
Metabolic and Cellular Compartments of Acetyl-CoA in the Healthy and Diseased Brain
 , and
, and
Abstract
:1. Introduction
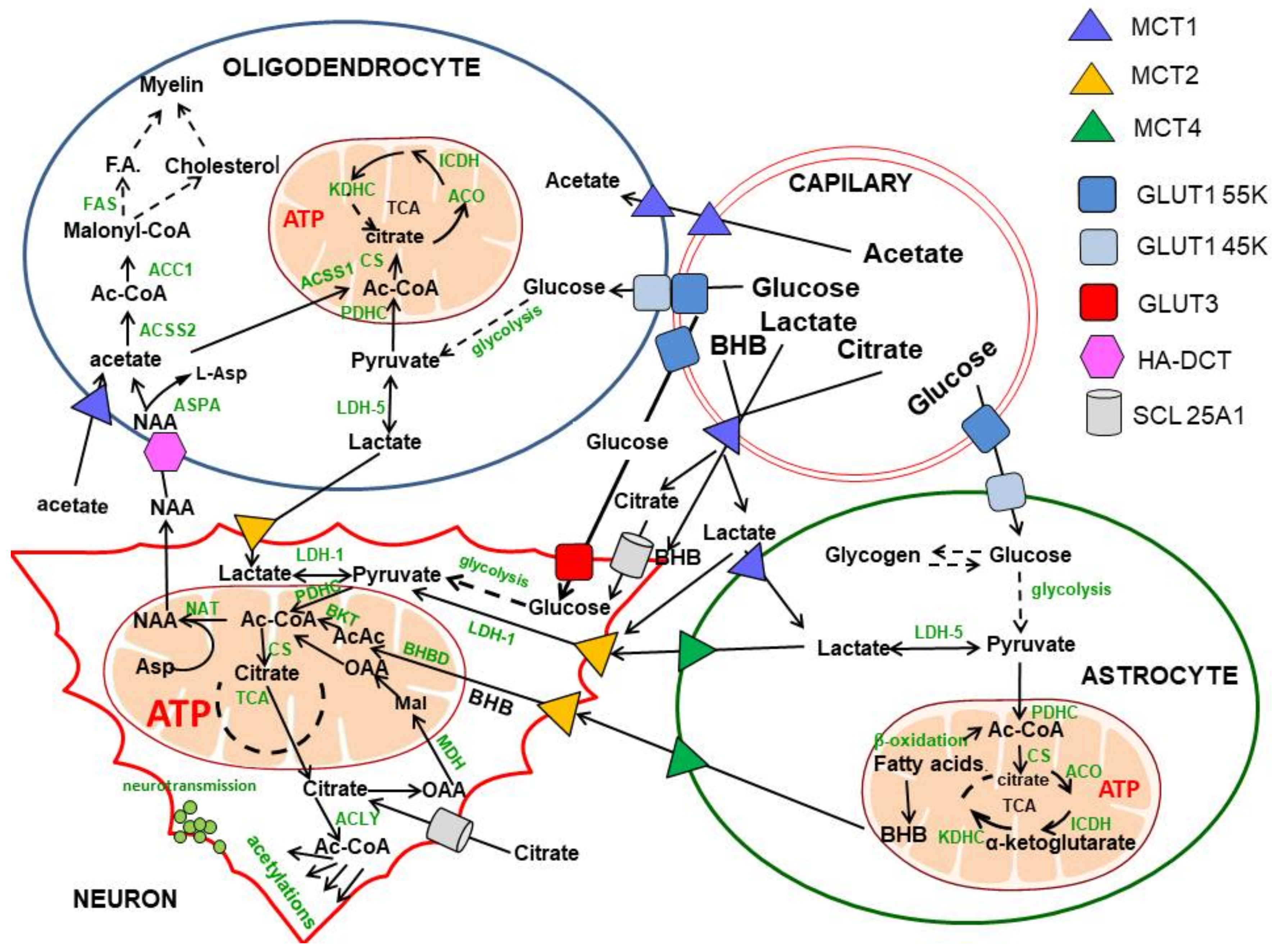
2. Glucose and Lactate—The Key Precursors of Brain Acetyl-CoA in Health and Disease
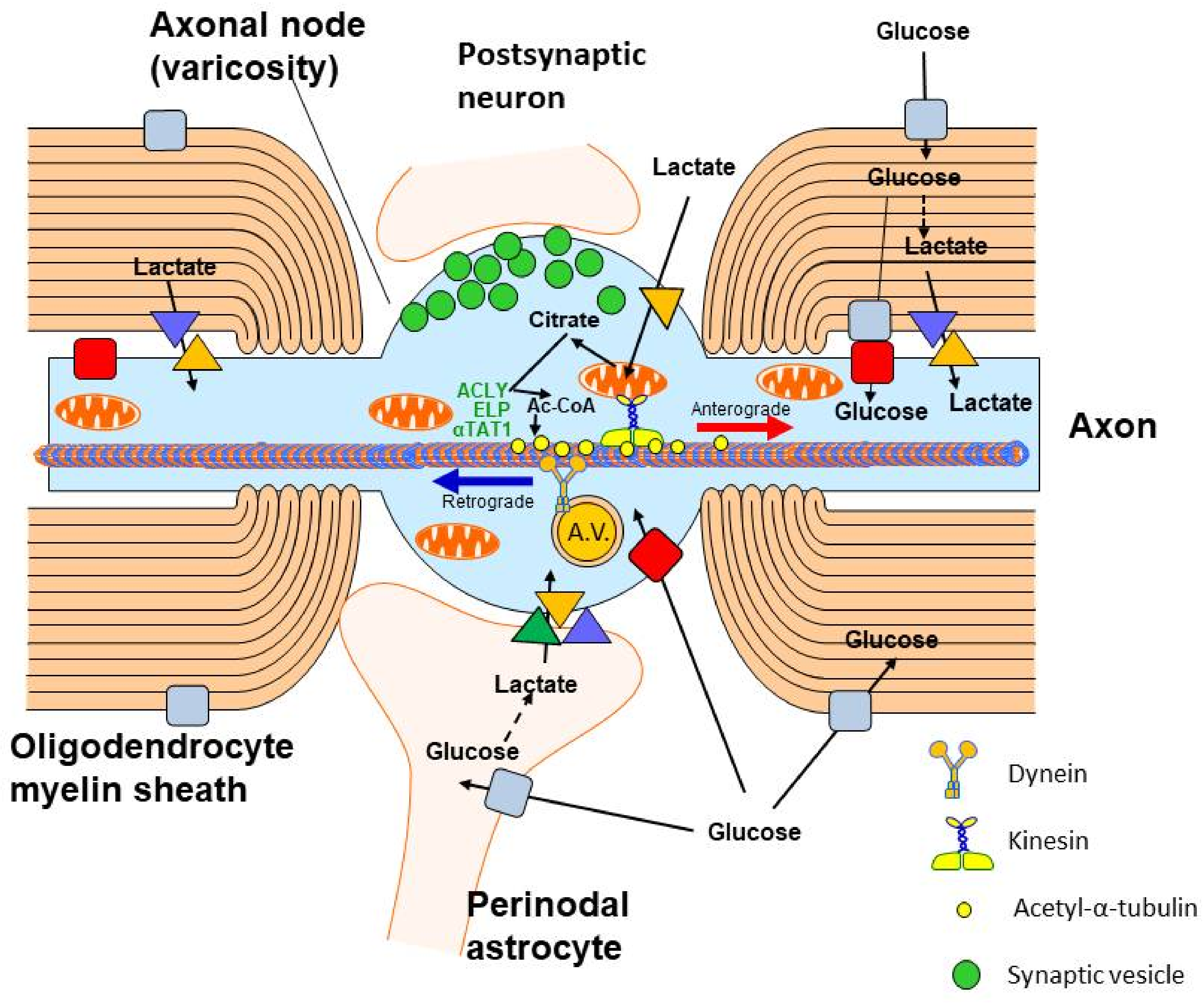
3. Origin and Metabolic Role of Axonal Acetyl-CoA
4. Glucose and Pyruvate-Derived Acetyl-CoA Metabolism in Cholinergic Neurons
4.1. Acetyl-CoA Compartments in Cholinergic and Noncholinergic Neurons
4.2. Zinc and Cholinergic Acetyl-CoA Metabolism
4.3. Thiamine Deficiency and Cholinergic Acetyl-CoA Metabolism
4.4. Nerve Growth Factor (NGF) and Acetyl-CoA in Cholinergic Cells
4.5. Citrate and ACLY Key Players in Cholinergic Acetyl-CoA Metabolism
5. Compartmentation of Brain N-Acetylaspartate and Acetyl-CoA Metabolism
6. The Contribution of Acetate to Brain Acetyl-CoA Metabolism
7. Fatty Acids and Ketone Bodies in Brain Acetyl-CoA Metabolism
8. Endoplasmic Reticulum and Nuclear Pools of Acetyl-CoA
8.1. Endoplasmic Reticulum Acetyl-CoA
8.2. Nuclear Acetyl-CoA
9. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | Signal/Conditions | Acetyl-CoA Level/Relative Change | Reference/Comments |
|---|---|---|---|
| Rat brain | Hypoxia in vivo | Whole tissue (nmol/g tissue) | [17] |
| Control | 5.4 | ||
| Hypoxia 100N2 90 s | 6.7 ** | ||
| Rat brain | Brain region (whole tissue) | Whole tissue (nmol/g tissue) | [224] |
| Thalamus | 9.1 | ||
| Hippocampus | 7.1 | ||
| Cortex | 6.2 | ||
| Cerebellum | 6.1 | ||
| Rat brain slices | 60 min. incubation 31.2 mM K+ | Brain slices (nmol/g tissue) | [18] |
| Control | 5.04 | ||
| +3-bromorypuvate 0.25 mM | 2.45 | ||
| Rat brain synaptosomes | 30 min. incubation 30 mM K+ | Synaptosomes Mitochondria Cytoplasm | [19] |
| (pmol/mg protein) | |||
| Control | 12.3 46.8 | ||
| +3-bromopyruvate 0.25 mM | 0 7.4 ** | ||
| Healthy adult rat brain synaptosomes | Healthy control | Whole synaptosomes (pmol/mg protein) | [143] different from pyruvate alone, † p < 0.05 |
| Substrate used (mM) | |||
| Pyr. 2.5 BHB 2.5 Pyr. + BHB | |||
| 24.3 7.1 22.8 | |||
| STZ diabetes 10 d | 31.3 * 10.5 * 29.4 * | ||
| Streptozotocin-diabetic rat brain synaptosomes | STZ diabetes 10 d + Insulin 5 d | 30.6 * 10.0 *† 35.6 *† | |
| Cholinergic neuroblastoma cell culture: nondifferentiated (NC) and differentiated (DC, db-cAMP 1 mM + retinoic acid (RA) 0.001 mM 48 h) | Control | Cellular compartment levels (pmol/mg protein) | [98] from respective NC, † p < 0.05, †† p < 0.01 |
| Mitochondria Cytoplasm | |||
| NC DC NC DC | |||
| 71 22 † 13 50 † | |||
| +NGF 100 ng/mL 24 h | 55 42 ** 71 ** 29 *† | ||
| Native SN56TrkA-/p75NTR+ | Control | 95 23 † 13 49 † | |
| Tg T17 SN56TrkA+/p75NTR+ | +NGF 100 ng/mL 24 h | 59 * 39 * 129 ** 48 † | |
| Cholinergic neuroblastoma cell culture Tg T17 SN56TrkA+/p75NTR+ NC, and DC | 24 h cell culture with: | Relative change against no addition control (%) | [97,126] Different from respective NC, † p < 0.05, †† p < 0.01; from Aβ (25–35) alone, ‡ p < 0.05, ‡‡ p < 0.01 |
| Mitochondria Cytoplasm | |||
| NC DC NC DC | |||
| Aβ25-35 0.001 mM | 10 −23 −17 −58 ** | ||
| Acetyl-carnitine 0.1 mM | +39 ** 0 † 0 +54 **†† | ||
| Aβ + acetyl-carnitine | +22 ‡‡ 0 0 0 ‡‡ | ||
| ILβ 10 ng/mL | −11 −18 +38 * −42 *†† | ||
| Aβ + ILβ | −18 −1 +1 +3 ‡‡ | ||
| Cholinergic neuroblastoma cell culture | ChAT (nmol/min/mg protein) NC DC | Whole cells (pmol/mg protein) NC DC | [100] Different from respective native SN56, † p < 0.05, †† p < 0.01 |
| Native SN56 TrkA-/p75NTR+ | 0.22 0.79 *** | 31.2 21.9 *** | |
| Tg T17 TrkA+/p75NTR+ | 0.19 0.47 *** | 39.7 † 26.8 ***† | |
| Tg ChAT2 TrkA-/p75NTR+ | 3.80 ††† 6.80 ***††† | 15.5 †† 11.2 *** †† | |
| Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR + DC | 24 h cell culture with: Control | Mitochondria Cytoplasm (pmol/mg protein) | [40] † different from ZnCl2 0.10 mmol/L |
| 11.8 20.9 | |||
| ZnCl2 0.10 mM | 9.3 19.6 | ||
| ZnCl2 0.15 mM | 11.4 13.5 *† | ||
| Cholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR + NC and DC | 30 min incubation (protein free medium) with: Zn 0.1 mM | Relative change vs. no Zn control (%) Mitochondria Cytoplasm NC DC NC DC −5 −35 ** −100 ** −80 ** | [110] |
| Subcutaneous pyrithiamine (PT) 0.025 mg/kg b.w./day and thiamine free diet 14 d Rat forebrain synaptosomes | PT synaptosomes vs. no PT control | Forebrain synaptosomesRelative change against no PT control (%) | [119] |
| Mitochondria Cytoplasm | |||
| No Ca Ca 1.0mM no Ca Ca1.0mM | |||
| −53 *** −35 *** −43 *** −24 * | |||
| Subcutaneous PT 0.025 mg/kg b.w./day and thiamine-free diet 14 d. Rat forebrain whole mitochondria | PT whole forebrain mitochondria vs. no PT control | Forebrain whole mitochondria Relative change vs. no PT control (%) | [120] |
| No Ca Ca 0.01 mM ADP/HX | |||
| −62 *** −62 *** −52 *** | |||
| Cholinergic neuroblastoma cell culture Native SN56 TrkA-/p75NTR+ | Thiamine-free culture medium 48 h +Amprolium 2 mM | Relative change vs. no amprolium NC control (%) | [121] Amprolium suppressed TPP level—28% vs. control |
| Mitochondria Cytoplasm. | |||
| NC DC NC DC | |||
| −43 −57 −58 *** −50 ** | |||
| Endoplasmic reticulum from WT and AT 1-1S113R/+ mice | Mutation AT 1-1S113R/+ | Acetyl-CoA transport (pmol/mg/5 min.) | [206] |
| WT 370 | |||
| AT-1S113R/+ 142 *** | |||
| N9 microglioma cells culture | 24 h culture with: LPS 0.01 µg/mL | Relative change against respective no addition control (%) | [38] ‡‡ different from SNP 0.4 mM, p < 0.01 ††† different from N9 cells, p < 0.001 |
| Whole cells | |||
| N9 SN56 | |||
| −23 * +4 | |||
| SynchronizedCholinergic neuroblastoma cells Native SN56 TrkA-/p75NTR+ DC | SNP 0.4 mM | −3 −38 * | |
| LPS + SNP | −6 92 ***†††‡‡ | ||
| WT 14–16 mos mouse brain AβPP-Tg 2576 14-16 m mouse brain | Accumulation about 0.6 μM Aβ1-42 in Tg brain | Relative change vs. WT control (%) Mitochondria Cytoplasm ** | [62] Acetyl-CoA—control WT mice Synapt. mitoch. 39 pmol/mg prot. Synapt. cytopl. 90 pmol/mg prot. Whole brain mitoch. 45 pmol/mg. |
| Forebrain synaptosomes | −44 ** −34 | ||
| Forebrain whole mitochondria | +5 - | ||
| WT mouse brain AT1 Tg mouse brain (overexpression) | Hippocampus Isolated adult neurons H4 neuroglioma | AT1 Tg vs. WT Relative difference (%) Cytoplasm | [207] |
| −41 * | |||
| −45 * | |||
| −43 * | |||
| WT 9 d postnatal mouse brain | 24 h post hypoxia/ischemia | Relative change vs. control (%) Mitochondrial fraction Vehicle-treated DCA-treated +6 +27 * | [34] |
| Cell line cultures WT SN56 TrkA-/p75NTR NC | Intracellular Zn accumulation of 5 nmol/mg protein at extracellular Zn in culture medium: 0.125 mM | Relative change vs. no Zn control (%) SN56 NC −54 *** | [11] † different from NC, p < 0.05 |
| DC | 0.110 mM | SN56 DC −48 ***† | |
| SHSY5Y dopaminergic neurons | 0.150 mM | SHSY5Y −31 * | |
| C6 astroglioma | 0.200 mM | C6 −44 ** | |
| 3XTg AD 16.5 mos mouse brain | 8 mos ketone ester-feeding | Relative change vs. non-ketotic control (%) Hippocampus +79 * | [134] Acetyl-CoA no ketone control: 17 μmol/g tissue |
| Mouse BV2 microglial cells culture | Dimethylsulfoxide-induced 6 h hypoxia | Relative change vs. no hypoxia control (%) +79 ** | [30] |
| Hypoxia + Lonidamine 0.05 mM | −58 * | ||
| Hypoxia + 3-Bromopuryvate | −42 * | ||
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC | 30 min incubation (protein-free medium) with: Control Nifedipine 0.01 mM GVIA 0.0005 mM MVIIC 0.0002 mM | Whole cells (pmol/mg protein) No Zn Zn 0.15 mM 30.5 13.8 * 30.7 29.2 † 28.8 21.6 *† 28.1 20.5 *† | [105] Compounds used here are inhibitors of different types of calcium channels. * p < 0.01 vs. no Zn control; † < 0.01 vs. 0.15 mM Zn. |
| SAMP8 mice brain cortex | 13 mos vs. 9 mos change No treatment Fed with CMS121 4 mos Fed with J147 4 mos | Relative change 13 mos vs. 9 mos | [175] CMS121, J147 are acetyl-CoA carboxylase inhibitors. |
| (%) | |||
| −41 **** | |||
| −12 | |||
| −6 | |||
HT22 hippocampal neuronal cell culture Primary E21 mice neuronal culture | 24 h culture with: | Relative change vs. no addition control (%) | [10,175] Compounds used here inhibit acetyl-CoA carboxylase by different mechanisms. |
| +ACC1 siRNA | +114 *** | ||
| +TOFA 0.01 mM | +178 *** | ||
| +CMS 121 0.001 mM | +140 *** | ||
| +J147 0.001 mM | +100 ** | ||
| +CAD031 0.001 mM | +177 *** | ||
| +CMS 121 0.001 mM | +57 *** | ||
| +J147 0.001 mM | +29 | ||
| +CAD031 0.001 mM | +108 *** | ||
| Brain-specific pdha1flox8/wt deficient mice (PDHD) | PDHD | Relative change vs. control (%) −12 | [54] |
| 3xTgAD mice WT control mice | Ageing—2, 11, 21 mos hippocampus whole tissue Control | 2 mos 11 mos 21 mos | [179] Different from the corresponding 2 mos mice, † p < 0.05, ††† p < 0.001 |
| (Arbitrary units) | |||
| Male | |||
| 0.5 1.1† 1.3 ††† | |||
| 3XTgAD | 1.2 * 1.6 * 2.6 **††† | ||
| Female | |||
| Control | 0.5 0.8 1.0 † | ||
| 3XTgAD | 0.5 1.3 *† 1.2 † | ||
| Rat permanent middle cerebral artery occlusion model of brain stroke (pMCAO) | Shengui Shanseng San (SSS) extraction feeding per os 3 d before and 7 d after pMCAO | Relative change vs. sham control | [35] Absolute sham control value of infarct-corresponding control region equal to 24.4 µmol/µL tissue is 106 times higher than those reported elsewhere. |
| In brain infarcted region (%) pMCAO −80 *** | |||
| Low dose SSS + pMCAO −52 *** | |||
| Middle dose SSS +pMCAO −44 *** | |||
| High dose SSS +pMCAO −4 | |||
| Closed-head impact acceleration model of mild or severe traumatic rat brain injury (mTBI/sTBI) | mTBI/sTBI | Relative change vs. control (%) Whole brain extracts | [58] Absolute control value about 39 nmol/g wet weight is about 10 times higher than values reported elsewhere. Different from the corresponding of post mTBI time, † p < 0.005 |
| Post mTBI 6 h −13 | |||
| 24 h −22 | |||
| 48 h −24 | |||
| 120 h −13 | |||
| Post sTBI 6 h −34 * | |||
| 24 h −56 *† | |||
| 48 h −47 *† | |||
| 120 h −58 *† | |||
| HEK293 cell culture | DIP2A overexpression | Relative change DIP2A vs. no insert control (%) +120 * | [208] |
| Traumatic brain injury/control cortical impact rat brain (TBI/CCI) | TBI/CCI | Peri-contusional brain cortex acetyl-CoA (ng/mg protein) Early immediate 3 h i.v. administration | [41] Absolute control value is about 34.5 pmol/mg protein. |
| Sham (saline 0.9%) 27 | |||
| Control 38 | |||
| Glucose 30% 57 * | |||
| Lactate 100 mM 29 | |||
| BHB 2M 52 * | |||
| Late (6 h post impact) 3 h i.v. administration | |||
| Glucose 30% 38 | |||
| Lactate 100 mM 21 | |||
| BHB 2M 38 | |||
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC | 30 min incubation (protein-free medium) with: | Relative change vs. control (%) Mitochondria Cytoplasm | [160] Mecamylamine is a nonselective antagonist of nicotinic receptors. 2APB is inhibitor of IP3 receptors and TRP channels. |
| Mecamylamine 0.002 mM | −36 ** +7 | ||
| Nifedipine 0.01 mM | 0 +28 | ||
| 2-Aminoethoxydiphenyl borate (2-APB) 0.05 mM | +43 -56 ** | ||
| Zn 0.15 mM | −64 *** −39 ** | ||
| Human fibroblastoma HT1080 cell line ACLY WT ACLY-WT ACLY KO | 4 h incubation with or without 20 mM acetate ACLY-WT ACLY-KO | Relative change vs. WT-acetate control (%) | [215] Absolute control value for ACLY-WT is 6.1 μM (normalised to internal standard) |
| acetate 20 mM No acetate | |||
| 0 −14 | |||
| −67 *** −95 *** | |||
| E18 C57BL/6J mice model of AD | 24 h culture with Aβ1-42 10µM | Relative change vs. control (%) | [183] Absolute control values for neurons and microglia are 0.45 and 0.75 μM, respectively |
| Neurons Microglia ** | |||
| 0 −31 | |||
| 5XFAD 9 mos mouse brain | 5XFAD control 5XFAD + efavirenz 0.1 mg/kg b.w./d in drinking water from 3 to 9 mos of life | Whole brain Mitochondria (pmols/mg protein) | [225] Efavirenz is an inhibitor of reverse transcriptase. Acetyl-CoA control levels reported here are about 10 times higher than reported elsewhere. |
| 145 87 | |||
| 351 *** 352*** | |||
| B6SJ/L 9 months mouse brain | B6SJ/L control | 361 157 | |
| Tg Cyp46a1+/+ | Tg Cyp46a1+/+ | 257 *** 128 | |
| Tg Cyp46a1−/− | Tg Cyp46a1−/− | 143 *** 100 *** | |
| Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ NC and DC | 24 h culture in thiamine-free medium with: +Zn 0.1 mM +Amprolium 5 mmol/L +Zn +Amprolium | Relative change vs. no Zn, and amprolium control (%) Mitochondria | [45] Absolute control acetyl-CoA levels in NC and DC mitochondria were: 11.6 and 11.9 pmol/mg protein, respectively. Absolute control acetyl-CoA levels in NC and DC cytoplasm were: 13.6 and 11.7 pmol/mg protein, respectively. †† different from NC/DC Zn, p < 0.0.1 different from NC/DC amprolium, ‡ p < 0.05, ‡‡ p < 0.01 |
| NC DC | |||
| −5 −23 ** | |||
| −5 −16 * | |||
| −45 ***††‡‡ -50 **††‡‡ | |||
| Cytoplasm | |||
| +Zn 0.1 mM | −4 −12 | ||
| +Amprolium 5 mmol/L | −17 −12 | ||
| Thiamine-deficient culture medium | +Zn +Amprolium | −54 **††‡‡ −53 **††‡‡ | |
| C6 astroglioma cells Cholinergic neuroblastoma cells WT SN56 TrkA-/p75NTR+ DC | 24 h culture C6 in thiamine-free or thiamine-supplemented medium with: Amprolium 10 mM Zn 0.15 mM Zn 0.20 mM 24 h culture SN56 in thiamine-free medium in co-culture with C6 C6 co-culture Amprolium 5 mM Zn 0.1 mM Amprolium + Zn Amprolium + Zn+C6 co-culture | Relative change vs. no Zn, no amprolium control (%) Thiamine deficient Thiamine suppl. | [39] Absolute control levels of acetyl-CoA in SN56 and C6 cells were: 27.2 and 14.6 pmol/mg protein, respectively. † different from Amprolium+Zn, p < 0.05 |
| −26 ** 0 | |||
| −28 −16 | |||
| −68 ** −56 ** | |||
| Relative change vs. no co-culture, Zn, and no amprolium control (%) | |||
| +10 | |||
| −26 | |||
| −29 | |||
| −64 * | |||
| −10 † | |||
| WT mouse brain C57BL/6J mouse brain | Glycerol triacetate 3 g/kg b.w./d 10 d by gavage, and euthanised 60 min. post last gavage | Hippocampus Relative change vs. control (%) | [178] |
| Whole tissue Nuclei Cytoplasm | |||
| +171 * +19 *** +13 ** | |||
| Non-fasted mouse brain | Sacrificed 30 min. post oral ketone esters (KE) administration 3 mg/g b.w | Relative change KE vs. control (%) Brain cortex +114 *** | [226] |
| Cultured primary neurons (E17 C57BL/6J mice) | Astrocyte-derived ApoE particles Astrocyte-derived medium (ADM)Apo E enriched ADMApo E depleted ADM | Relative change vs. no ApoE control (%) Acetyl-CoA/CoA ratio Whole cells Nuclei +86 * +175 *** Acetyl-CoA/CoA ratio +200 *** +40 | [32] |
| WT mouse brain Elp3 conditional KO mouse brain | Lack Elongator to Atat1 activity | Relative change vs. WT control (%) Cortical neurons−72 | [75] |
| WT mouse brain C57Bl/6J mouse brain | Acute stress | Relative change vs.no stress control (%) Prefrontal cortex +113 * | [192] Absolute acetyl-CoA level, 0.37 pmol/μg |
| C57BL/6J mice—stroke and hypoxia | 12 wk post-stroke oral administration p75 NTR modulator (LM11A-31) | Relative change vs. sham control (%) Brain infarcted region None LM11A-31 −32 +36 * | [23] |
| Primary astrocytes—0–1-day-old mice cerebral cortex U87 human glioblastoma cells U87FABP7wt U87FABPmut. U251human glioma cells U251 FABP7KO | FABP7-KO vs. WT cells FABP7wt vs. control FABP7mut vs. control FABP7KO vs. control | Relative change vs. WT control (%) Whole cells Isolated nuclei −34 * −28 * +87 * +74 * −10 −39 −48 * −70 * | [213,214] Absolute acetyl-CoA for control WT cells is 450 pmol/106, and 74 pmol/107 nuclei. |
| WT mouse brain SLC25A1 nTg mouse brain | Hippocampus and cortex cytoplasm Lumen of the endoplasmic reticulum | Relative change vs. WT control (%) Cytoplasm ER +58 *** +72 **** | [130] SLC25A1 nTg—mitochondrial citrate carrier |
Author Contributions
Funding
Conflicts of Interest
References
- Belanger, M.J.; Allman, I.; Magistretti, P.J. Brain energy metabolism. Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Dienel, C.A.; Sonnewald, H.S.; Waagepetersen, H.S.; Schousboe, A. Energy metabolism of the brain. In Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology, 8th ed.; Brady, S., Siegel, G., Albers, R.W., Donald, L., Price, D.L., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; pp. 200–231. [Google Scholar]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C.H. Astrocytes as key regulators of brain energy metabolism: New therapeutic perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Zhang, S.; Lachance, B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef]
- Sun, W.; Cornwell, A.; Li, J.; Peng, S.; Osorio, J.; Aslling, N.; Wang, S.; Benraiss, A.; Lou, N.; Goldman, S.A.; et al. S0X9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci. 2017, 37, 4493–4507. [Google Scholar] [CrossRef]
- Popov, A.; Branze, N.; Fedotova, A.; Tiaglik, A.; Bychkov, M.; Morozova, N.; Branze, A.; Aronov, D.; Lyukmanova, E.; Lazareva, N.; et al. A high-fat diet changes astrocytic metabolism to promote synaptic plasticity and behavior. Acta Physiol. 2022, 236, e13847. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Rosenberger, T.A. Acetate treatment increases fatty acid content in LPS-stimulated BV2 microglia. Lipids 2014, 49, 621–631. [Google Scholar] [CrossRef]
- Currais, A.; Huang, L.; Petrascheck, M.; Maher, P.; Schubert, D. A chemical biology approach to identifying molecular pathways associated with aging. Geroscience 2021, 43, 353–365. [Google Scholar] [CrossRef]
- Zyśk, M.; Bielarczyk, H.; Gul-Hinc, S.; Dyś, A.; Gapys, S.; Ronowska, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A. Phenotype-dependent interaction between N-acetyl-L-aspartate and acetyl CoA in septal SN56 cholinergic cells exposed to an excess of zinc. J. Alzheimer Dis. 2017, 56, 1145–1158. [Google Scholar] [CrossRef]
- Janssen, L.; Ai, X.; Zheng, X.; Wei, W.; Caglayan, A.B.; Kilic, E.; Wang, Y.-C.; Hermann, D.M.; Venkataramani, V.; Bähr, M.; et al. Inhibition of fatty acid synthesis aggravates brain injury, reduces blood-brain barrier integrity and impairs neurological ecoverry in a murine stroke model. Front. Cell. Neurosci. 2021, 15, 327. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hic, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The regulatory effects of acetyl-CoA distribution in the healthy and diseased brain. Front. Cell. Neurosci. 2018, 12, 169–189. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Zyśk, M.; Dyś, A.; Ronowska, A.; Gul-Hinc, S.; Klimaszewska-Łata, J. Early and late pathomechanisms in Alzheimer’s disease: From zinc to amyloid-β neurotoxicity. Neurochem. Res. 2017, 42, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.C. Acetyl-CoA metabolism and histone acetylation in the regulation of aging and lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- Schuberth, J.; Sollenberg, J.; Sundvall, A.; Sörbo, B. Acetylcoenzyme A in brain. J. Neurochem. 1966, 13, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Ičny, J.; Tuček, S. Acetyl-coenzyme and acetylcholine in slices of rat caudate nuclei incubated in the presence of metabolic inhibitors. J. Biol. Chem. 1981, 256, 4919–4923. [Google Scholar]
- Bielarczyk, H.; Szutowicz, A. Evidence for the regulatory function of synaptoplasmic acetyl-CoA in acetylcholine synthesis in nerve endings. Biochem. J. 1989, 262, 337–380. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose transporters in brain: In health and in Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Crumpacker, R.H.; Calderon, K.E.; Garcia, F.G.; Zbesko, J.C.; Frye, J.B.; Gonzalez, S.; Becktel, D.A.; Yang, T.; Tavera-Garcia, M.A.; et al. Post-stroke administration of the p75 neurotrophin receptor modulator, LM11A-31, attenuates chronic changes in brain metabolism, increases neurotransmitter levels, and improves recovery. J. Pharmacol. Exp. Ther. 2022, 380, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Sharma, V.; Singh, T.G. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem. Res. 2022, 47, 2173–2186. [Google Scholar] [PubMed]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Roosterman, D.; Cottrell, G.S. Astrocytes and neurons communicate via a monocarboxylic acid shuttle. AIMS Neurosci. 2020, 7, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I.A. cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Wohnsland, S.; Bürgers, H.F.; Kuschinsky, W.; Maurer, M.H. Neurons and neuronal stem cells survive in glucose-free lactate and in high glucose cell culture medium during normoxia and anoxia. Neurochem. Res. 2010, 35, 1635–1642. [Google Scholar] [CrossRef]
- Li, Y.; Lu, B.; Sheng, L.; Zhu, Z.; Sun, H.; Zhou, Y.; Yang, Y.; Xue, D.; Chen, W.; Tian, X.; et al. Heksokinase 2-dependent hyperglycolisis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 2018, 144, 186–200. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Li, D.; He, C.H.; He, K.; Xue, T.; Wan, L.; Zhang, C.H.; Liu, Q.; Wan, L.; et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 2021, 109, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhao, T.; Du, J.; Ji, G.; Li, X.; Ji, S.; Tian, W.; Wang, X.; Hao, A. TIGAR promotes neural stem cell differentiation through acetyl-CoA-mediated histone acetylation. Cell Death Dis. 2019, 10, 198. [Google Scholar] [CrossRef]
- Sun, Y.; Li, T.; Xie, C.; Zhang, Y.; Zhou, K.; Wang, X.; Blomgren, K.; Zhu, C.H. Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget 2016, 7, 31708–31722. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Bian, X.; Zhang, Q.; Xia, Z.; Liu, B.; Chn, Q.; Ke, C.; Wu, J.L.; Zhao, Y. Shengui sansheng san ameliorates cerebral energy deficiency via citrate cycle after ischemic stroke. Front. Pharmacol. 2019, 10, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Khoury, N.; Xu, J.; Stegelman, S.D.; Jackson, C.W.; Koronowski, K.B.; Dave, K.R.; Young, J.I.; Perez-Pinzon, M.A. Resveratrol reconditioning induces genomic and metabolic adaptations with long-term window of cerebral ischemic tolerance leading to bioenergetic efficiency. Mol. Neurobiol. 2019, 56, 4549–4565. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Khoury, N.; Saul, N.; Loriz, Z.B.; Cohan, C.H.; Stradecki-Cohan, H.M.; Dave, K.R.; Young, J.I. Neuronal SIRT1 (silent information regulator 2 homologue 1) regulates glycolysis and mediates resveratrol-induced ischemic tolerance. Stroke 2017, 48, 3117–3125. [Google Scholar] [CrossRef]
- Klimaszewska-Łata, J.; Gul-Hinc, S.; Bielarczyk, H.; Ronowska, A.; Zyśk, M.; Grużewska, K.; Pawełczyk, T.; Szutowicz, A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J. Neurochem. 2015, 133, 284–297. [Google Scholar] [CrossRef]
- Gul-Hinc, S.; Michno, A.; Zyśk, M.; Szutowicz, A.; Jankowska-Kulawy, A.; Ronowska, A. Protection of cholinergic neurons against zinc toxicity by glial cells in thiamine-deficient media. Int. J. Mol. Sci. 2021, 22, 13337–13357. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Bielarczyk, H.; Pawełczyk, T.; Szutowicz, A. Effects of zinc on SN56 cholinergic neuroblastoma cells. J. Neurochem. 2007, 103, 972–983. [Google Scholar] [CrossRef]
- Greco, T.; Vespa, P.M.; Prins, M.L. Alternative substrate metabolism depends on cerebral metabolic state following traumatic brain injury. Exp. Neurol. 2020, 329, 113289. [Google Scholar] [CrossRef]
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 35, 43542–43552. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W. Zinc and traumatic brain injury: From chelation to supplementation. Med. Sci. 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Gul-Hinc, S.; Michno, A.; Bizon-Zygmańska, D.; Zyśk, M.; Bielarczyk, H.; Szutowicz, A.; Gapys, B.; Jankowska-Kulawy, A. Aggravated effects of coexisting marginal thiamine deficits and zinc excess on SN56 neuronal cells. Nutr. Neurosci. 2021, 6, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Annoni, F.; Peluso, L.; Bogossian, E.G.; Creteur, J.; Zanier, E.R.; Taccone, F.S. Brain protection after anoxic brain injury: Is lactate supplementation helpful ? Cells 2021, 10, 1714. [Google Scholar] [CrossRef] [PubMed]
- Duhaut, D.E.; Heurteaux, C.; Gandin, C.; Ichai, C.; Quintard, H. The antiedematous effect of exogenous lactate therapy in traumatic brain injury: A physiological and mechanistic approach. Neurocrit. Care 2021, 35, 747–755. [Google Scholar] [CrossRef]
- Wang, P.; Chen, M.; Yang, Z.; Yu, T.; Zhu, J.; Zhou, L.; Lin, J.; Fang, X.; Huang, Z.; Jiang, L.; et al. Activation of pyruvate dehydrogenase activity by dichloroacetate improves survival and neurologic outcomes after cardiac arrest in rats. Shock 2018, 49, 704–711. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Skulimowska, H. Effect of dichloroacetate on acetyl-CoA content and acetylcholine synthesis in rat brain synaptosomes. Neurochem. Res. 1994, 19, 1107–1112. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Kubler, M.M.; Donnino, M.W.; Sharp, W.W. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE 2017, 12, e0185046. [Google Scholar] [CrossRef]
- Ikeda, K.; Liu, X.; Kida, K.; Marutani, E.; Hirai, S.; Sakaguchi, M.; Andersen, L.W.; Bagchi, A.; Cocchi, M.N.; Berg, K.M.; et al. Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation 2016, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The effects of sodium dichloroacetate on mitochondrial dysfunction and neuronal death following hypoglycemia-induced injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.H.; Hooshyar, M.A.; Sundarrajan, C.H.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457. [Google Scholar] [CrossRef] [PubMed]
- Jakkamsetti, V.; Ma, Q.; Pascual, J.M. A subset of synaptic transmission events is coupled to acetyl coenzyme A production. J. Neurophysiol. 2022, 127, 623–636. [Google Scholar] [CrossRef]
- Chevalier, A.C.; Rosenberger, T.A. Increasing acetyl-CoA metabolism attenuates injury and alters spinal cord lipid content in mice subjected to experimental autoimmune encephalomyelitis. J. Neurochem. 2017, 141, 721–737. [Google Scholar] [CrossRef]
- Della-Flora Nunes, G.; Mueller, L.; Silvestri, N.; Patel, M.S.; Wrabetz, L.; Feltri, M.L.; Poitelon, Y. Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia 2017, 65, 1626–1639. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Musumeci, G.; Lazzarino, G.; Caruso, G.; Pastore, F.S. Pyruvate dehydrogenase and tricarboxylic acid cycle enzymes are sensitive targets of traumatic brain injury induced metabolic derangement. Int. J. Mol. Sci. 2019, 20, 5774. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Huang, Z.; Yan, Q.; Wang, Y.; Zou, Q.; Li, J.; Liu, Z.; Cai, Z. Role of mitochondrial dysfunction in the pathology of amyloid-β. J. Alzheimers Dis. 2020, 78, 505–514. [Google Scholar] [CrossRef]
- Hoshi, M.; Takashima, A.; Murayama, M.; Yoshida, N.; Hohino, T. Nontoxic amyloid beta peptide 1–42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer’s disease. J. Biol. Chem. 1997, 272, 2038–2041. [Google Scholar]
- Bielarczyk, H.; Jankowska-Kulawy, A.; Höfling, C.; Ronowska, A.; Gul-Hinc, S.; Roβner, S.; Schliebs, R.; Pawełczyk, T.; Szutowicz, A. AβPP-transgenic 2576 mice mimic cell type-specific aspects of acetyl-CoA-linked metabolic deficits in Alzheimer’s disease. J. Alzheimer Dis. 2015, 48, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Gandbhir, O.; Sundaram, P. Effect of AmyTrap, an amyloid-β binding drug, on Aβ induced mitochondrial dysfunction and tau phosphorylation in cultured neuroblastoma cells. Metab. Brain Dis. 2020, 35, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport:driving synaptic function. Cellular Neurosci. 2019, 366, 6462. [Google Scholar]
- Zahavi, E.E.; Hoogenraad, C.C. Multiple layers of spatial regulation coordinate axonal cargo transport. Curr. Opin. Neurobiol. 2021, 69, 241–246. [Google Scholar] [CrossRef]
- Babetto, E.; Beirowski, B. Of axons that struggle to make ends meet: Linking axonal bioenergetic failure to programmed axon degeneration. Biochim. Biophys. Acta (BBA)-Bioenerg. 2022, 1863, 148545. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Huang, N.; Sheng, Z.-H. Programming axonal mitochondrial maintenance and bioenergetics in neurodegeneration and regeneration. Neuron 2022, 110, 1899–1923. [Google Scholar] [CrossRef]
- Henn, R.E.; Noureldein, M.H.; Elzinga, S.E.; Kim, B.; Savelieff, M.G.; Feldman, E.L. Glial-neuron crosstalk in health and disease: A focus on metabolism, obesity, and cognitive impairment. Neurobiol. Dis. 2022, 170, 105766. [Google Scholar] [CrossRef]
- Roney, J.C.; Cheng, X.T.; Sheng, Z.H. Neuronal endolysosomal transport and lysosomal functionality in maintaining axonostasis. J. Cell Biol. 2022, 221, e202111077. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; van der Pol, S.M.; van Het Hof, B.; Reijerkerk, A.; Pellerin, L.; van der Valk, P.; de Vries, H.E.; et al. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.J.; Simkins, T.J.; Emery, B. Neuron-oligodendrocyte interaction in the structure and integrity of axons. Front. Cell Dev. Biol. 2021, 9, 653101. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Sterling, N.A.; Junker, I.P.; Helm, C.T.; Smith, G.M. Effects of αTAT1 and HDAC5 on axonal regeneration in adult neuron. PLoS ONE 2017, 12, e0177496. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.S.C.; Picci, C.; Swift, M.; Levinson, M.; Willis, D.; Langley, B. α-tubulin acetyltransferase is a novel target mediating neurite growth inhibitory effects of chondroitin sulfate proteoglycans and myelin-associated glycoprotein. eNeuro 2018, 5, e0240-17. [Google Scholar] [CrossRef]
- Even, A.; Morelli, G.; Tuchetto, S.; Shilian, M.; Le Bail, R.; Laguesse, S.; Krusy, N.; Brisker, A.; Brandis, A.; Inbar, S.; et al. ATP-citrate lyase promotes axonal transport across species. Nature Commun. 2021, 12, 5878. [Google Scholar] [CrossRef]
- Kojic, M.; Wainwright, B. The many faces of elongator in neurodevelopment and disease. Front. Mol. Neurosci. 2016, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.Y.; Knowles, R.; Dehorter, N. New insights into cholinergic neuron diversity. Front. Mol. Neurosci. 2019, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, W.; Tian, I.; Shi, J. Sirtuin 3 mitochondrial permeability transition pore (mPTP): A systemetic review. Mitochondrion 2022, 64, 103–111. [Google Scholar] [CrossRef]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. Activation of c-jun-N-terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 2009, 583, 1132–1140. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of cholinergic signaling in Alzheimer’s disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Diaz Brinton, R. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Baslow, H.M. N-acetylaspartate, and N-acetylaspartyglutamate. In Handbook of Neurochemistry and Molecular Biology: Amino Acids and Peptides in Nervous System, 3rd ed.; Lajtha, A., Oja, S.S.A., Saransaari, P., Schousboe, A., Eds.; Springer Science & Business Media: Berlin, Germany, 2007; pp. 305–356. [Google Scholar]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Laursen, B.; Mørk, A.; Plath, N.; Kristiansen, U.; Bastlund, J.F. Impaired hippocampal acetylcholine release parallels spatial memory deficits in Tg2576 mice subjected to basal forebrain cholinergic degeneration. Brain Res. 2014, 1543, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Cranston, A.L.; Wysocka, A.; Steczkowska, M.; Zadrożny, M.; Palasz, E.; Harrington, C.R.; Theuring, F.; Wischik, C.M.; Riedel, G.; Niewiadomska, G. Cholinergic and inflammatory phenotypes in transgenic tau mouse models of Alzheimer’s disease and frontotemporal lobar degeneration. Brain Commun. 2020, 2, fcaa033. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, N.; Hu, N.; Jiang, R.; Lu, H.; Xuan, A.; Long, D.; Chen, Y. Neural stem cell transplantation improves learning and memory by protecting cholinergic neurons and restoring synaptic impairment in an amyloid precursor protein/presenilin 1 transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 2020, 21, 1172–1180. [Google Scholar] [CrossRef]
- Chen, Y.; Han, S.; Huang, X.; Ni, J.; He, X. The protective effect of icariin on mitochondrial transport and distribution in primary hippocampal neurons from 3× Tg-AD mice. Int. J. Mol. Sci. 2016, 17, 163. [Google Scholar] [CrossRef]
- Park, D.; Choi, E.K.; Cho, T.H.; Joo, S.S.; Kim, Y.B. Human neural stem cells encoding ChAT gene restore cognitive function via acetylcholine synthesis, Aβ elimination, and neuroregeneration in APPswe/PS1dE9 mice. Int. J. Mol. Sci. 2020, 21, 3958. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Gissen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18 F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcón, F.; de la Peña, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic mitochondria: An early target of amyloid-β and tau in Alzheimer’s disease. J. Alzheimers Dis. 2021, 84, 1391–1414. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yamagata, N.; Takasaki, K.; Sano, K.; Hayakawa, K.; Katsurabayashi, S.; Egashira, N.; Mishima, K.; Iwasaki, K.; Fujiwara, M. Decreased acetylcholine release is correlated to memory impairment in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res. 2009, 16, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.J.; Kirchner, T.; Narkhede, A.; Wyss, M.; Van Bergen, J.M.G.; Steininger, S.C.; Gietl, A.; Leh, S.E.; Treyer, V.; Buck, A.; et al. Brain amyloid burden and cerebrovascular disease are synergistically associated with neurometabolism in cognitively unimpaired older adults. Neurobiol. Aging 2018, 63, 152–161. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, A.; Längströ, M.B.; Darreh-Shori, T. Discovery of novel choline acetyltransferase inhibitors using structure-based virtual screening. Sci. Rep. 2017, 7, 16287–16304. [Google Scholar] [CrossRef]
- Bielarczyk, H.; Jankowska-Kulawy, A.; Gul, S.; Pawełczyk, T.; Szutowicz, A. Phenotype dependent differential effects of interleukin-1β and amyloid-β on viability and cholinergic phenotype of T17 neuroblastoma cells. Neurochem. Int. 2005, 47, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Szutowicz, A.; Madziar, B.; Pawełczyk, T.; Tomaszewicz, M.; Bielarczyk, H. Effects of NGF on acetylcholine, acetyl-CoA metabolism and viability of differentiated and non-differentiated cholinergic neuroblastoma cells. J. Neurochem. 2004, 90, 952–961. [Google Scholar] [CrossRef]
- Xu, J.; Patassini, S.; Begley, P.; Church, S.; Waldvogel, H.J.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Cerebral deficiency of vitamin B5 (d-pantothenic acid; pantothenate) as a potentially-reversible cause of neurodegeneration and dementia in sporadic Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2020, 527, 676–681. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Gul, S.; Ronowska, A.; Pawełczyk, T.; Jankowska-Kulawy, A. Phenotype-dependent susceptibility of cholinergic neuroblastoma cells to neurotoxic inputs. Met. Brain Dis. 2006, 21, 149–161. [Google Scholar] [CrossRef]
- Bagga, S.; Kumar, M. Current status of Alzheimer’s disease and pathological mechanisms investigating the therapeutic molecular targets. Curr. Mol. Med. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Liu, H.Y.; Gale, J.R.; Reynolds, I.J.; Weiss, J.H.; Aizenman, E. The multifaceted roles of zinc in neuronal mitochondrial dysfunction. Biomedicines 2021, 9, 489. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc signal in brain diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Li, B.; Xia, M.; Zorec, R.; Parpura, V.; Verkhratsky, A. Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res. 2021, 1752, 147234. [Google Scholar] [CrossRef] [PubMed]
- Zyśk, M.; Gapys, B.; Ronowska, A.; Gul-Hinc, S.; Erlandsson, A.; Iwanicki, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A.; Bielarczyk, H. Protective effects of voltage-gated calcium channel antagonist against zinc toxicity in SN56 neuroblastoma cholinergic cells. PLoS ONE 2018, 13, e0209363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M. N-Acetyl-Cysteine:modulating the cysteinę redox proteome in neurodegenerative disease. Antioxidants. 2022, 11, 416. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Bader Lange, M.L. Oxidatively modified glyceraldehyde-3-pfosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. JAD 2010, 20, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Maczurek, A.; Hager, K.; Kenklies, M.; Sharman, M.; Martins, R.; Engel, J.; Carlson, D.A.; Münch, G. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv. Drug Deliv. Rev. 2008, 60, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef]
- Ronowska, A.; Dyś, A.; Jankowska-Kulawy, A.; Klimaszewska-Łata, J.; Bielarczyk, H.; Romianowski, P.; Pawełczyk, T.; Szutowicz, A. Short-term effects of zinc on acetylcholine metabolism and viability of SN56 cholinergic neuroblastoma cells. Neurochem. Int. 2010, 56, 143–151. [Google Scholar] [CrossRef]
- Xue, Y.N.; Liu, Y.N.; Su, J.; Li, J.L.; Wu, Y.; Guo, R.; Yu, B.B.; Yan, X.Y.; Zhang, L.C.; Sun, L.K.; et al. Zinc cooperates with p53 to inhibit the activity of mitochondrial aconitase through reactive oxygen species accumulation. Cancer Med. 2019, 8, 2462–2473. [Google Scholar] [CrossRef]
- Liu, W.; Fan, Z.; Gao, F.; Ou, L.; Li, M.; Zhou, X.; Luo, W.; Wei, P.; Miao, F. Emodin inhibits zinc-induced neurotoxicity in neuroblastoma SH-SY5Y cells. Biosci. Rep. 2019, 39, 1–9. [Google Scholar] [CrossRef]
- Ji, S.G.; Weiss, J.H. Zn2+-induced disruption of neuronal mitochondrial function: Synergism with Ca2+, critical dependence upon cytosolic Zn2+ buffering, and contributions to neuronal injury. Exp. Neurol. 2018, 302, 181–195. [Google Scholar] [CrossRef]
- Moriyama, M.; Fujitsuka, S.; Kawabe, K.; Takano, K.; Nakamura, Y. Zinc potentiates lipopolysaccharide-induced nitric oxide production in cultured primary rat astrocytes. Neurochem. Res. 2018, 43, 363–374. [Google Scholar] [CrossRef]
- Butterworth, R.F. Thiamine deficiency-related brain dysfunction in chronic liver failure. Metab. Brain. Dis. 2009, 24, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Smith, T.J.; Johnson, C.R.; Koshy, R.K.; Hess, S.Y.; Qureshi, U.A.; Mynak, M.L.; Fischer, P.R. Thiamine deficiency disorders: A clinical perspective. Ann. N. Y. Acad. Sci. 2021, 1498, 9–28. [Google Scholar] [CrossRef]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Wróblewska, M.; Szutowicz, A. Acetyl-CoA and acetylcholine metabolism in nerve terminal compartment of thiamine deficient rat brain. J. Neurochem. 2010, 115, 333–342. [Google Scholar] [CrossRef]
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Wróblewska, M.; Szutowicz, A. Acetyl-CoA deficit in brain mitochondria in experimental thiamine deficiency encephalopathy. Neurochem. Int. 2010, 57, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Bizon-Zygmańska, D.; Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Ronowska, A.; Marszałł, M.; Szutowicz, A. Acetyl-CoA metabolism in amprolium-evoked thiamine pyrophosphate deficits in cholinergic SN56 neuroblastoma cells. Neurochem. Int. 2011, 59, 208–216. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Üçal, M.; Müllebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 925–931. [Google Scholar] [CrossRef]
- Boskovic, Z.; Meier, S.; Wang, Y.; Milne, M.R.; Onraet, T.; Tedoldi, A.; Coulson, E.J. Regulation of cholinergic basal forebrain development, connectivity, and function by neurotrophin receptors. Neuronal Signal 2019, 1, NS20180066. [Google Scholar] [CrossRef]
- Latina, V.; Caioli, S.; Zona, C.; Ciotti, M.T.; Amadoro, G.; Calissano, P. Impaired NGF/TrkA signaling causes early AD-linked presynaptic dysfunction in cholinergic primary neurons. Front. Cell. Neurosci. 2017, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Raya, J.; Cruz-Raya, U.; López-Martínez, M.; Picazo, O. Choline acetyltransferase and TrkA expression, as well as the improvement in cognition produced by E2 and P4 in ovariectomized rats, are blocked by ICI 182 780 and RU486. Behav. Pharmacol. 2018, 29, 457–461. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Gul, S.; Zieliński, P.; Pawełczyk, T.; Tomaszewicz, M. Nerve growth factor and acetyl-L-carnitine evoked shifts in acetyl-CoA and cholinergic SN56 cell vulnerability to neurotoxic inputs. J. Neurosci. Res. 2005, 79, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Brann, A.B.; Tcherpakov, M.; Williams, I.M.; Futerman, A.H.; Fainzilber, M. Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J. Biol. Chem. 2002, 277, 9812–9818. [Google Scholar] [CrossRef] [PubMed]
- Barrett, G.L.; Naim, T.; Trieu, J.; Huang, M. In vivo knockdown of basal forebrain p75 neurotrophin receptor stimulates choline acetyltransferase activity in the mature hippocampus. J. Neurosci. Res. 2016, 94, 389–400. [Google Scholar] [CrossRef]
- Nguyen, Y.T.K.; Ha, H.T.T.; Nguyen, T.H.; Nguyen, L.N. The role of SLC transporters for brain health and disease. Cell. Mol. Life Sci. 2021, 79, 20. [Google Scholar] [CrossRef]
- Rigby, M.J.; Orefice, N.S.; Lawton, A.J.; Ma, M.; Shapiro, S.L.; Yi, S.Y.; Dieterich, I.A.; Frelka, A.; Miles, H.N.; Pearce, R.A.; et al. Increased expression of SLC25A1/CIC causes an autistic-like phenotype with altered neuron morphology. Brain 2022, 145, 500–516. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Pawełczyk, T.; Ronowska, A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative disease. Neurochem. Res. 2013, 38, 1523–1542. [Google Scholar] [CrossRef]
- Patel, M.S.; Owen, O.E. Lipogenesis from ketone bodies in rat brain. Evidence for conversion of acetoacetate into acetyl-coenzyme A in the cytosol. Biochem. J. 1976, 156, 603–607. [Google Scholar]
- Amaral, A.I.; Hadera, M.G.; Kotter, M.; Sonnewald, U. Oligodendrocytes do not export NAA-derived aspartate in vitro. Neurochem. Res. 2017, 4, 827–837. [Google Scholar] [CrossRef]
- Szutowicz, A.; Stępień, M.; Łysiak, W.; Angielski, S. Effect of (-) hydroxycitrate on the activities of ATP citrate lyase and the enzymes of acetyl-CoA metabolism in rat brain. Acta Biochim. Pol. 1976, 23, 227–234. [Google Scholar] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R. Effects of a dietary ketone esters on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Peterson, C. Acetylcholine and oxidative metabolism in septum and hippocampus in vitro. J. Biol. Chem. 1983, 258, 1142–1145. [Google Scholar] [CrossRef]
- Szutowicz, A.; Stępień, M.; Bielarczyk, H.; Kabata, J.; Łysiak, W. ATP citrate lyase in cholinergic nerve endings. Neurochem. Res. 1982, 7, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewicz, M.; Roβner, S.; Schliebs, R.; Ćwikowska, J.; Szutowicz, A. Changes in cortical acetyl-CoA metabolism after selective basal forebrain cholinergic degeneration by 192IgG-saporin. J. Neurochem. 2002, 87, 318–324. [Google Scholar] [CrossRef]
- Sun, J.; Pan, K.Q.; Chew, T.W.; Liang, F.; Burmeister, M.; Low, B.C. BNIP-H recruits the cholinergic machinery to neurite terminals to promote acetylcholine signaling and neuritogenesis. Dev. Cell 2015, 34, 555–568. [Google Scholar] [CrossRef]
- Choi, Y.S.; Song, J.E.; Lee, J.F.; Kim, E.; Kim, C.H.; Kim, D.H.; Song, H.T. Hyperpolarized [1-13C] lactate flux increased in hippocampal region in diabetic mice. Mol. Brain 2019, 12, 1–10. [Google Scholar] [CrossRef]
- Eiβing, A.; Fischer, D.; Rauch, I.; Baumann, A.; Schebb, N.H.; Karst, U.; Rose, K.; Klumpp, S.; Krieglstein, J. Acetylocholine content and viability of cholinergic neurons are influenced by the activity of protein histidine phosphatase. BMC Neurosci. 2012, 13, 31–37. [Google Scholar]
- Krieglstein, J.; Lehmann, M.; Maurer, A.; Gudermann, T.; Pinkenburg, O.; Wieland, T.; Litterscheid, S.; Klumpp, S. Reduced viability of neuronal cells after overexpression of protein histidine phosphatase. Neurochem. Int. 2008, 53, 132–136. [Google Scholar] [CrossRef]
- Szutowicz, A.; Tomaszewicz, M.; Jankowska, A.; Kisielevski, J. Acetylocholine synthesis in nerve terminals of diabetic rats. Neuroreport 1994, 5, 2421–2424. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma membrane Na+-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules 2017, 22, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Vigers, T.; Vinovskis, C.; Li, L.P.; Prasad, P.; Heerspink, H.; D’Alessandro, A.; Reisz, J.A.; Piani, F.; Cherney, D.Z.; van Raalte, D.H.; et al. Plasma levels of carboxylic acids are markers of early kidney dysfunction in young people with type 1 diabetes. Pediatr. Nephrol. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Jordan, K.; Stanton, E.H.; Milenkovic, V.M.; Federlin, M.; Drexler, K.; Buchalla, W.; Gaumann, A.; Adamski, J.; Proesccholdt, M.; Haferkamp, S.; et al. Potential involvement of extracellular citrate in brain tumor progression. Curr. Mol. Med. 2021, 21, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cordes, T.; Thalacker-Mercer, A.E.; Pajor, A.M.; Murphy, A.N.; Metallo, C.M. NaCT/SLC13A5 facilitates citrate import and metabolism under nutrient-limited conditions. Cell Rep. 2021, 36, 109701. [Google Scholar] [CrossRef] [PubMed]
- Pajor, A.M.; de Oliviera, C.A.; Song, K.; Huard, K.; Shanmugasundaram, V.; Erion, D.M. Molecular basis for inhibition of the Na+/citrate transporter NaCT (SCL13A5) by dicarboxylate inhibitors. Mol. Pharmacol. 2019, 90, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Ronowska, A.; Gul-Hinc, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Pawełczyk, T. Intracellular redistribution of acetyl-CoA, the pivotal point in differential susceptibility of cholinergic neurons and glial cells to neurodegenerative signals. Biochem. Soc. Trans. 2014, 42, 1101–1106. [Google Scholar] [CrossRef]
- Westergaard, N.; Waagepetersen, H.S.; Belhage, B.; Schousboe, A. Citrate, a ubiquitous key metabolite with regulatory function. Neurochem. Res. 2017, 42, 1583–1588. [Google Scholar] [CrossRef]
- Icard, P.; Wu, Z.; Fournel, L.; Coquerel, A.; Lincet, H.; Alifano, M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett. 2020, 471, 125–134. [Google Scholar] [CrossRef]
- Granchi, C. ATP-citrate lyase (ACLY) inhibitors as therapeutic agents: A patenting perspective. Expert opinion on therapeutic patients. Expert Opin. Ther. Pat. 2022, 32, 731–742. [Google Scholar] [CrossRef]
- Selch, S.; Chafai, A.; Sticht, H.; Birkenfeld, A.L.; Fromm, M.F.; König, J. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci. Rep. 2018, 8, 11330. [Google Scholar] [CrossRef]
- Klotz, J.; Porter, B.E.; Coals, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22, 310–321. [Google Scholar] [CrossRef]
- Yang, Q.Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG phenotype of SLC12A5 citrate transporter disorder. Child Neurol. Open. 2020, 7, 2329048X20931361. [Google Scholar] [CrossRef] [PubMed]
- Henke, C.; Töllner, K.; Van Dijk, R.M.; Miljanovic, N.; Cordes, T.; Twele, F.; Bröer, S.; Ziesak, V.; Rohde, M.; Hauck, M.; et al. Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol. Dis. 2020, 143, 105018. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Arun, P.; Ariyannur, P.S.; Namboodiri, A.M.A. N-acetylaspartate reductions in brain injury: Impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Front. Neuroenergetics 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Madhavarao, C.N.; Chinopoulos, C.; Chandrasekaran, K.; Namboodiri, M.A.A. Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain. J. Neurochem. 2013, 86, 824–835. [Google Scholar] [CrossRef]
- Papazisis, G.; Pourzitaki, C.; Sardeli, C.; Lallas, A.; Amaniti, E.; Kouvelas, D. Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol. Res. 2008, 57, 73–78. [Google Scholar] [CrossRef]
- Zyśk, M.; Sakowicz-Burkiewicz, M.; Pikul, P.; Kowalski, R.; Michno, A.; Pawełczyk, T. The impact of acetyl-CoA and aspartate shortages on the N-acetylaspartate level in different models of cholinergic neurons. Antioxidants 2020, 9, 522–545. [Google Scholar] [CrossRef] [PubMed]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte bioenergetics in health and disease. Neuroscientist 2019, 25, 334–343. [Google Scholar] [CrossRef]
- Li, S.; Clements, R.; Sulak, M.; Gregory, R.; Freeman, E.; McDonough, J. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem. Res. 2013, 38, 2385–2396. [Google Scholar] [CrossRef]
- Nordengen, K.; Heuser, C.; Rinholm, J.E.; Matalon, R.; Gundersen, V. Localisation of N-acetylaspartate in oligodendrocytes/myelin. Brain Struct. Funct. Action 2015, 220, 899–917. [Google Scholar] [CrossRef]
- Bhakoo, K.K.; Pearce, D. In vitro expression of N-acetylaspartate by oligodendrocytes: Implications for proton magnetic resonance spectroscopy signal in vivo. J. Neurochem. 2000, 74, 254–262. [Google Scholar] [CrossRef]
- Nešuta, O.; Thomas, A.G.; Alt, J.; Hin, N.; Neužilová, A.; Long, S.; Tsukamoto, T.; Rojas, C.; Wei, H.; Slusher, B.S. High throughput screening cascade to identify human aspartate N-acetyltransferase (ANAT) inhibitors for Canavan disease. ACS Chem. Neurosci. 2021, 12, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Bannerman, P.; Guo, F.; Burns, T.; Miers, L.; Croteau, C.; Singhal, N.K.; McDonough, J.A.; Pleasure, D. Suppressing N-acetyl-L-aspartate synthesis prevents loss of neurons in a murine model of Canavan leukodystrophy. J. Neurosci. 2017, 37, 413–421. [Google Scholar] [CrossRef] [PubMed]
- von Jonquieres, G.; Spencer, Z.H.T.; Rowlands, B.D.; Klugmann, C.B.; Bongers, A.; Harasta, A.E.; Parley, K.E.; Cederholm, J.; Teahan, O.; Pickford, R.; et al. Uncoupling N-acetylaspartate from brain pathology: Implications for Canavan disease gene therapy. Acta Neuropathol. 2018, 135, 95–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Jonquieres, G.; Rae, C.D.; Housley, G.D. Emerging concepts in vector development for glial gene therapy: Implications for leukodystrophies. Front. Cell. Neurosci. 2021, 15, 213. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Yang, M.; Ke, X.; Dai, Y.; Lin, H.; Wang, S.; Chen, L.; Tao, J. Chemical genetic activation of the cholinergic basal forebrain hippocampal circuit rescues memory loss in Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 53–73. [Google Scholar] [CrossRef]
- Klyuyeva, A.; Tuganova, A.; Kedishvili, N.; Popov, K.M. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J. Biol. Chem. 2019, 294, 838–851. [Google Scholar] [CrossRef]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate revisited: A key biomolecule at the nexus of metabolism, epigenetics and oncogenesis-part 1: Acetyl-CoA, acetogenesis and acyl-CoA short-chain synthetases. Front. Physiol. 2020, 11, 580167. [Google Scholar] [CrossRef]
- Deelchand, D.K.; Shestov, A.A.; Koski, D.M.; Uğurbil, K.; Henry, P.G. Acetate transport and utilization in the rat brain. J. Neurochem. 2009, 109, 46–54. [Google Scholar] [CrossRef]
- Waniewski, R.A.; Martin, D.L. Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci. 1998, 18, 5225–5233. [Google Scholar] [CrossRef]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 299–307. [Google Scholar] [CrossRef]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife 2019, 8, e47866. [Google Scholar] [PubMed]
- He, W.; Zhou, X.; Wu, Q.; Zhou, L.; Zhang, Z.; Zhang, R.J. Acetyl-CoA synthase 2 potentiates ATG5-induced autophagy against neuronal apoptosis after subarachnoid hemorrhage. J. Mol. Hist. 2022, 53, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Hu, W.; Cai, L.; Zeng, G.; Fang, W.; Dai, X.; Ye, Q.; Chen, X.; Zhang, J. Acetate supplementation produces antidepressant-like effect via enhanced histone acetylation. J. Affect. Disord. 2021, 281, 51–60. [Google Scholar] [CrossRef]
- Dong, Y.; Brewer, G.J. Global metabolic shifts in age and Alzheimer’s disease mouse brains pivot at NAD+/NADH redox sites. J. Alzheimers Dis. 2019, 71, 119–140. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, T.; Kang, Y.; Zhang, G.; Ji, X.; Feng, X.; Shi, G. Testosterone ameliorates age-related brain mitochondrial dysfunction. Aging 2021, 13, 16229–16247. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Srere, P.A. Kinetic studies of citrate synthase from rat kidney and rat brain. J. Biol. Chem. 1973, 248, 8022–8030. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Baek, B.; Song, I.S.; Choi, M.K.; Takuwa, Y.; Ryu, H.; Kim, S.H.; He, X.; et al. Neuronal SphK1 acetylates COX2 and contributes to pathogenesis in a model of Alzheimer’s disease. Nat. Commun. 2018, 9, 1479. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Song, I.S.; Choi, M.K.; Yu, E.; Park, C.M.; Kim, H.J.; Kim, S.H.; Schuchman, E.H.; et al. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2358. [Google Scholar] [CrossRef]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar]
- Blazquez, C.; Sanchez, C.; Velasco, G.; Guzman, M. Role of carnitine palmitoyltransferase I in the control of ketogenesis in primary cultures of rat astrocytes. J. Neurosci. 1998, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Long, A.; Scafidi, S.; Kristian, T. Interplay between NAD+ and acetyl CoA metabolism in ischemia-induced mitochondrial pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2060–2067. [Google Scholar] [PubMed]
- Eraso-Pichot, A.; Brasó-Vives, M.; Golbano, A.; Menacho, C.; Claro, E.; Galea, E.; Masgrau, R. GSEA of mouse and human mitochondrial reveals fatty acid oxidation in astrocytes. Glia 2018, 66, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yu, T.; Fang, X.; Ge, Q.; Song, F.; Huang, Z.; Jiang, L.; Wang, P. Short-term dietary restriction ameliorates brain injury after cardiac arrest by modulation of mitochondrial biogenesis and energy metabolism in rats. Ann. Transl. Med. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef]
- Altayyar, M.; Nasser, J.A.; Thomopoulos, D.; Bruneau, M. The implication of physiological ketosis on the cognitive brain: A narrative review. Nutrients 2022, 14, 513–535. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353. [Google Scholar]
- Hyeonwi-Son, H.; Baek, J.H.; Kang, J.S.; Jung, S.; Chung, H.J.; Kim, H.J. Acutely increased b-hydroxybutyrate plays a role in the prefrontal cortex to escape stressful conditions during the acute stress response. Biochem. Biophys. Res. Commun. 2021, 554, 19–24. [Google Scholar]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar]
- Andersen, J.V.; Westi, E.W.; Jakobsen, E.; Urruticoechea, N.; Borges, K.; Aldana, B.I. Astrocyte metabolism of the medium-chain fatty acids octanoic acid and decanoic acid promotes GABA synthesis in neurons via elevated glutamine supply. Mol. Brain 2021, 14, 132. [Google Scholar]
- Fochel, F. Triheptanoin for the treatment of brain energy deficit: A 14-year experience. J. Neurosci. Res. 2017, 95, 2236–2243. [Google Scholar]
- Dabke, P.; Das, A.M. Mechanism of action of ketogenic diet treatment: Impact of decanoic acid and beta-hydroxybutyrate on sirtuins and energy metabolism in hippocampal murine neurons. Nutrients 2020, 12, 2379–2398. [Google Scholar]
- Linn, T.C.; Srere, P.A. Binding of ATP citrate lyase to the microsomal fraction of rat liver. J. Biol. Chem. 1984, 259, 13379–13384. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Nomura, K.H.; Nomura, K. The acetyl-CoA transporter family SLC33. Mol. Aspects Med. 2013, 34, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Constantini, C.; Ko, M.H.; Jonas, M.C.; Puglielli, L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 2007, 407, 383–395. [Google Scholar] [CrossRef]
- Rigby, M.J.; Yun Ding, Y.; Farrugia, M.A.; Feig, M.; Cortese, G.P.; Mitchell, H.; Burger, C.; Puglielli, L. The endoplasmic reticulum acetyltransferases ATase1/NAT8B and ATase2/NAT8 are differentially regulated to adjust engagement of the secretory pathway. J. Neurochem. 2020, 154, 404–423. [Google Scholar]
- Farrugia, M.A.; Puglielli, L. Nε-lysine acetylation in the endoplasmic reticulum—A novel cellular mechanism that regulates proteostasis and autophagy. J. Cell Sci. 2019, 131, jcs221747. [Google Scholar]
- Jonas, M.C.; Pehar, M.; Puglielli, L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 2010, 123, 3378–3388. [Google Scholar]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Pehar, M.; Puglielli, L. Lysine acetylation in the lumen of ER: A novel and essential function under the control of the UPR. Biochim. Biophys. Acta 2013, 1833, 686–697. [Google Scholar]
- Rigby, M.J.; Orefice, N.S.; Lawton, A.J.; Ma, M.; Shapiro, S.L.; Yi, S.Y.; Dieterich, I.A.; Frelka, A.; Miles, H.N.; Pearce, R.A.; et al. SLC13A5/sodium-citrate co-transporter overexpression causes disrupted white matter integrity and an autistic-like phenotype. Brain Commun. 2022, 4, fcac002. [Google Scholar] [PubMed]
- Peng, Y.; Li, M.; Clarkson, B.D.; Pehar, M.; Lao, P.J.; Hillmer, A.T.; Barnhart, T.E.; Christian, B.T.; Mitchell, H.A.; Bendlin, B.B.; et al. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 2014, 34, 6772–6789. [Google Scholar] [CrossRef] [PubMed]
- Hullinger, R.; Li, M.; Wang, J.; Peng, Y.; Dowell, J.A.; Bomba-Warczak, E.; Mitchell, H.A.; Burger, C.; Chapman, E.R.; Denu, J.M.; et al. Increased expression of AT-1/SL33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J. Exp. Med. 2016, 213, 1267–1284. [Google Scholar] [PubMed]
- Ma, Y.; Chen, L.; He, X.X.; Wang, Y.J.; Yu, H.L.; He, Z.X.; Zhang, L.Q.; Zheng, Y.W.; Zhu, X.J. Functional prediction and characterization of Dip2gene in mice. Cell Biol. Int. 2019, 43, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.Q.; He, Z.X.; He, X.X.; Wang, Y.J.; Jian, Y.L.; Wang, X.; Zhang, B.B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar]
- Kim, J.Y.; Hwang, H.G.; Lee, J.Y.; Kim, M.; Kim, J.Y. Cortactin deacetylation by HDAC6 and SIRT2 regulates neuronal migration and dendrite morphogenesis during cerebral cortex development. Mol. Brain 2020, 13, 105. [Google Scholar]
- Zervopoulos, S.D.; Boukouris, A.E.; Saleme, B.; Haromy, A.; Tejay, S.; Sutendra, G.; Michelakis, E.D. MFN2-driven mitochondria to nucleus tethering allows a non-canonical nuclear entry pathway of the mitochondrial pyruvate dehydrogenase complex. Mol. Cell 2022, 82, 1066–1077. [Google Scholar]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef]
- Kagawa, Y.; Umaru, B.A.; Shima, H.; Ito, R.; Zama, R.; Islam, A.; Kanno, S.I.; Yasui, A.; Sato, S.; Jozaki, K.; et al. FABP7 regulates acetyl-CoA metabolism through the interaction with ACLY in the nucleus of astrocytes. Mol. Neurobiol. 2020, 57, 4891–4910. [Google Scholar] [CrossRef]
- Kagawa, Y.; Umaru, B.A.; Kanamori, M.; Zama, R.; Shil, S.K.; Miyzaki, H.; Kobayashi, S.; Wannakul, T.; Yang, S.; Tominaga, T.; et al. Nuclear FABP7 regulates cell proliferation of wild-type IDH1 glioma through caveolae formation. Mol. Oncol. 2022, 16, 289–306. [Google Scholar] [CrossRef]
- Houston, R.; Sekine, S.; Calderon, M.J.; Seifuddin, F.; Wang, G.; Kawagishi, H.; Malide, D.A.; Li, Y.; Gucek, M.; Pirooznia, M.; et al. Acetylation-mediated remodeling of the nucleolus regulates cellular acetyl-CoA responses. PLoS Biol. 2020, 18, e3000981. [Google Scholar]
- Boyle, E.; Wilfling, F. Bypassing the nuclear gate: A non-canonical entry pathway for the mitochondrial pyruvate dehydrogenase complex. Mol. Cell 2022, 82, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Niu, Y.J.; Nie, Z.W.; Kim, J.Y.; Xu, Y.N.; Yan, C.G. Nuclear accumulation of pyruvate dehydrogenase alpha 1 promotes histone acetylation and is essential for zygotic genome activation in porcine embryos. Mol. Cell Res. 2020, 1867, 118648–118660. [Google Scholar]
- Madiraju, P.; Pande, S.V.; Prentki, M.; Madiraju, S.R.M. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 2009, 6, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H.J.; Dekker, F.J.; Aheng, G.Y. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding. Eur. J. Med. Chem. 2012, 47, 337–344. [Google Scholar] [PubMed]
- Wepenaar, H.; van der Wouden, P.E.; Groves, M.R.; Rotili, D.; Mai, A.; Dekker, F.J. Enzyme kinetics and inhibition of histone acetyltransferase KAT8. Eur. J. Med. Chem. 2015, 105, 289–296. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef]
- Crews, F.T.; Fisher, R.; Deason, C.; Vetreno, R.P. Loss of basal forebrain cholinergic neurons following adolescent binge ethanol exposure: Recovery with the cholinesterase inhibitor galantamine. Front. Behav. Neurosci. 2021, 15, 652494. [Google Scholar]
- de Diego, I.; Müller-Eigner, A.; Peleg, S. The brain epigenome goes drunk: Alcohol consumption alters histone acetylation and transcriptome. Trends Biochem. Sci. 2020, 45, 93–95. [Google Scholar] [PubMed]
- Shea, P.A.; Aprison, M.H. The distribution of acetyl-CoA in specific areas of the CNS of the rat as measured by a modification of a radio-enzymatic assay for acetylcholine and choline. J. Neurochem. 1977, 28, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mast, N.; Petrov, A.M.; Prendergast, E.; Bederman, I.; Pikuleva, I.A. Brain acetyl-CoA production and phosphorylation of cytoskeletal proteins are targets of CYP46A1 activity modulation and altered sterol flux. Neurotherapeutics 2021, 18, 2040–2206. [Google Scholar]
- Suissa, L.; Kotchetkov, P.; Guigonis, J.M.; Doche, E.; Osman, O.; Pourcher, T.; Lindenthal, S. Ingested ketone ester leads to a rapid rise of acetyl-CoA and competes with glucose metabolism in the brain of non-fasted mice. Int. J. Mol. Sci. 2021, 22, 524. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankowska-Kulawy, A.; Klimaszewska-Łata, J.; Gul-Hinc, S.; Ronowska, A.; Szutowicz, A. Metabolic and Cellular Compartments of Acetyl-CoA in the Healthy and Diseased Brain. Int. J. Mol. Sci. 2022, 23, 10073. https://doi.org/10.3390/ijms231710073
Jankowska-Kulawy A, Klimaszewska-Łata J, Gul-Hinc S, Ronowska A, Szutowicz A. Metabolic and Cellular Compartments of Acetyl-CoA in the Healthy and Diseased Brain. International Journal of Molecular Sciences. 2022; 23(17):10073. https://doi.org/10.3390/ijms231710073
Chicago/Turabian StyleJankowska-Kulawy, Agnieszka, Joanna Klimaszewska-Łata, Sylwia Gul-Hinc, Anna Ronowska, and Andrzej Szutowicz. 2022. "Metabolic and Cellular Compartments of Acetyl-CoA in the Healthy and Diseased Brain" International Journal of Molecular Sciences 23, no. 17: 10073. https://doi.org/10.3390/ijms231710073