
The GAG-Binding Peptide MIG30 Protects against Liver Ischemia-Reperfusion in Mice
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MIG30 Inhibits the Binding of CXCL6 to Heparan Sulfate GAGs
2.2. Treatment with MIG30 Protects Mice from Liver Ischemia-Reperfusion Injury (IRI)
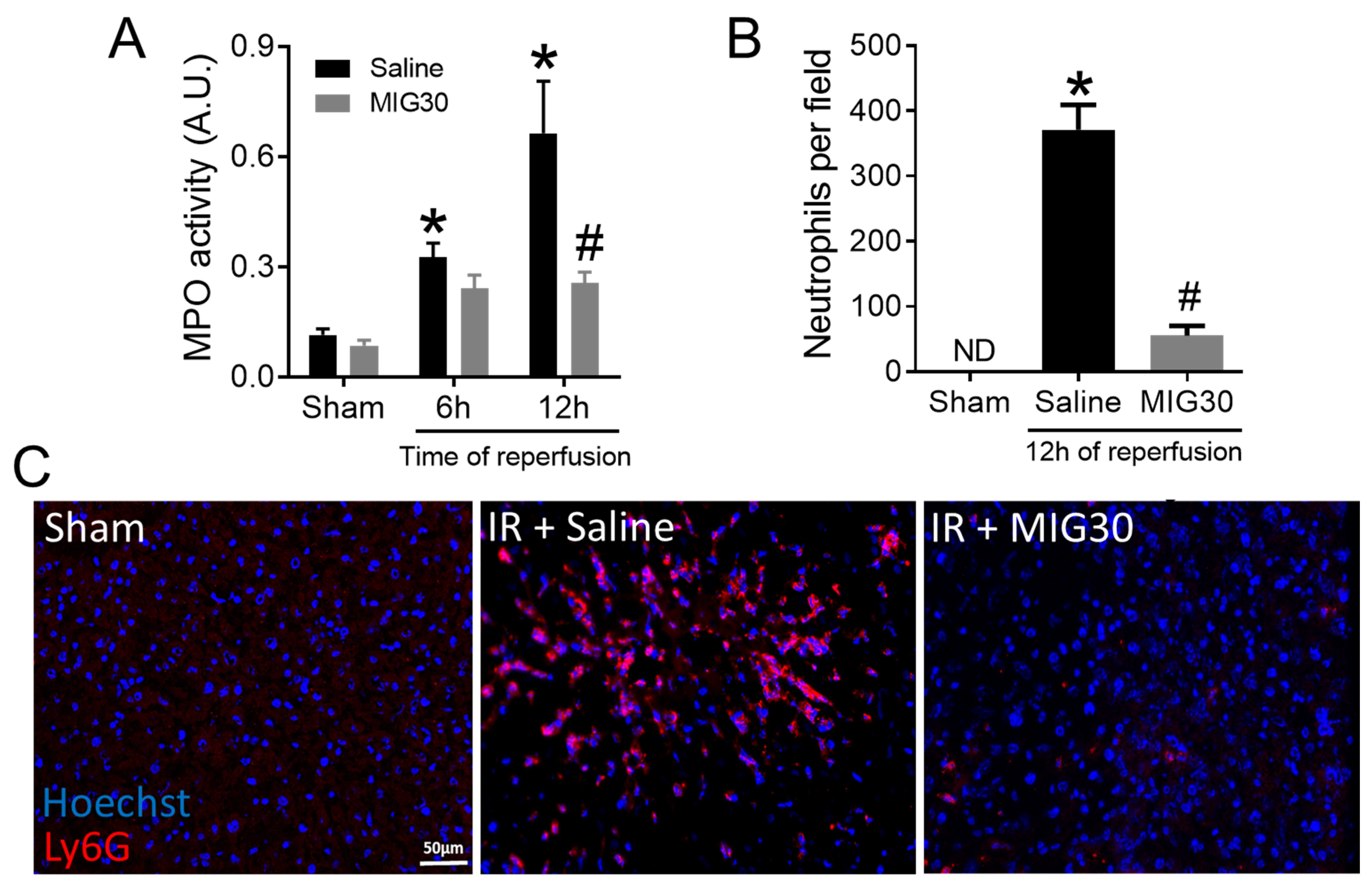
2.3. MIG30 Treatment Impairs Neutrophil Recruitment during Liver IRI
2.4. The Levels of CXC Chemokines and Proinflammatory Cytokines Are Reduced in MIG30-Treated Mice
2.5. The MIG30 Peptide Does Not Affect the Responsiveness or Activation Status of Leukocytes towards Stimuli
3. Discussion
4. Materials and Methods
4.1. MIG30 Peptide Synthesis
4.2. Competition Assay for Binding of CXCL6 to GAGs In Vitro
4.3. Model of Hepatic Ischemia-Reperfusion Injury (IRI)
4.4. Liver Intravital Microscopy
4.5. Histological Analysis
4.6. Myeloperoxidase (MPO) Activity Assay
4.7. Leukocyte Purification (Neutrophils and PBMCs)
4.8. Ca2+ Mobilization Assay
4.9. Neutrophil Flow Cytometry
4.10. Release of CXCL8, Elastase and MPO In Vitro
4.11. Immunofluorescence Staining
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellanti, F. Ischemia-reperfusion injury: Evidences for translational research. Ann. Transl. Med. 2016, 4 (Suppl. S1), S55. [Google Scholar] [CrossRef] [PubMed]
- Chouillard, E.K.; Gumbs, A.A.; Cherqui, D. Vascular clamping in liver surgery: Physiology, indications and techniques. Ann. Surg. Innov. Res. 2010, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, Q.H.; Zhou, C.J.; Hu, M.Z.; Qian, H.X. Protective effect of eNOS overexpression against ischemia/reperfusion injury in small-for-size liver transplantation. Exp. Ther. Med. 2016, 12, 3181–3188. [Google Scholar] [CrossRef]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Pina, E.; Geller, D.A. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef]
- Selzner, M.; Selzner, N.; Jochum, W.; Graf, R.; Clavien, P.A. Increased ischemic injury in old mouse liver: An ATP-dependent mechanism. Liver Transplant. 2007, 13, 382–390. [Google Scholar] [CrossRef]
- Guan, L.Y.; Fu, P.Y.; Li, P.D.; Li, Z.N.; Liu, H.Y.; Xin, M.G.; Li, W. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J. Gastrointest. Surg. 2014, 6, 122–128. [Google Scholar] [CrossRef]
- Honda, M.; Takeichi, T.; Asonuma, K.; Tanaka, K.; Kusunoki, M.; Inomata, Y. Intravital imaging of neutrophil recruitment in hepatic ischemia-reperfusion injury in mice. Transplantation 2013, 95, 551–558. [Google Scholar] [CrossRef]
- Schoening, W.; Ariyakhagorn, V.; Schubert, T.; Olschewski, P.; Andreou, A.; Neuhaus, P.; Pratschke, J.; Puhl, G. Warm HTK donor pretreatment reduces liver injury during static cold storage in experimental rat liver transplantation. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 596–602. [Google Scholar] [CrossRef]
- de Rougemont, O.; Dutkowski, P.; Clavien, P.A. Biological modulation of liver ischemia-reperfusion injury. Curr. Opin. Organ Transplant. 2010, 15, 183–189. [Google Scholar] [CrossRef]
- Lee, W.Y.; Kubes, P. Leukocyte adhesion in the liver: Distinct adhesion paradigm from other organs. J. Hepatol. 2008, 48, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Ju, C.; Ramaiah, S.K.; Uetrecht, J.; Jaeschke, H. Mechanisms of immune-mediated liver injury. Toxicol. Sci. 2010, 115, 307–321. [Google Scholar] [CrossRef] [PubMed]
- van Golen, R.F.; van Gulik, T.M.; Heger, M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev. 2012, 23, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef]
- Bamboat, Z.M.; Balachandran, V.P.; Ocuin, L.M.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010, 51, 621–632. [Google Scholar] [CrossRef]
- Van Sweringen, H.L.; Sakai, N.; Tevar, A.D.; Burns, J.M.; Edwards, M.J.; Lentsch, A.B. CXC chemokine signaling in the liver: Impact on repair and regeneration. Hepatology 2011, 54, 1445–1453. [Google Scholar] [CrossRef]
- Konishi, T.; Lentsch, A.B. Hepatic Ischemia/Reperfusion: Mechanisms of Tissue Injury, Repair, and Regeneration. Gene Expr. 2017, 17, 277–287. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- de Oliveira, T.H.C.; Marques, P.E.; Poosti, F.; Ruytinx, P.; Amaral, F.A.; Brandolini, L.; Allegretti, M.; Proost, P.; Teixeira, M.M. Intravital Microscopic Evaluation of the Effects of a CXCR2 Antagonist in a Model of Liver Ischemia Reperfusion Injury in Mice. Front. Immunol. 2017, 8, 1917. [Google Scholar] [CrossRef]
- Vanheule, V.; Boff, D.; Mortier, A.; Janssens, R.; Petri, B.; Kolaczkowska, E.; Kubes, P.; Berghmans, N.; Struyf, S.; Kungl, A.J.; et al. CXCL9-Derived Peptides Differentially Inhibit Neutrophil Migration In Vivo through Interference with Glycosaminoglycan Interactions. Front. Immunol. 2017, 8, 530. [Google Scholar] [CrossRef] [Green Version]
- Vanheule, V.; Janssens, R.; Boff, D.; Kitic, N.; Berghmans, N.; Ronsse, I.; Kungl, A.J.; Amaral, F.A.; Teixeira, M.M.; Van Damme, J.; et al. The Positively Charged COOH-terminal Glycosaminoglycan-binding CXCL9(74-103) Peptide Inhibits CXCL8-induced Neutrophil Extravasation and Monosodium Urate Crystal-induced Gout in Mice. J. Biol. Chem. 2015, 290, 21292–21304. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Vandendriessche, S.; de Oliveira, T.H.C.; Crijns, H.; Lopes, M.E.; Blanter, M.; Schuermans, S.; Yu, K.; Poosti, F.; Vanheule, V.; et al. Inhibition of Drug-Induced Liver Injury in Mice Using a Positively Charged Peptide That Binds DNA. Hepatol. Commun. 2021, 5, 1737–1754. [Google Scholar] [CrossRef] [PubMed]
- Boff, D.; Russo, R.C.; Crijns, H.; de Oliveira, V.L.S.; Mattos, M.S.; Marques, P.E.; Menezes, G.B.; Vieira, A.T.; Teixeira, M.M.; Proost, P.; et al. The Therapeutic Treatment with the GAG-Binding Chemokine Fragment CXCL9(74-103) Attenuates Neutrophilic Inflammation and Lung Dysfunction during Klebsiella pneumoniae Infection in Mice. Int. J. Mol. Sci. 2022, 23, 6246. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Cesta, M.C.; Garin, A.; Proudfoot, A.E. Current status of chemokine receptor inhibitors in development. Immunol. Lett. 2012, 145, 68–78. [Google Scholar] [CrossRef]
- Adage, T.; Piccinini, A.M.; Falsone, A.; Trinker, M.; Robinson, J.; Gesslbauer, B.; Kungl, A.J. Structure-based design of decoy chemokines as a way to explore the pharmacological potential of glycosaminoglycans. Br. J. Pharmacol. 2012, 167, 1195–1205. [Google Scholar] [CrossRef]
- Johnson, Z.; Proudfoot, A.E.; Handel, T.M. Interaction of chemokines and glycosaminoglycans: A new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005, 16, 625–636. [Google Scholar] [CrossRef]
- Proost, P.; De Wolf-Peeters, C.; Conings, R.; Opdenakker, G.; Billiau, A.; Van Damme, J. Identification of a novel granulocyte chemotactic protein (GCP-2) from human tumor cells. In vitro and in vivo comparison with natural forms of GRO, IP-10, and IL-8. J. Immunol. 1993, 150, 1000–1010. [Google Scholar]
- Wuyts, A.; Haelens, A.; Proost, P.; Lenaerts, J.P.; Conings, R.; Opdenakker, G.; Van Damme, J. Identification of mouse granulocyte chemotactic protein-2 from fibroblasts and epithelial cells. Functional comparison with natural KC and macrophage inflammatory protein-2. J. Immunol. 1996, 157, 1736–1743. [Google Scholar]
- Bertini, R.; Allegretti, M.; Bizzarri, C.; Moriconi, A.; Locati, M.; Zampella, G.; Cervellera, M.N.; Di Cioccio, V.; Cesta, M.C.; Galliera, E.; et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: Prevention of reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 11791–11796. [Google Scholar] [CrossRef]
- Middleton, J.; Neil, S.; Wintle, J.; Clark-Lewis, I.; Moore, H.; Lam, C.; Auer, M.; Hub, E.; Rot, A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 1997, 91, 385–395. [Google Scholar] [CrossRef]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef]
- Wang, L.; Fuster, M.; Sriramarao, P.; Esko, J.D. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 2005, 6, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef]
- Falsone, A.; Wabitsch, V.; Geretti, E.; Potzinger, H.; Gerlza, T.; Robinson, J.; Adage, T.; Teixeira, M.M.; Kungl, A.J. Designing CXCL8-based decoy proteins with strong anti-inflammatory activity In Vivo. Biosci. Rep. 2013, 33, e00068. [Google Scholar] [CrossRef] [PubMed]
- Adage, T.; Del Bene, F.; Fiorentini, F.; Doornbos, R.P.; Zankl, C.; Bartley, M.R.; Kungl, A.J. PA401, a novel CXCL8-based biologic therapeutic with increased glycosaminoglycan binding, reduces bronchoalveolar lavage neutrophils and systemic inflammatory markers in a murine model of LPS-induced lung inflammation. Cytokine 2015, 76, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Mortier, A.; Janssens, R.; Boff, D.; Vanbrabant, L.; Lamoen, N.; Van Damme, J.; Teixeira, M.M.; De Meester, I.; Amaral, F.A.; et al. Glycosaminoglycans Regulate CXCR3 Ligands at Distinct Levels: Protection against Processing by Dipeptidyl Peptidase IV/CD26 and Interference with Receptor Signaling. Int. J. Mol. Sci. 2017, 18, 1513. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Graham, G.J. Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef]
- Robinson, D.E.; Buttle, D.J.; Short, R.D.; McArthur, S.L.; Steele, D.A.; Whittle, J.D. Glycosaminoglycan (GAG) binding surfaces for characterizing GAG-protein interactions. Biomaterials 2012, 33, 1007–1016. [Google Scholar] [CrossRef]
- Marques, P.E.; Antunes, M.M.; David, B.A.; Pereira, R.V.; Teixeira, M.M.; Menezes, G.B. Imaging liver biology In Vivo using conventional confocal microscopy. Nat. Protoc. 2015, 10, 258–268. [Google Scholar] [CrossRef]
- Antoine, D.J.; Williams, D.P.; Kipar, A.; Jenkins, R.E.; Regan, S.L.; Sathish, J.G.; Kitteringham, N.R.; Park, B.K. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol. Sci. 2009, 112, 521–531. [Google Scholar] [CrossRef]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015, 61, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Blanter, M.; Cambier, S.; De Bondt, M.; Vanbrabant, L.; Portner, N.; Salama, S.A.; Metzemaekers, M.; Marques, P.E.; Struyf, S.; Proost, P.; et al. Method Matters: Effect of Purification Technology on Neutrophil Phenotype and Function. Front. Immunol. 2022, 13, 820058. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, T.H.C.; Vanheule, V.; Vandendriessche, S.; Poosti, F.; Teixeira, M.M.; Proost, P.; Gouwy, M.; Marques, P.E. The GAG-Binding Peptide MIG30 Protects against Liver Ischemia-Reperfusion in Mice. Int. J. Mol. Sci. 2022, 23, 9715. https://doi.org/10.3390/ijms23179715
Oliveira THC, Vanheule V, Vandendriessche S, Poosti F, Teixeira MM, Proost P, Gouwy M, Marques PE. The GAG-Binding Peptide MIG30 Protects against Liver Ischemia-Reperfusion in Mice. International Journal of Molecular Sciences. 2022; 23(17):9715. https://doi.org/10.3390/ijms23179715
Chicago/Turabian StyleOliveira, Thiago Henrique Caldeira, Vincent Vanheule, Sofie Vandendriessche, Fariba Poosti, Mauro Martins Teixeira, Paul Proost, Mieke Gouwy, and Pedro Elias Marques. 2022. "The GAG-Binding Peptide MIG30 Protects against Liver Ischemia-Reperfusion in Mice" International Journal of Molecular Sciences 23, no. 17: 9715. https://doi.org/10.3390/ijms23179715