1. Introduction
Stroke represents one of the main causes of disability and death worldwide. The pathological subtypes of stroke comprise ischemic stroke and hemorrhagic stroke, in particular, 87% of which is ischemic stroke [
1,
2]. Oxidative stress and apoptosis are considered to be the key pathogenic mechanisms of the brain injury caused by ischemic stroke [
3,
4,
5]. Though oxidative metabolism is highly essential for the survival of neurons, the consequence of oxidative stress is oxidative damage that occurs when there is an excess of oxygen free radicals (oxidants) or depletion of antioxidants, which can potentially lead to cellular dysfunction, apoptosis, necrosis and/or cell death [
3,
6]. In physiological conditions, excessive reactive oxygen species (ROS) could be scavenged by endogenous antioxidant defense systems, such as catalase (CAT), superoxide dismutase (SOD) and the glutathione system (GSH), to maintain homeostasis [
7,
8,
9]. Thus, antioxidants and antioxidant defense systems play a critical role in exerting a protective effect on tissues and cells from oxidative damage.
Nitric oxide (NO), a free radical, is produced by nitric oxide synthases (NOS), which converts
L-arginine to
L-citrulline with the consequent release of NO. Three isoforms of NOS including neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS) were identified. Both nNOS and eNOS are considered constitutively expressed (also known as cNOS), modulated by Ca
2+ activated calmodulin, whereas iNOS is calcium-independent [
10]. NO rapidly reacts with the superoxide anion radical [O
2−] to yield the peroxynitrite anion [ONOO
−], an important component of ROS, which can induce not only oxidative stress but also nitrative stress. Normal production of ONOO
− is involved in growth, proliferation and differentiation, whereas abnormal production may cause lipid peroxidation, protein oxidation and DNA damage, leading to cell death via either apoptosis or necrosis depending on the nature and extent of oxidative or nitrative stress [
8]. Recently, many studies have evidenced the implication of NO/NOS in a variety of pathophysiological symptoms, including cerebral ischemic stroke, cardiovascular disease and neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease and hypertension [
4,
10,
11]. Therefore, the regulatory balance in the production of NO and its derived species depends not only on the activity of the NOS isoforms but also on the oxidative state of the cell, with both determining the cell fate.
According to the aforementioned, inhibiting the excessive production of ROS and/or regulating the NO/NOS signaling pathway would be a suitable strategy to decrease oxidative stress and apoptosis induced by ischemic stroke. However, few therapeutic drugs and strategies show an effective potential for ischemic stroke to date. Thus, it is necessary to investigate the underlying mechanisms of ischemic stroke and develop new therapeutic drugs to ameliorate the consequences of cerebral ischemia injury.
Herbal medicine has been reported as a promising alternative choice for treating ischemic cerebral injury and thereby, greater attention should be given to natural compounds with wide therapeutic windows, clear pharmacological targets and fewer side effects [
12]. Leonurine (C
14H
21N
3O
5, Leo), one of the active alkaloid compounds purified from Herba Leonuri, is a traditional Chinese herbal medicine with a great deal of biochemical activities including antioxidant, anti-inflammatory and antiapoptotic properties [
13,
14,
15,
16]. However, whether Leo plays a neuroprotective role through the NO/NOS pathway and the exact mechanism regarding how the NO/NOS signaling pathway exerts its impact in ischemic stroke are still to be elucidated.
In the present study, given the key role of the NO/NOS signaling pathway in cerebral ischemia events, we hypothesize that Leo, a substance capable of reducing oxidative stress, can exert its neuroprotective effect after cerebral ischemia, at least in part, by modulating the NO/NOS system response. We established an in vivo model of cerebral ischemia stimulated by photochemistry and an oxygen–glucose deprivation (OGD)-induced PC12 cells model in vitro, combined with the pretreatment of L-NAME, an inhibitor of NOS, to explore the effects of NO/NOS signaling on oxidative stress and apoptosis in vivo and in vitro. We then investigated the protective effects of Leo against OGD-induced oxidative stress in PC12 cells and evaluated the molecular mechanism underlying its neuroprotection related to the NO/NOS pathway.
3. Discussion
Our current research suggests that Leo ameliorated OGD-induced injury, apoptosis and oxidative stress-related factor levels in PC12 cells. The mechanistic study demonstrates that the protective properties of Leo on OGD-stimulated changes may occur through inhibiting the NO/NOS signaling pathways. In addition, the data from L-NAME, a NOS inhibitor, also illustrated the neuroprotective effect of Leo.
Stroke is considered as the third leading cause of death in adults, with a high rate of morbidity, mortality and disability [
21]. Increasing evidence shows that cerebral ischemia is a frequent symptom of stroke [
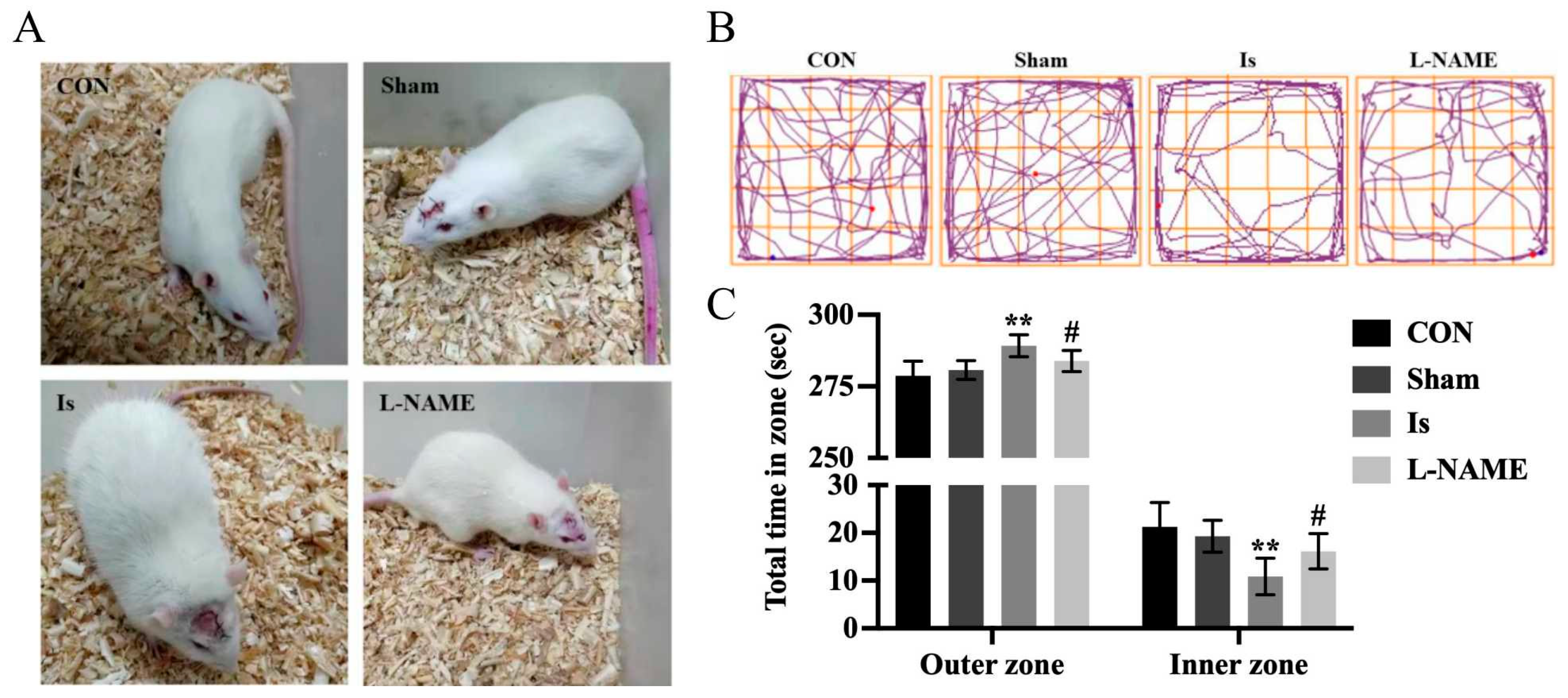
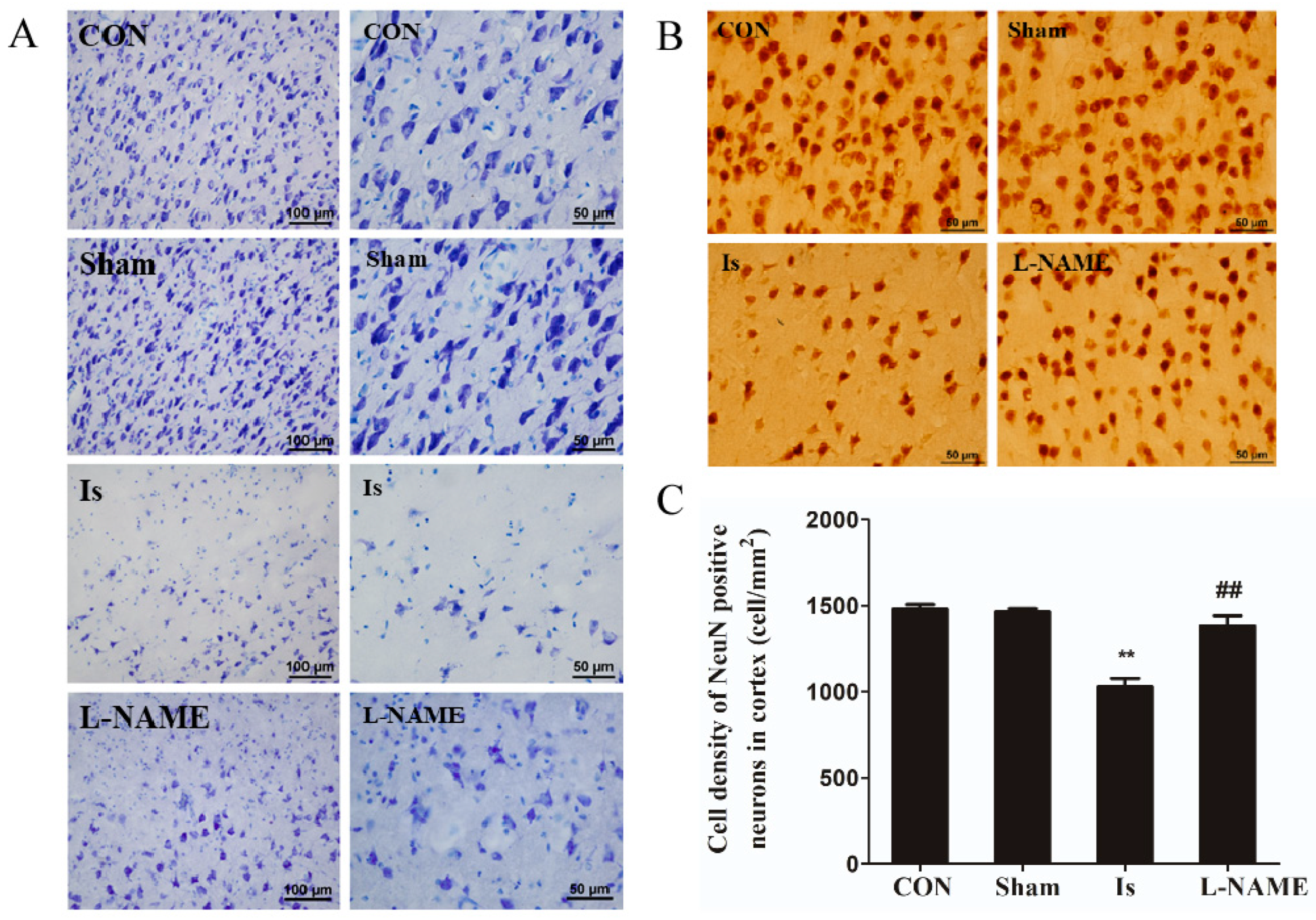
22]. It is well known that ischemia can cause an abnormal behavior in humans as well as in animals, including anxious performance and neuronal damage. In the present research, results from the OFT indicate that ischemic brain injury in rats exhibited insufficient interest and spent more time in the outer zone, suggesting that anxiety-like behaviors were induced following cerebral ischemia. Moreover, Nissl staining and immunohistochemical staining results reveal that in the Is group, a great number of Nissl bodies and NeuN-positive neurons were significantly lost, indicating neuronal damage of a high degree after ischemia induction. However, the detrimental effect caused by cerebral ischemia was obviously restored by pretreatment with L-NAME, since it has a potent neuroprotective activity in vivo. Meanwhile, an OGD-stimulated model of PC12 cells was established to simulate ischemic-like conditions in vitro to explore the potential mechanism by which the signaling pathway was involved after ischemia. Interestingly, Leo significantly ameliorated cell viability in a concentration-dependent manner without obvious cytotoxicity in the OGD-stimulated PC12 cells. A similar effect was also observed by preadministration with L-NAME. The results above therefore show that Leo may have a potential neuroprotective potency in ischemic brain injury.
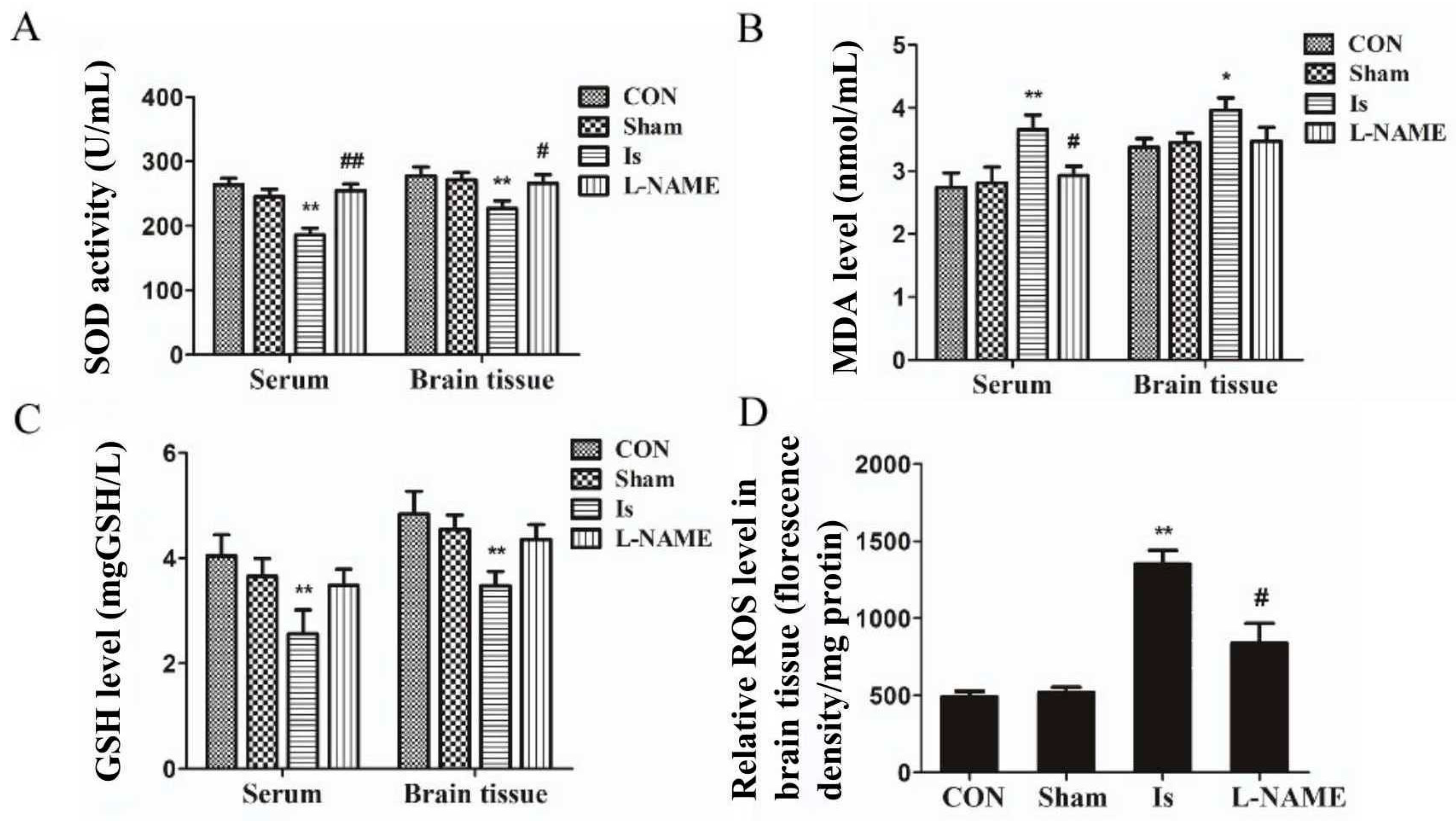
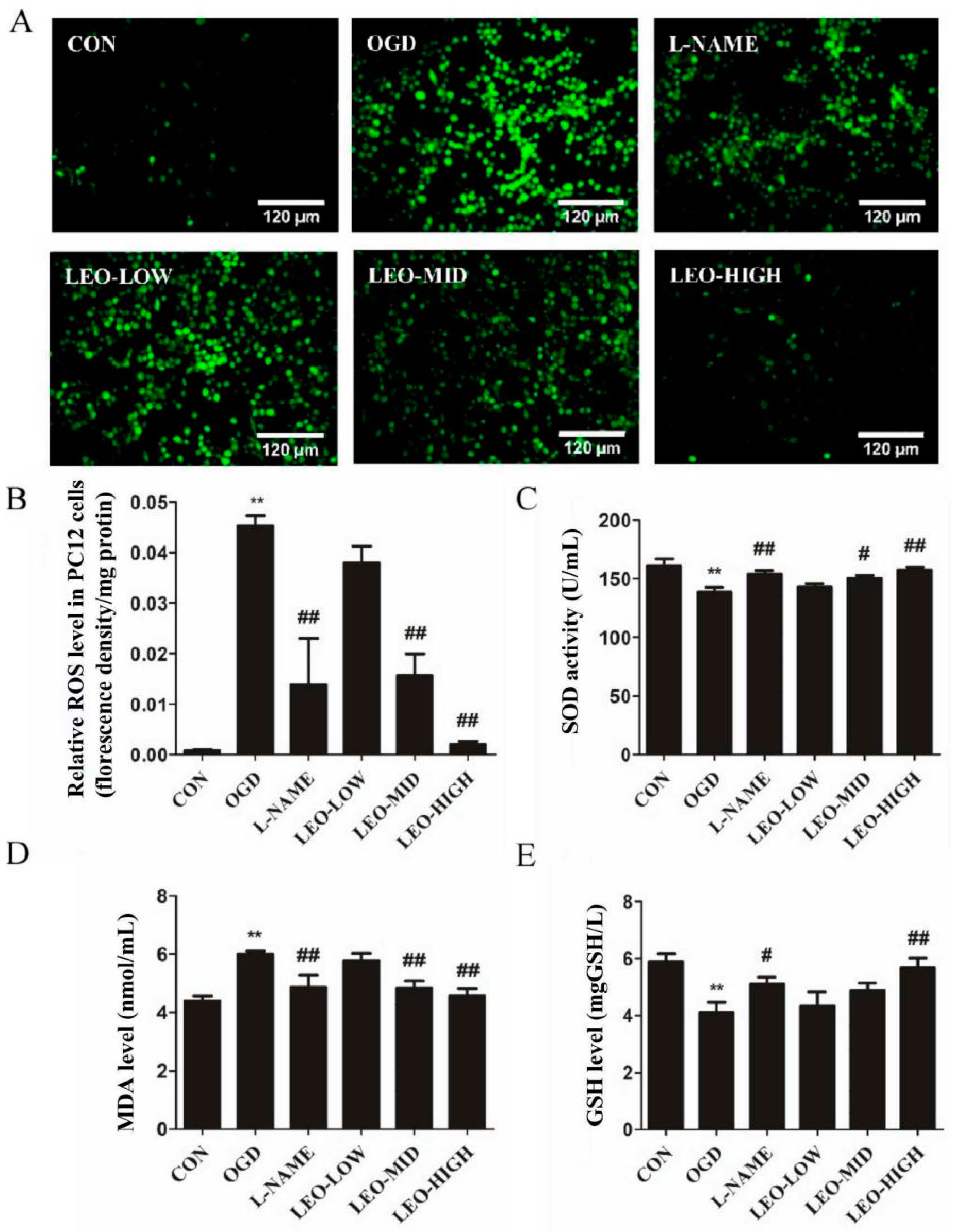
The results from various investigations show that, oxidative stress plays a crucial role in the ischemia/reoxygenation process [
23]. It is widely accepted that SOD is frequently regarded as an antioxidative enzyme in antioxidant defense systems against cellular and tissue damage induced by cytotoxic reactive oxygen species and is usually utilized to evaluate the extent of damage in these processes in vivo [
24]. It is also reported that SOD shows the function to catalyze the reduction in O
− to H
2O
2 [
25]. MDA is a lipid peroxide product of reactive oxygen species attacking protein, DNA and polyunsaturated fatty acid PUFA, and therefore commonly acts as a biomarker of oxidative stress [
26]. Glutathione, an antioxidant, consists of tripeptides of cysteine, glutamic acid and glycine, and the most common form is GSH, which can scavenge free radicals by reducing hydrogen peroxide and lipid peroxide [
27]. Thus, SOD, MDA and GSH play a central role in the oxidative stress response. In our observations, the ischemia-induced rats as well as OGD-stimulated PC12 cells exhibited a decline in SOD and GSH expression and an augment in the MDA contents compared with the normal controls. In contrast, preadministration with L-NAME and Leo showed potent functions against ischemia- and/or OGD-induced changed antioxidant enzyme (SOD) activities and maintained the generation of MDA and GSH at normal levels. It is worthy to note that the protective effect at the highest dose of Leo is higher than L-NAME, a well-accepted NO blocker agent. In the literature, it is well reported that ROS, a group of mediators of mitochondria, DNA repair enzymes and transcription factors, are conducive to oxidative damage [
28,
29,
30]. ROS generation reduction is one of the strategies to prevent neurons from damage after an oxidative stress event. As mentioned in our data, oxidative stress caused excessive ROS production both in rat brain tissue and PC12 cells. However, ROS levels significantly down-regulated in a dose-dependent trend by pretreatment with Leo, especially in the LEO-HIGH group as well as in the L-NAME group. According to the data above, we therefore propose that Leo can exert antioxidant properties to improve oxidative stress-induced injury.
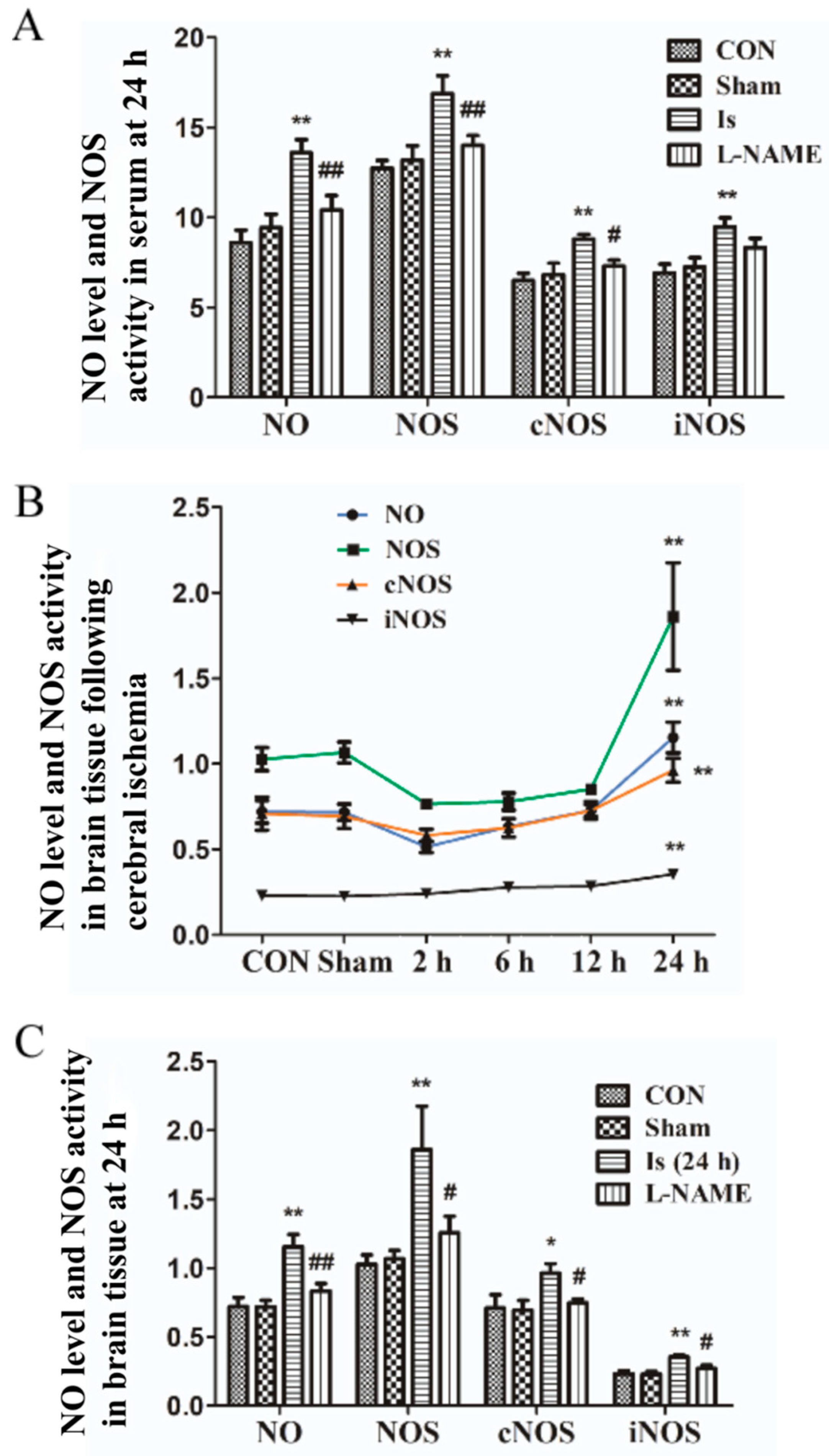
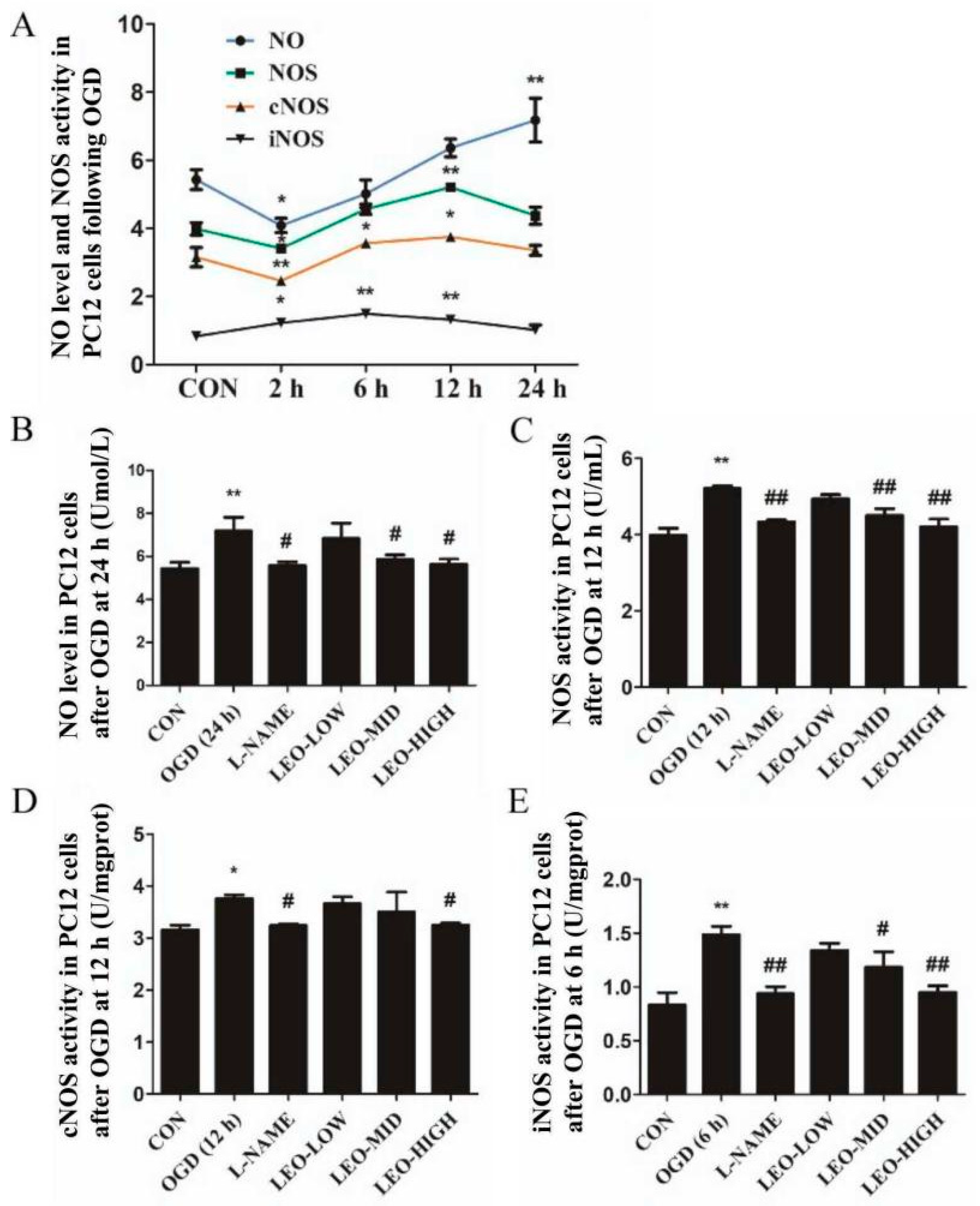
To the best of our knowledge, nitric oxide (NO) liberated from L-arginine in a condition catalyzed by NO synthases (NOS) react with O
2− to generate peroxynitrite (ONOO
−) [
31]. Under oxidative stress situations, the pathway of activation of NOS and generation of NO are involved. Within the heart, the isoform of NOS contains at least two types, including cNOS and iNOS [
32]. nNOS and eNOS, dependent on a rise in tissue calcium concentration for activity, are regarded as constitutively expressed proteins (cNOS). A large number of studies from animal and cell models show that NO is mainly generated by nNOS in the early phase of cerebral ischemia to induce brain insult while, in the late stage, NO is predominantly induced by iNOS. A previous study investigated that 7-NI, an inhibitor of nNOS, alleviated the blood–brain barrier disorder after transient focal ischemic brain injury, and it could also ameliorate neuronal damage as well as reduce the region of brain infarction [
33]. In the recent experiment, the expressions of NOS/iNOS/cNOS were detected in brain tissue and PC12 cells at different time points (2 h, 6 h, 12 h and 24 h) and in serum at 24 h after oxidative stress. Consistent with previous work [
34,
35], the NO/NOS/iNOS/cNOS levels under oxidative stress conditions were eventually increased at 24 h in all groups compared with the controls, although the reduced expression in brain tissue and PC12 cells were determined at the first 2 h. Interestingly, in our observations, the expression of NO/NOS/iNOS/cNOS in serum were markedly higher than that in brain tissue at 24 h after ischemia, and thus in the future, we have to explore the potential mechanism accounting for these events. Additionally, another attractive finding obtained was that the NOS/iNOS/cNOS levels in OGD-stimulated PC12 cells eventually reduced (higher than controls) after they reached their peak activity, which is inconsistent with the data of brain tissue in rats following ischemia. Treatment by L-NAME significantly decreased the NO content and NOS/iNOS/cNOS expressions both in ischemia-induced rats and OGD-stimulated PC12 cells. In in vitro experiments, we show Leo preadministration dramatically down-regulated NO/NOS/iNOS/cNOS contents in a dose-dependent manner at their peak time points, especially in the LEO-HIGH group, which nearly returned to the normal control level. The results above indicate that, in line with the in vivo experiments, Leo possibly showed its antioxidant functions via inhibiting the NO/NOS signaling pathway.
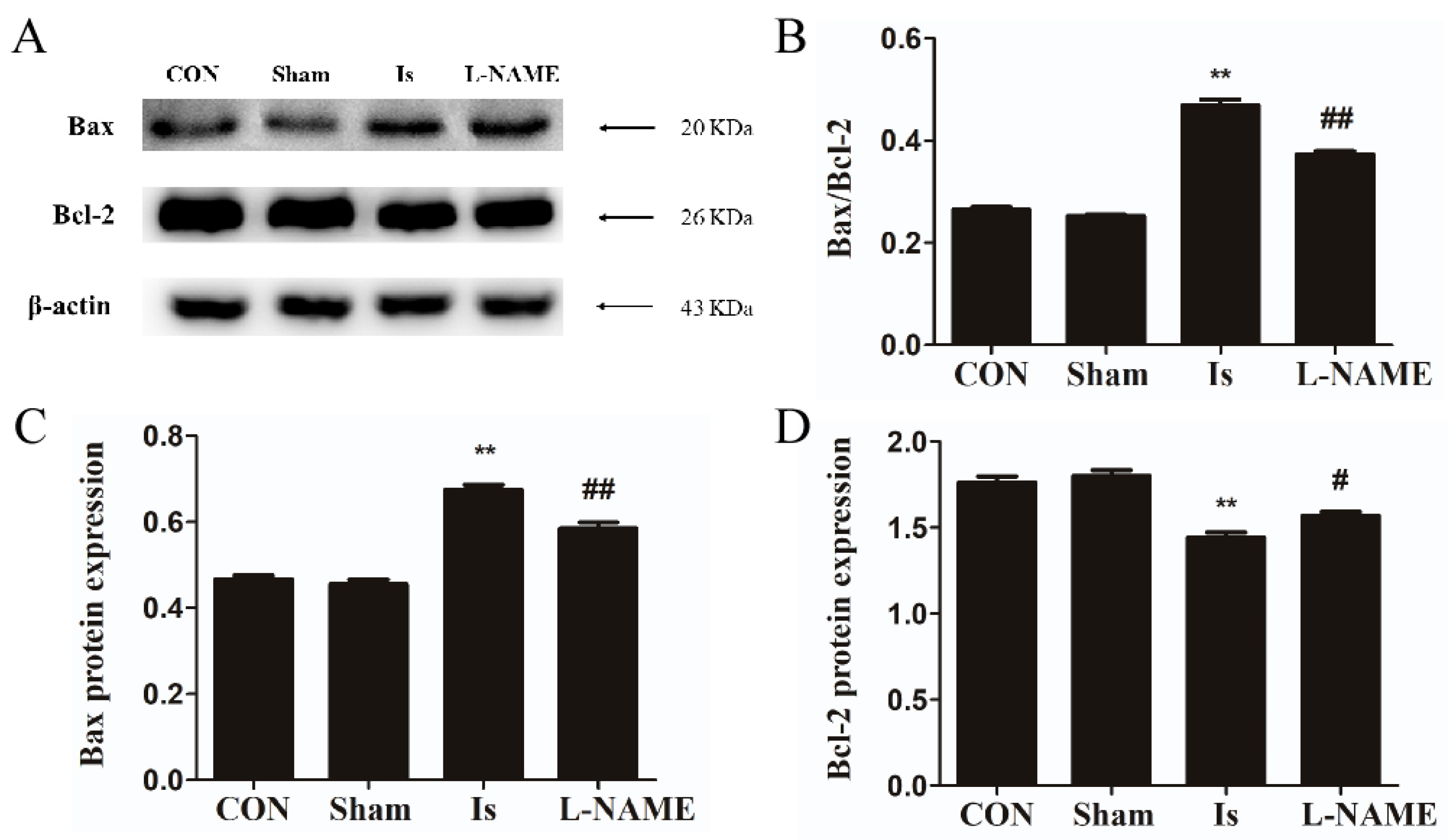
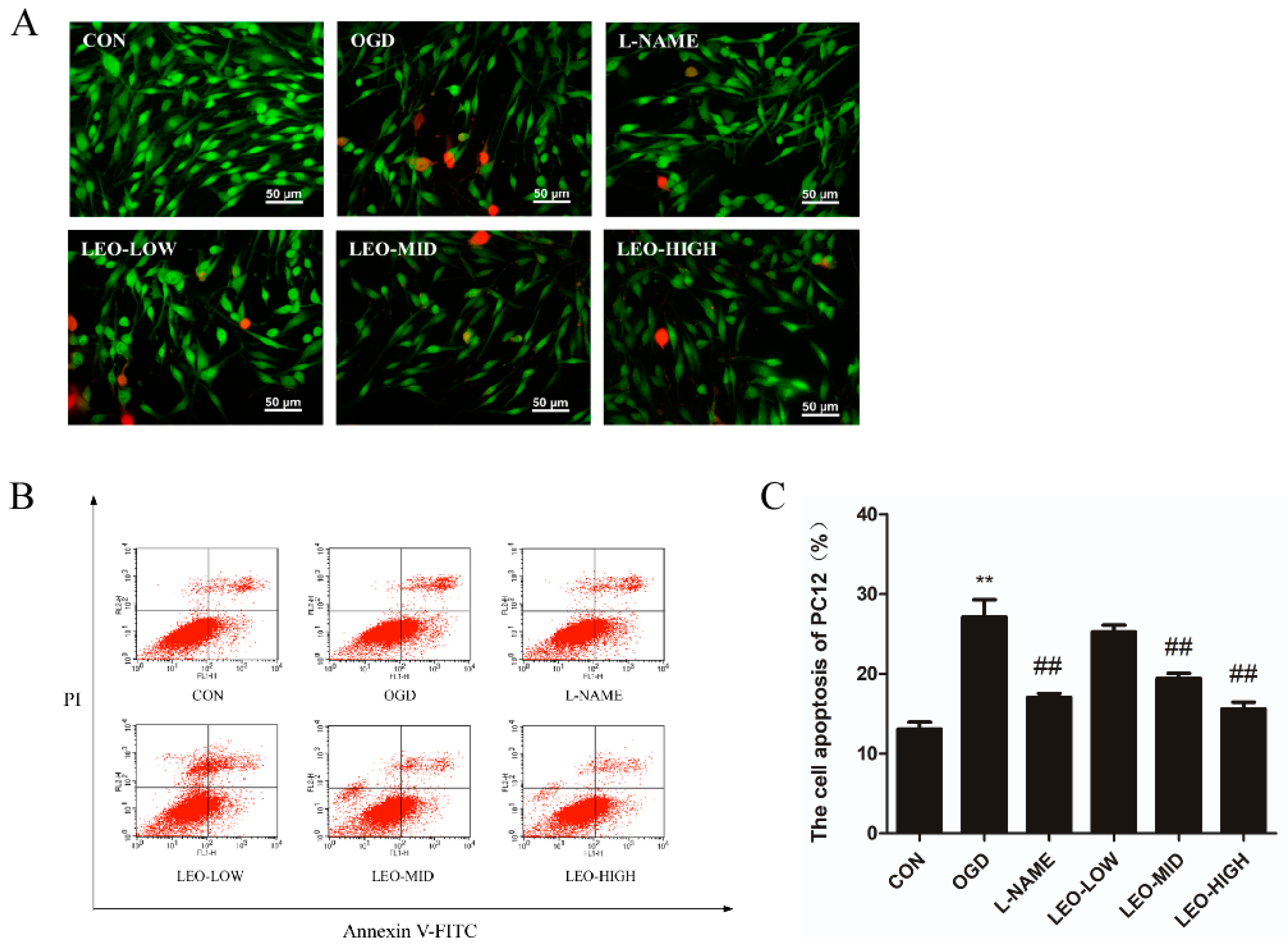
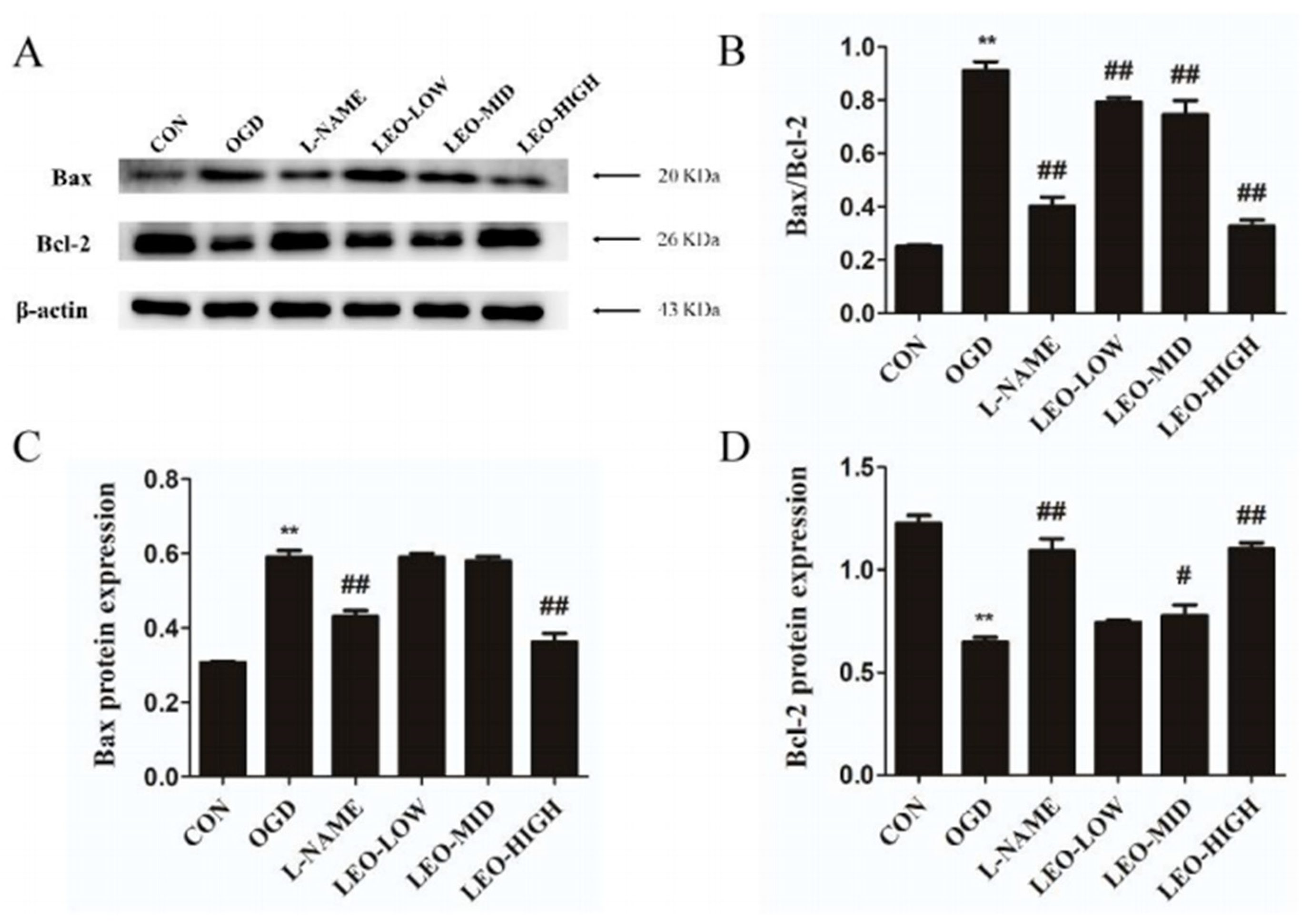
Existing studies have shown that oxidative stress and ROS production are related to the initiation of apoptosis, which contains obvious morphological and biochemical characteristics [
36]. Although the mechanism of apoptosis during ischemia is still not yet fully clear, recent animal studies and clinical observations have shown that the balance between antiapoptotic protein Bcl-2 and proapoptotic protein Bax are associated with the regulation of apoptosis in the brain following ischemia [
37,
38]. Previous research has suggested that Leo alleviated OGD-induced PC12 cell death via targeting the Cx36/CaMKII signaling pathway [
14]. In our present observations, Leo showed alleviative functions on the reduction in cell survival and the enhancement of apoptosis caused by OGD in PC12 cells. In addition, treatment by Leo restored the disturbed expression of Bax and Bcl-2 proteins, especially in the LEO-HIGH group, which was comparable to the control group. Preadministration with L-NAME in OGD-induced PC12 cells showed similar effects when compared with the LEO-HIGH group. The findings from the present study suggest that Leo may act as a neuroprotective agent by modulating apoptosis after oxidative stress.
There are also some limitations in our present study. Firstly, Leo pretreatment was found to regulate the levels of oxidative stress-related factors by inhibiting NO/NOS expression. However, it was not fully clear whether other molecules and pathways were involved. Secondly, inflammatory factors were also associated with cerebral ischemia. We are yet to demonstrate whether Leo administration simultaneously blocked the NO/NOS pathway and alleviated inflammation such as TLR/TRAF/NF-κB pathways to improved cell apoptosis. This will be explored in future experiments. Thirdly, we only used the OGD-induced PC12 cell model in vitro to determine the antioxidant and antiapoptotic functions of Leo, as verified by L-NAME at the same time. Consequently, further research is necessary to estimate the neuroprotective effect of Leo on in vivo ischemia. Moreover, the therapeutic efficacy and time window of Leo in an ischemic animal model also need to be assessed in future observations.
4. Materials and Methods
4.1. Animals and Focal Cerebral Ischemia Model
Adult male Sprague Dawley rats (8 weeks old; weighing 200–230 g; Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) were employed in the present study. The rats were individually housed in cages with a controlled temperature (20 ± 3 °C), 60 ± 5% humidity, 12 h light/dark cycle and ad libitum access to water and food. All experimental protocols were approved by the Committee for the Care and Use of Experimental Animals, China Agricultural University. Every effort was made to minimize the suffering and number of animals used.
The rats were randomly divided into four groups: (1) CON: control; (2) Sham: control treated with Rose Bengal, submitted to surgery without laser illumination; (3) Is: submitted to surgery for cerebral ischemia model; (4) L-NAME: treated with L-NAME, submitted to cerebral ischemia model. Focal cerebral ischemia was induced by a slight modification of photochemical model as per the previous description [
39]. Briefly, a light-sensitive dye Rose Bengal (Sigma, Burlington, MA, USA) was freshly prepared by dissolving in saline (20 mg/mL saline) and kept protected from light until use. Rats were slowly injected through a tail vein with Rose Bengal dye (50 mg/kg body weight) prior to being anaesthetized with inhaled isoflurane (ZS Dichuang Science Technology Development Co., Ltd., Beijing, China) and secured in a stereotaxic frame in which isoflurane was continuously delivered via a nose cone. After a small incision was made on the scalp, a craniotomic window (3 mm × 4 mm) was made over the motor cortex with the center at the coordinate of 2 mm posterior to the bregma and 2 mm lateral to the midline. A cold laser beam of 1.5 mm diameter and 560 nm wavelength (Bjtoptime Science Technology Co., Ltd., Beijing, China) was stereotactically positioned at the middle of the craniotomic window and illuminated for 25 min to induce focal cerebral ischemia. The Sham were subjected to the same procedures except for laser illumination. In the L-NAME group, after the rats were intraperitoneally injected with a dose of 1% L-NAME (10 mg/kg body weight, diluted with saline) for 20 min, they were treated for cerebral ischemia, and other groups were intraperitoneally pretreated with saline instead of L-NAME. All rats were able to survive until they were sacrificed in the present study.
4.2. Behavioral Test
Neurobehavioral test was determined by using the open-field test (OFT) 24 h after the operation. OFT was conducted as previously described [
40]. The rats were transferred to the testing apparatus, which was an illuminated, soundproofed box (100 cm × 100 cm × 50 cm) with black inner walls. The bottom surface of the box was divided into 25 squares (20 cm × 20 cm). The 16 squares adjacent to the walls were defined as outer zone, whereas the other nine squares were defined as inner zone. Using 75% alcohol as disinfectant, the floor surfaces and walls of the apparatus were carefully sterilized and then dried prior to the next animal test. Each rat was placed onto a corner square of the arena and allowed to freely explore the open field for 5 min per trial. The total distance traveled in the OFT and time spent in inner zone and outer zone were quantified by the ANY-maze video tracking system (Stoelting Co., Wood Dale, IL, USA), and general behaviour was evaluated as spending time in inner and outer zone of the testing apparatus.
4.3. Cell Culture and OGD Model
The neuron-like rat pheochromocytoma cell line PC12 cell was obtained from Boster Biological Technology Co., Ltd. (Wuhan, China) and cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco, Waltham, MA, USA) containing 10% fetal bovine serum and 100 U/mL antibiotics (penicillin and streptomycin) at 37 °C in 5% CO
2 in a humidified incubator. To mimic cerebral ischemic-like conditions, a model of OGD-induced PC12 cells was established as previously described [
14]. Briefly, the PC12 cells were incubated in glucose-free DMEM, in a hypoxia chamber (Jinfeng Science and Technology Co., Ltd., Beijing, China) containing 95% N
2 and 5% CO
2. After incubation for 2 h, the cells were moved to normal cultured conditions (glucose-free DMEM was replaced with DMEM containing 4500 mg/L of glucose) and cultivated for a short period for the subsequent experiments. According to the different evaluation items, the appropriate culture time for PC12 cells after OGD induction was chosen. The PC12 cells were separated into 6 groups: (1) CON: control group with PC12 cells cultured in normal condition; (2) OGD: submitted to OGD model; (3) L-NAME: treated with L-NAME (1 mmol/L), submitted to OGD model; (4) LEO-LOW: treated with the low dose of Leo (50 μg/mL), submitted to OGD model; (5) LEO-MID: treated with the middle dose of Leo (100 μg/mL), submitted to OGD model; (6) LEO-HIGH: treated with the high dose of Leo (200 μg/mL), submitted to OGD model.
4.4. Sample Collection and Treatment
In in vivo experiments, a methodology for collection and processing of blood and brain tissue samples following behavioral tests was used as previously described [
41]. Two-, six-, twelve- and twenty-four hours later, six rats from each group were humanely sacrificed under isoflurane anesthesia. The blood samples were collected and centrifuged at 2000×
g for 10 min at 4 °C, and then the serum samples were stored at −20 °C for measurement of oxidative stress index and NO/NOS production. After euthanizing the rats, tissues of the cerebral cortex were dissected, frozen in liquid nitrogen and stored at −80 °C for analysis of oxidative stress and NO/NOS signaling, as well as for Western blot analysis. The other six rats in each group were also deeply anesthetized with chloral hydrate, perfused transcardially with 200 mL of 5 mmol·L
−1 sodium phosphate (pH 7.4)-buffered 0.9% (
w/
v) saline (PBS), followed by 300 mL of 4% (
w/
v) formaldehyde in 0.1 mol·L
−1 sodium phosphate buffer (pH 7.4). The brains were removed to cut into several blocks and postfixed with the same fixative for one day at 4 °C. After cryoprotection with 30% (
w/
v) sucrose in PBS, the blocks were sectioned into 30-µm-thick coronal slices on a freezing microtome, which were placed in PBS for subsequent histochemistry and immunohistochemistry. In in vitro experiments, the method of sample collection was the same as that previously described [
14].
4.5. Oxidation and Antioxidant Test
The degree of lipid peroxidation in the serum and brain tissue of rats and PC12 cells was determined by detecting the ROS and MDA levels. The antioxidant capacity of the serum, brain tissue and cell was evaluated by analyzing the SOD activity and GSH content. The fluorescence value of ROS production was measured by chemiluminescence method, using ROS detection kit (E004, NJJCBio Inc., Nanjing, China). MDA content was measured by the thiobarbituric acid method, using MDA detection kit (A003-2, NJJCBio Inc.). SOD activity was determined by the xanthine oxidase method, using SOD detection kit (A001-1, NJJCBio Inc.). GSH content was quantified by the colorimetric analysis, using GSH detection kit (A006-1, NJJCBio Inc.). All assays were conducted according to manufacturer instructions.
4.6. Measurement of NO and NOS Content
NO production in serum, brain tissue and cell were detected, respectively, by using the nitrate reductase method and a NO assay kit (A012, NJJCBio Inc.), according to manufacturer instructions. The level was expressed as µmol/g protein. The activities of total NOS, cNOS and iNOS were measured using NOS assay kit (A014-1, NJJCBio Inc.) in accordance with manufacturer instructions, and the results were expressed as U/mg protein.
4.7. Histochemical and Immunohistochemical Procedure
Nissl staining was conducted to examine the neuronal damage or survival in cerebral cortex. After rinsing with 5 mM PBS, the frozen sections were mounted on gelatin-coated glass slides, air-dried, then dehydrated with a series of 50, 70, 95 and 100% alcohol solutions and stored in 100% alcohol overnight. After defatting by xylene for 24 h, the slides were rehydrated and rinsed in distilled water, subsequently stained using 0.1% cresyl violet solution for 30 min, then differentiated in 95% ethyl alcohol for 2–3 min, dehydrated again in 100% alcohol and finally cleared in xylene.
Immunohistochemical staining was employed to assess the change in morphology of cerebral cortex in rats. Frozen sections were rinsed in PBS and then incubated in 1% hydrogen peroxide solution for 30 min to quench the endogenous peroxidase reactivity. All the following incubations described hereafter were performed at room temperature and followed by a rinse with 5 mM PBS at pH 7.4 containing 0.3% Triton X-100 (PBS-X). The sections were incubated overnight in a humidified chamber with a mouse monoclonal antibody against NeuN (1:500, Millipore, Billerica, MA, USA) in PBS-X containing 0.12% lambda-carrageenan, 0.02% sodium azide and 1% donkey serum (PBS-XCD). After a rinse with PBS-X, the sections were incubated for 2 h with biotinylated donkey anti-mouse IgG (1:100; Jackson, West Grove, PA, USA) in PBS-XCD and then for 1 h with ABC (1:50; Vector, Torrance, CA, USA). After a rinse with PBS-X, the bound peroxidase was finally developed as brown by reaction for 5 min with 0.02% diaminobenzidine-4HCl (DAB) (Sigma, Burlington, MA, USA) and 0.0001% H2O2 in 50 mM Tris-HCl (pH 7.6). All the stained sections were mounted onto the gelatinized glass slides, dried in an ethanol series, cleared in xylene and finally, coverslipped.
The sections were observed under a microscope (Ni-U, Nikon, Tokyo, Japan), and the immunoreactivity intensities of NeuN were determined using Image-Pro plus 6.0 (Media Cybernetics, Bethesda, MD, USA). The cytoarchitectonic areas of cerebral cortex were determined using the rat brain atlas [
42]. Three slices were randomly selected for each group (five regions per slice) to count the neuronal numbers.
4.8. Western Blot Analysis
Protein expressions of Bax/Bcl-2 in cerebral cortex and PC12 cells were assessed using Western blot analysis as previously described [
14,
43]. Briefly, total protein was extracted using a total protein extraction kit (Biochain, Hayward, CA, USA) and quantified using a bicinchoninic acid (BCA) protein assay kit (78510, Pierce, Rockford, IL, USA). Following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 8–12%), equivalent amounts of proteins were transferred onto polyvinyl difluoridine (PVDF) membranes (Millipore, Billerica, MA, USA) at 250 mA for 90 min. After rinsing 3 times with TBST, the polyvinylidene fluoride membranes were placed in a 5% nonfat milk blocking solution on a shaking table at room temperature for 2 h. Subsequently, the membranes were incubated overnight at 4 °C with one of the following primary antibodies: rabbit polyclonal antibody against Bax (1:1000; Sigma, Burlington, MA, USA), rabbit polyclonal antibody against Bcl-2 (1:1000; Sigma, USA) or mouse monoclonal antibody against β-actin (1:1000; 50201; Kemei Borui Science and Technology Co., Ltd., Beijing, China). The membranes were then incubated for 1 h at 37 °C with horseradish peroxidase-conjugated secondary antibodies (HRP-Goat-anti-Rabbit IgG, 1:5000, Kemei Borui Science and Technology Co., Ltd., Beijing, China; HRP-Goat-anti-mouse IgG, 1:10,000, Earth-OX, Millbrae, CA, USA, respectively), and proteins were detected using enhanced chemiluminescence (ECL) substrate (Millipore, Billerica, MA, USA) according to manufacturer instructions. Finally, protein bands were visualized using a chemiluminescence system (5200, Tanon Science & Technology Co., Ltd., Shanghai, China).
4.9. AO/EB Staining
Cell apoptosis was detected by double staining with acridine orange (AO) and ethidium bromide (EB). The PC12 cells were cultured on glass coverslips as previously described and then washed with PBS. A mixture of 100 μg/mL AO and 100 μg/mL EB (AO/EB, Sigma, St. Louis, MO, USA) was added to each coverslip, and each coverslip was covered on a slide and kept in a dark place for 15 min, according to manufacturer instructions. The morphology of the apoptotic cells was observed under a fluorescent microscope (Ni-U, Nikon, Tokyo, Japan).
4.10. Flow Cytometry Analysis
Cell apoptotic rate was evaluated using an Annexin V-FITC/PI apoptosis detection kit (KeyGen BioTech Co., Ltd., Nanjing, China). After trypsinization, single-cell suspensions were extracted and washed with PBS. The cells were resuspended in 500 µL of binding buffer and stained with Annexin V-FITC and PI for 15 min according to the manufacturer instructions. The samples were then analyzed using a flow cytometer (BD, Franklin Lakes, NJ, USA) with a maximal excitation wavelength at 488 nm and emission at 530 nm.
4.11. Statistics
Statistically, no less than six rats in each group were required for statistical significance, and each in vitro experiment was repeated three times. The data were evaluated using one-way ANOVA, followed by Tukey’s post hoc test with SPSS 21.0 (IBM Inc., Chicago, IL, USA). Before analysis, the normal distribution of all data and homogeneity of variances were verified. All the data were expressed as mean ± standard error mean (SEM). A p value < 0.05 was considered statistically significant.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








