J.; Jijon, H. Th17 Plasticity and Its Changes Associated with Inflammatory Bowel Disease. World J. Gastroenterol. 2015, 21, 12283–12295. [Google Scholar] [CrossRef]
- Dziąbowska-Grabias, K.; Sztanke, M.; Zając, P.; Celejewski, M.; Kurek, K.; Szkutnicki, S.; Korga, P.; Bulikowski, W.; Sztanke, K. Antioxidant Therapy in Inflammatory Bowel Diseases. Antioxidants 2021, 10, 412. [Google Scholar] [CrossRef]
- Gomollón, F.; Dignass, A.; Annese, V.; Tilg, H.; Van Assche, G.; Lindsay, J.O.; Peyrin-Biroulet, L.; Cullen, G.J.; Daperno, M.; Kucharzik, T.; et al. 3rd European Evidence-Based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 1: Diagnosis and Medical Management. J. Crohns Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef]
- Harbord, M.; Eliakim, R.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnár, T.; Raine, T.; Sebastian, S.; et al. Third European Evidence-Based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 2: Current Management. J. Crohns Colitis 2017, 11, 769–784. [Google Scholar] [CrossRef]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef]
- Lee, J.; Aoki, T.; Thumkeo, D.; Siriwach, R.; Yao, C.; Narumiya, S. T Cell-Intrinsic Prostaglandin E2-EP2/EP4 Signaling Is Critical in Pathogenic TH17 Cell-Driven Inflammation. J. Allergy Clin. Immunol. 2019, 143, 631–643. [Google Scholar] [CrossRef]
- Li, H.; Edin, M.L.; Gruzdev, A.; Cheng, J.; Bradbury, J.A.; Graves, J.P.; DeGraff, L.M.; Zeldin, D.C. Regulation of T Helper Cell Subsets by Cyclooxygenases and Their Metabolites. Prostaglandins Other Lipid. Mediat. 2013, 104–105, 74–83. [Google Scholar] [CrossRef]
- Fasching, P.; Stradner, M.; Graninger, W.; Dejaco, C.; Fessler, J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules 2017, 22, 134. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, Synthesis, Biological Evaluation and in Silico Studies of Novel Pyrrolo[3,4-d]Pyridazinone Derivatives Withpromising Anti-Inflammatory and Antioxidant Activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Wakulik, K.; Wiatrak, B.; Szczukowski, Ł.; Bodetko, D.; Szandruk-Bender, M.; Dobosz, A.; Świątek, P.; Gąsiorowski, K. Effect of Novel Pyrrolo[3,4-d]Pyridazinone Derivatives on Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2020, 21, 2575. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Merwid-Ląd, A.; Wiatrak, B.; Danielewski, M.; Dzimira, S.; Szkudlarek, D.; Szczukowski, Ł.; Świątek, P.; Szeląg, A. Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]Pyridazinone Exert Anti-Inflammatory Activity without Acute Gastrotoxicity in the Carrageenan-Induced Rat Paw Edema Test. J. Inflamm. Res. 2021, 14, 5739–5756. [Google Scholar] [CrossRef]
- Masciocchi, D.; Gelain, A.; Porta, F.; Meneghetti, F.; Pedretti, A.; Celentano, G.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; et al. Synthesis, Structure–Activity Relationships and Stereochemical Investigations of New Tricyclic Pyridazinone Derivatives as Potential STAT3 Inhibitors. Med. Chem. Commun. 2013, 4, 1181–1188. [Google Scholar] [CrossRef]
- Dell’Orto, S.; Masciocchi, D.; Villa, S.; Meneghetti, F.; Celentano, G.; Barlocco, D.; Colombo, D.; Legnani, L.; Toma, L.; Jeon, Y.J.; et al. Modeling, Synthesis and NMR Characterization of Novel Chimera Compounds Targeting STAT3. Med. Chem. Commun. 2014, 5, 1651–1657. [Google Scholar] [CrossRef]
- Khanam, R.; Hejazi, I.I.; Shahabuddin, S.; Bhat, A.R.; Athar, F. Pharmacokinetic Evaluation, Molecular Docking and in Vitro Biological Evaluation of 1, 3, 4-Oxadiazole Derivatives as Potent Antioxidants and STAT3 Inhibitors. J. Pharm. Anal. 2019, 9, 133–141. [Google Scholar] [CrossRef]
- Masciocchi, D.; Villa, S.; Meneghetti, F.; Pedretti, A.; Barlocco, D.; Legnani, L.; Toma, L.; Kwon, B.-M.; Nakano, S.; Asai, A.; et al. Biological and Computational Evaluation of an Oxadiazole Derivative (MD77) as a New Lead for Direct STAT3 Inhibitors. Med. Chem. Commun. 2012, 3, 592–599. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Wiatrak, B.; Szczukowski, Ł.; Świątek, P.; Rutkowska, M.; Dzimira, S.; Merwid-Ląd, A.; Danielewski, M.; Szeląg, A. Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]Pyridazinone Exert Antinociceptive Activity in the Tail-Flick and Formalin Test in Rodents and Reveal Reduced Gastrotoxicity. Int. J. Mol. Sci. 2020, 21, 9685. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically Induced Mouse Models of Acute and Chronic Intestinal Inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Silva, I.; Pinto, R.; Mateus, V. Preclinical Study in Vivo for New Pharmacological Approaches in Inflammatory Bowel Disease: A Systematic Review of Chronic Model of TNBS-Induced Colitis. J. Clin. Med. 2019, 8, 1574. [Google Scholar] [CrossRef] [PubMed]
- Szandruk-Bender, M.; Rutkowska, M.; Merwid-Ląd, A.; Wiatrak, B.; Szeląg, A.; Dzimira, S.; Sobieszczańska, B.; Krzystek-Korpacka, M.; Kucharska, A.Z.; Matuszewska, A.; et al. Cornelian Cherry Iridoid-Polyphenolic Extract Improves Mucosal Epithelial Barrier Integrity in Rat Experimental Colitis and Exerts Antimicrobial and Antiadhesive Activities In Vitro. Oxid. Med. Cell. Longev. 2020, 2020, 7697851. [Google Scholar] [CrossRef]
- Goyal, N.; Rana, A.; Ahlawat, A.; Bijjem, K.R.V.; Kumar, P. Animal Models of Inflammatory Bowel Disease: A Review. Inflammopharmacology 2014, 22, 219–233. [Google Scholar] [CrossRef]
- Jin, X.; Bai, X.; Zhao, Y.; Dong, Z.; Pang, J.; Liu, M.; Liu, X. Nrf2 Participates in M2 Polarization by Trichinella Spiralis to Alleviate TNBS-Induced Colitis in Mice. Front. Immunol. 2021, 12, 698494. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, M.; Bindas, J.; Schwarzenberger, P.; Kolls, J.K. Critical Role of IL-17 Receptor Signaling in Acute TNBS-Induced Colitis. Inflamm. Bowel. Dis. 2006, 12, 382–388. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When Worlds Collide: Th17 and Treg Cells in Cancer and Autoimmunity. Cell Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef]
- Abraham, C.; Dulai, P.S.; Vermeire, S.; Sandborn, W.J. Lessons Learned From Trials Targeting Cytokine Pathways in Patients With Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 374–388.e4. [Google Scholar] [CrossRef]
- Buchele, V.; Konein, P.; Vogler, T.; Kunert, T.; Enderle, K.; Khan, H.; Büttner-Herold, M.; Lehmann, C.H.K.; Amon, L.; Wirtz, S.; et al. Th17 Cell-Mediated Colitis Is Positively Regulated by Interferon Regulatory Factor 4 in a T Cell-Extrinsic Manner. Front. Immunol. 2021, 11, 590893. [Google Scholar] [CrossRef]
- Chen, Q.-Q.; Yan, L.; Wang, C.-Z.; Wang, W.-H.; Shi, H.; Su, B.-B.; Zeng, Q.-H.; Du, H.-T.; Wan, J. Mesenchymal Stem Cells Alleviate TNBS-Induced Colitis by Modulating Inflammatory and Autoimmune Responses. World J. Gastroenterol. 2013, 19, 4702–4717. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R.; Stonesifer, E.; Small, J.S.; Liby, K.T. The Synthetic Triterpenoid (CDDO-Im) Inhibits STAT3, as Well as IL-17, and Improves DSS-Induced Colitis in Mice. Inflammopharmacology 2014, 22, 341–349. [Google Scholar] [CrossRef]
- Izzo, R.; Bevivino, G.; Monteleone, G. Tofacitinib for the Treatment of Ulcerative Colitis. Expert Opin. Investig. Drugs 2016, 25, 991–997. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-M.; Zhao, Y.; Zeng, Y.; Yan, L.; Chen, B.-L.; Leng, A.-M.; Mu, Y.-B.; Zhang, G.-Y. Inhibition of Pim-1 Kinase Ameliorates Dextran Sodium Sulfate-Induced Colitis in Mice. Dig. Dis. Sci. 2012, 57, 1822–1831. [Google Scholar] [CrossRef]
- Pagnini, C.; Pizarro, T.T.; Cominelli, F. Novel Pharmacological Therapy in Inflammatory Bowel Diseases: Beyond Anti-Tumor Necrosis Factor. Front. Pharmacol. 2019, 10, 671. [Google Scholar] [CrossRef]
- Danese, S.; Vermeire, S.; Hellstern, P.; Panaccione, R.; Rogler, G.; Fraser, G.; Kohn, A.; Desreumaux, P.; Leong, R.W.; Comer, G.M.; et al. Randomised Trial and Open-Label Extension Study of an Anti-Interleukin-6 Antibody in Crohn’s Disease (ANDANTE I and II). Gut 2019, 68, 40–48. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; D’Haens, G.; Panés, J.; Kaser, A.; Ferrante, M.; Louis, E.; Franchimont, D.; Dewit, O.; Seidler, U.; et al. Induction Therapy with the Selective Interleukin-23 Inhibitor Risankizumab in Patients with Moderate-to-Severe Crohn’s Disease: A Randomised, Double-Blind, Placebo-Controlled Phase 2 Study. Lancet 2017, 389, 1699–1709. [Google Scholar] [CrossRef]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A Pilot Randomized Trial of a Human Anti-Interleukin-6 Receptor Monoclonal Antibody in Active Crohn’s Disease. Gastroenterology 2004, 126, 989–996; discussion 947. [Google Scholar] [CrossRef]
- Sands, B.E.; Chen, J.; Feagan, B.G.; Penney, M.; Rees, W.A.; Danese, S.; Higgins, P.D.R.; Newbold, P.; Faggioni, R.; Patra, K.; et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017, 153, 77–86.e6. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.-L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Bassolas-Molina, H.; Raymond, E.; Labadia, M.; Wahle, J.; Ferrer-Picón, E.; Panzenbeck, M.; Zheng, J.; Harcken, C.; Hughes, R.; Turner, M.; et al. An RORγt Oral Inhibitor Modulates IL-17 Responses in Peripheral Blood and Intestinal Mucosa of Crohn’s Disease Patients. Front. Immunol. 2018, 9, 2307. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R.; Small, J.; O’Connell, R.; Talbott, G.; Alton, G.; Zapf, J. VPR-254: An Inhibitor of ROR-Gamma T with Potential Utility for the Treatment of Inflammatory Bowel Disease. Inflammopharmacology 2020, 28, 499–511. [Google Scholar] [CrossRef]
- Shibata, A.; Uga, K.; Sato, T.; Sagara, M.; Igaki, K.; Nakamura, Y.; Ochida, A.; Kono, M.; Shirai, J.; Yamamoto, S.; et al. Pharmacological Inhibitory Profile of TAK-828F, a Potent and Selective Orally Available RORγt Inverse Agonist. Biochem. Pharmacol. 2018, 150, 35–45. [Google Scholar] [CrossRef]
- Withers, D.R.; Hepworth, M.R.; Wang, X.; Mackley, E.C.; Halford, E.E.; Dutton, E.E.; Marriott, C.L.; Brucklacher-Waldert, V.; Veldhoen, M.; Kelsen, J.; et al. Transient Inhibition of ROR-Γt Therapeutically Limits Intestinal Inflammation by Reducing TH17 Cells and Preserving Group 3 Innate Lymphoid Cells. Nat. Med. 2016, 22, 319–323. [Google Scholar] [CrossRef]
- Danese, S.; Grisham, M.; Hodge, J.; Telliez, J.-B. JAK Inhibition Using Tofacitinib for Inflammatory Bowel Disease Treatment: A Hub for Multiple Inflammatory Cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G155–G162. [Google Scholar] [CrossRef]
- Tan, X.; Li, C.; Yang, R.; Zhao, S.; Li, F.; Li, X.; Chen, L.; Wan, X.; Liu, X.; Yang, T.; et al. Discovery of Pyrazolo[3,4-d]Pyridazinone Derivatives as Selective DDR1 Inhibitors via Deep Learning Based Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2022, 65, 103–119. [Google Scholar] [CrossRef]
- Khader, S.A.; Gaffen, S.L.; Kolls, J.K. Th17 Cells at the Crossroads of Innate and Adaptive Immunity against Infectious Diseases at the Mucosa. Mucosal. Immunol. 2009, 2, 403–411. [Google Scholar] [CrossRef]
- Uchida, K.; Koike, Y.; Hashimoto, K.; Saigusa, S.; Inoue, M.; Otake, K.; Tanaka, K.; Matsushita, K.; Okita, Y.; Fujikawa, H.; et al. The Increased Expression of CCL20 and CCR6 in Rectal Mucosa Correlated to Severe Inflammation in Pediatric Ulcerative Colitis. Gastroenterol. Res. Pract. 2015, 2015, 856532. [Google Scholar] [CrossRef]
- Katchar, K.; Kelly, C.P.; Keates, S.; O’brien, M.J.; Keates, A.C. MIP-3alpha Neutralizing Monoclonal Antibody Protects against TNBS-Induced Colonic Injury and Inflammation in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1263–G1271. [Google Scholar] [CrossRef]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef]
- Neurath, M. Current and Emerging Therapeutic Targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 688. [Google Scholar] [CrossRef]
- Lee, Y.; Kuchroo, V. Defining the Functional States of Th17 Cells. F1000Research 2015, 4, 132. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a Human Anti-IL-17A Monoclonal Antibody, for Moderate to Severe Crohn’s Disease: Unexpected Results of a Randomised, Double-Blind Placebo-Controlled Trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Napolitani, G.; Acosta-Rodriguez, E.V.; Lanzavecchia, A.; Sallusto, F. Prostaglandin E2 Enhances Th17 Responses via Modulation of IL-17 and IFN-Gamma Production by Memory CD4+ T Cells. Eur. J. Immunol. 2009, 39, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Chicheportiche, R.; Alvarez, M.; de Rham, C.; Roux-Lombard, P.; Ferrari-Lacraz, S.; Dayer, J.-M. Prostaglandin E2 Synergistically with Interleukin-23 Favors Human Th17 Expansion. Blood 2008, 112, 3696–3703. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; de Waal Malefyt, R. Prostaglandin E2 Regulates Th17 Cell Differentiation and Function through Cyclic AMP and EP2/EP4 Receptor Signaling. J. Exp. Med. 2009, 206, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Muramoto, K.; Masaaki, N.; Ding, Y.; Yang, H.; Mackey, M.; Li, W.; Inoue, Y.; Ackermann, K.; Shirota, H.; et al. A Novel Antagonist of the Prostaglandin E(2) EP(4) Receptor Inhibits Th1 Differentiation and Th17 Expansion and Is Orally Active in Arthritis Models. Br. J. Pharmacol. 2010, 160, 292–310. [Google Scholar] [CrossRef]
- Clough, J.N.; Omer, O.S.; Tasker, S.; Lord, G.M.; Irving, P.M. Regulatory T-Cell Therapy in Crohn’s Disease: Challenges and Advances. Gut 2020, 69, 942–952. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of Regulatory T Cell in the Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [CrossRef]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef]
- Maruyama, T.; Konkel, J.E.; Zamarron, B.F.; Chen, W. The Molecular Mechanisms of Foxp3 Gene Regulation. Semin. Immunol. 2011, 23, 418–423. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, X.; Zhu, Y.; Ma, J.; Ma, H.; Zhang, H. Probiotic Mixture Protects Dextran Sulfate Sodium-Induced Colitis by Altering Tight Junction Protein Expressions and Increasing Tregs. Mediat. Inflamm. 2018, 2018, e9416391. [Google Scholar] [CrossRef]
- Chen, Z.; Lin, F.; Gao, Y.; Li, Z.; Zhang, J.; Xing, Y.; Deng, Z.; Yao, Z.; Tsun, A.; Li, B. FOXP3 and RORγt: Transcriptional Regulation of Treg and Th17. Int. Immunopharmacol. 2011, 11, 536–542. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.; Pathak, M.; Lal, G. Role of Chemokine Receptors and Intestinal Epithelial Cells in the Mucosal Inflammation and Tolerance. J. Leukoc. Biol. 2017, 101, 377–394. [Google Scholar] [CrossRef]
- Meitei, H.T.; Jadhav, N.; Lal, G. CCR6-CCL20 Axis as a Therapeutic Target for Autoimmune Diseases. Autoimmun. Rev. 2021, 20, 102846. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of Interleukin-17-Producing Regulatory T Cells in Inflamed Intestinal Mucosa from Patients with Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.-X.; Wang, B.; Li, B. IL-10 and IL-22 in Mucosal Immunity: Driving Protection and Pathology. Front. Immunol. 2020, 11, 1315. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 Signaling in Regulatory T Cells Is Required for Suppression of Th17 Cell-Mediated Inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef]
- Stumhofer, J.S.; Silver, J.S.; Laurence, A.; Porrett, P.M.; Harris, T.H.; Turka, L.A.; Ernst, M.; Saris, C.J.M.; O’Shea, J.J.; Hunter, C.A. Interleukins 27 and 6 Induce STAT3-Mediated T Cell Production of Interleukin 10. Nat. Immunol. 2007, 8, 1363–1371. [Google Scholar] [CrossRef]
- Morris, G.P.; Beck, P.L.; Herridge, M.S.; Depew, W.T.; Szewczuk, M.R.; Wallace, J.L. Hapten-Induced Model of Chronic Inflammation and Ulceration in the Rat Colon. Gastroenterology 1989, 96, 795–803. [Google Scholar] [CrossRef]
- Gálvez, J.; Coelho, G.; Crespo, M.E.; Cruz, T.; Rodríguez-Cabezas, M.E.; Concha, A.; Gonzalez, M.; Zarzuelo, A. Intestinal Anti-Inflammatory Activity of Morin on Chronic Experimental Colitis in the Rat. Aliment. Pharmacol. Ther. 2001, 15, 2027–2039. [Google Scholar] [CrossRef]
- Arribas, B.; Suárez-Pereira, E.; Ortiz Mellet, C.; García Fernández, J.M.; Buttersack, C.; Rodríguez-Cabezas, M.E.; Garrido-Mesa, N.; Bailon, E.; Guerra-Hernández, E.; Zarzuelo, A.; et al. Di-D-Fructose Dianhydride-Enriched Caramels: Effect on Colon Microbiota, Inflammation, and Tissue Damage in Trinitrobenzenesulfonic Acid-Induced Colitic Rats. J. Agric. Food Chem. 2010, 58, 6476–6484. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Group | Value | p-Value (vs. Control) | p-Value (vs. TNBS) | p-Value (vs. Comp. 10b-10) | p-Value (vs. Comp. 10b-20) | p-Value (vs. Comp. 13b-10) | p-Value (vs. Comp. 13b-20) |
|---|---|---|---|---|---|---|---|---|
| Body weight loss (g) | Control | 20.50 ± 2.25 | - | <0.0001 | <0.0001 | 0.0097 | 0.0001 | 0.0187 |
| TNBS | −10.78 ± 1.39 | <0.0001 | - | NS | NS | NS | 0.0355 | |
| 7b-10 | 10.80 ± 3.55 | NS | 0.0006 | 0.0047 | NS | NS | NS | |
| 7b-20 | 25.00 ± 4.64 | NS | <0.0001 | <0.0001 | 0.0003 | <0.0001 | 0.0025 | |
| 10b-10 | −7.78 ± 3.96 | <0.0001 | NS | - | NS | NS | NS | |
| 10b-20 | 2.00 ± 1.36 | 0.0097 | NS | NS | - | NS | NS | |
| 13b-10 | −3.60 ± 2.36 | 0.0001 | NS | NS | NS | - | NS | |
| 13b-20 | 5.00 ± 1.91 | 0.0187 | 0.0355 | NS | NS | NS | - | |
| DAI | Control | 0.00 ± 0.00 | - | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0011 |
| TNBS | 5.70 ± 1.25 | <0.0001 | - | NS | NS | NS | 0.0040 | |
| 7b-10 | 3.40 ± 1.35 | 0.0040 | 0.0011 | NS | NS | NS | NS | |
| 7b-20 | 1.65 ± 0.82 | NS | <0.0001 | 0.0001 | 0.0005 | 0.0005 | 0.0390 | |
| 10b-10 | 4.60 ± 1.35 | <0.0001 | NS | - | NS | NS | NS | |
| 10b-20 | 4.10 ± 1.29 | <0.0001 | NS | NS | - | NS | NS | |
| 13b-10 | 4.10 ± 1.60 | <0.0001 | NS | NS | NS | - | NS | |
| 13b-20 | 3.60 ± 1.27 | 0.0011 | 0.0040 | NS | NS | NS | - | |
| Colon index | Control | 0.002 ± 0.000 | - | <0.0001 | <0.0001 | 0.0004 | 0.0009 | NS |
| TNBS | 0.007 ± 0.002 | <0.0001 | - | NS | NS | NS | 0.0001 | |
| 7b-10 | 0.003 ± 0.001 | NS | <0.0001 | 0.0005 | 0.0211 | NS | NS | |
| 7b-20 | 0.002 ± 0.000 | NS | <0.0001 | <0.0001 | 0.0002 | 0.0004 | NS | |
| 10b-10 | 0.007 ± 0.002 | <0.0001 | NS | - | NS | NS | NS | |
| 10b-20 | 0.005 ± 0.001 | 0.0004 | NS | NS | - | NS | NS | |
| 13b-10 | 0.005 ± 0.002 | 0.0009 | NS | NS | NS | - | NS | |
| 13b-20 | 0.004 ± 0.001 | NS | 0.0001 | NS | NS | NS | - |
| Parameter | Group | Value | p-Value (vs. Control) | p-Value (vs. TNBS) | p-Value (vs. Comp. 10b-10) | p-Value (vs. Comp. 10b-20) | p-Value (vs. Comp. 13b-10) | p-Value (vs. Comp. 13b-20) |
|---|---|---|---|---|---|---|---|---|
| Macroscopic damage score (0–5 points) | Control | 0.00 ± 0.00 | - | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0009 |
| TNBS | 3.50 ± 1.08 | <0.0001 | - | NS | NS | NS | 0.0016 | |
| 7b-10 | 1.93 ± 0.84 | 0.0003 | 0.0048 | NS | NS | NS | NS | |
| 7b-20 | 1.36 ± 0.69 | NS | <0.0001 | NS | NS | NS | NS | |
| 10b-10 | 3.00 ± 0.82 | <0.0001 | NS | - | NS | NS | NS | |
| 10b-20 | 2.36 ± 0.75 | <0.0001 | NS | NS | - | NS | NS | |
| 13b-10 | 2.71 ± 0.70 | <0.0001 | NS | NS | NS | - | NS | |
| 13b-20 | 1.79 ± 0.49 | 0.0009 | 0.0016 | NS | NS | NS | - | |
| Microscopic damage score, H&E staining (0–25 points) | Control | 0.00 ± 0.00 | - | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| TNBS | 15.78 ± 4.27 | <0.0001 | - | NS | NS | NS | 0.0018 | |
| 7b-10 | 9.89 ± 3.33 | <0.0001 | 0.0001 | 0.0004 | NS | NS | NS | |
| 7b-20 | 2.89 ± 1.27 | NS | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |
| 10b-10 | 15.44 ± 2.35 | <0.0001 | NS | - | NS | NS | 0.0045 | |
| 10b-20 | 12.11 ± 1.76 | <0.0001 | NS | NS | - | NS | NS | |
| 13b-10 | 12.11 ± 2.93 | <0.0001 | NS | NS | NS | - | NS | |
| 13b-20 | 10.78 ± 1.39 | <0.0001 | 0.0018 | 0.0045 | NS | NS | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szandruk-Bender, M.; Wiatrak, B.; Dzimira, S.; Merwid-Ląd, A.; Szczukowski, Ł.; Świątek, P.; Szeląg, A. Targeting Lineage-Specific Transcription Factors and Cytokines of the Th17/Treg Axis by Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone Attenuates TNBS-Induced Experimental Colitis. Int. J. Mol. Sci. 2022, 23, 9897. https://doi.org/10.3390/ijms23179897
Szandruk-Bender M, Wiatrak B, Dzimira S, Merwid-Ląd A, Szczukowski Ł, Świątek P, Szeląg A. Targeting Lineage-Specific Transcription Factors and Cytokines of the Th17/Treg Axis by Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone Attenuates TNBS-Induced Experimental Colitis. International Journal of Molecular Sciences. 2022; 23(17):9897. https://doi.org/10.3390/ijms23179897
Chicago/Turabian StyleSzandruk-Bender, Marta, Benita Wiatrak, Stanisław Dzimira, Anna Merwid-Ląd, Łukasz Szczukowski, Piotr Świątek, and Adam Szeląg. 2022. "Targeting Lineage-Specific Transcription Factors and Cytokines of the Th17/Treg Axis by Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone Attenuates TNBS-Induced Experimental Colitis" International Journal of Molecular Sciences 23, no. 17: 9897. https://doi.org/10.3390/ijms23179897