
A Long Noncoding RNA Derived from lncRNA–mRNA Networks Modulates Seed Vigor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
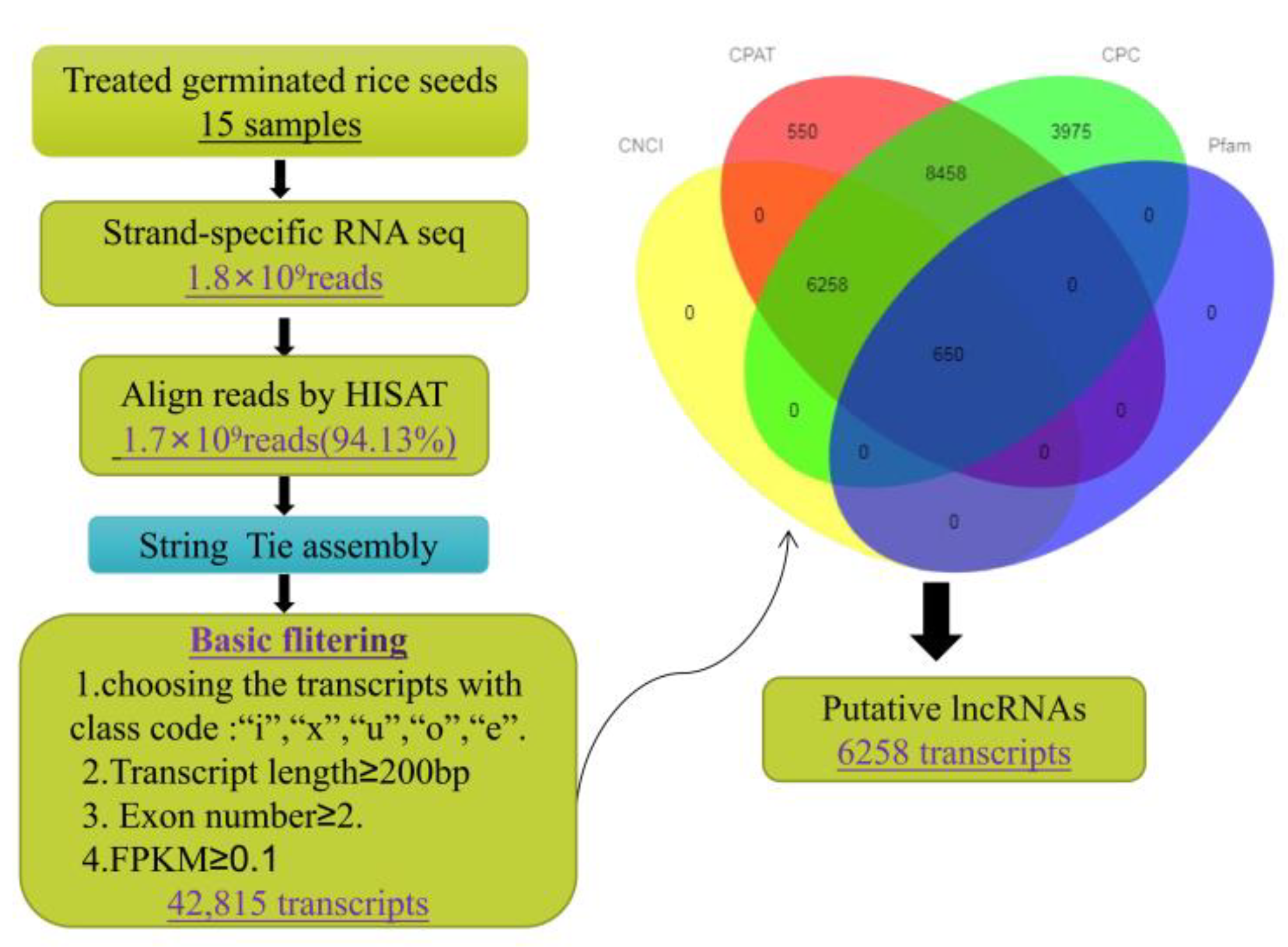
2.1. RNA Sequencing and Identification of lncRNAs
2.2. Structural Characterization of lncRNAs
2.3. More lncRNAs Than mRNAs Are Specifically Expressed at Different Stages
2.4. Analysis of Differentially Expressed lncRNAs
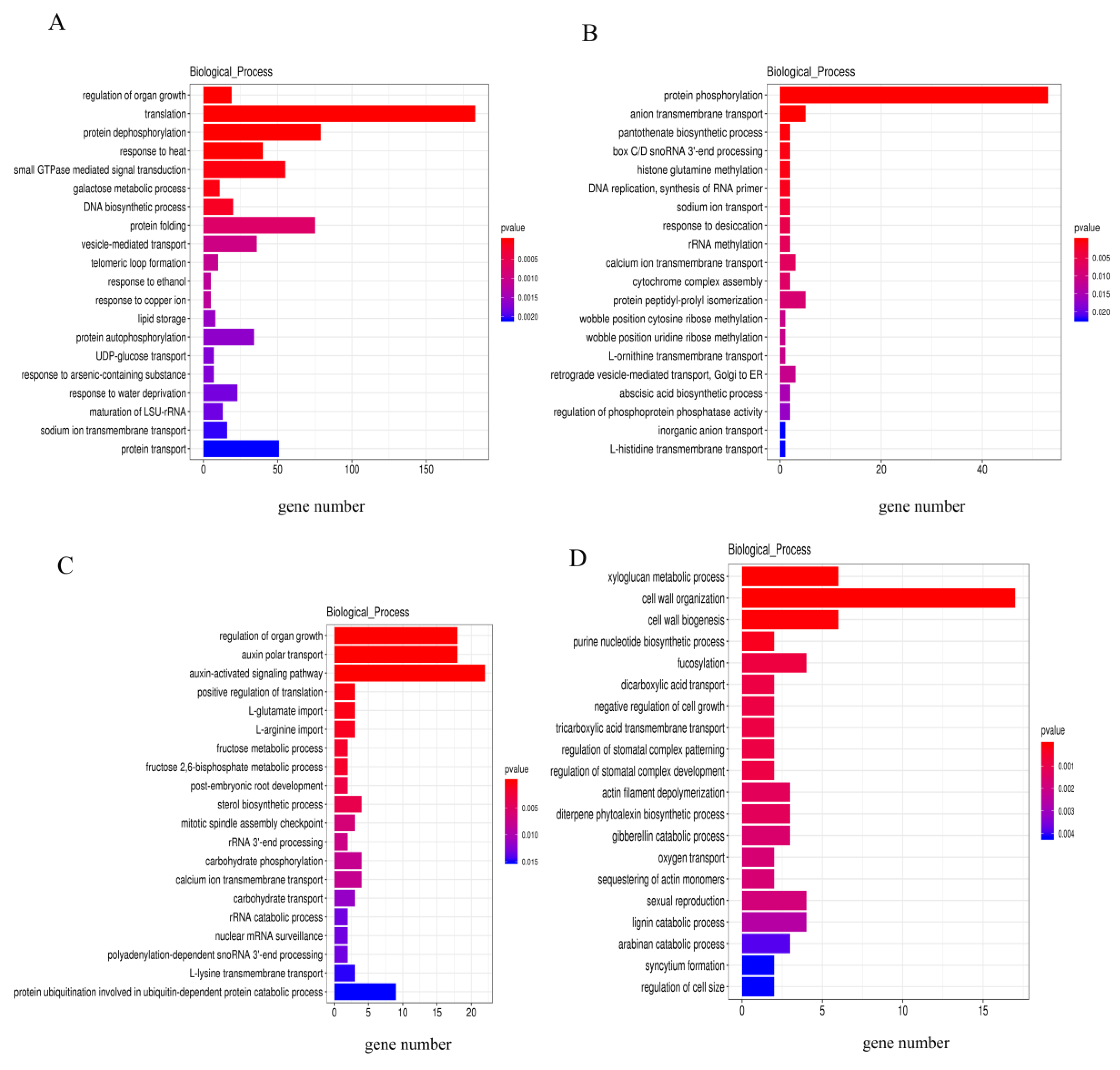
2.5. Prediction of Target Genes and Enrichment Analysis of DE lncRNAs
2.6. lncRNA Acting as Potential Regulator of the Auxin-Activated Signaling Pathway
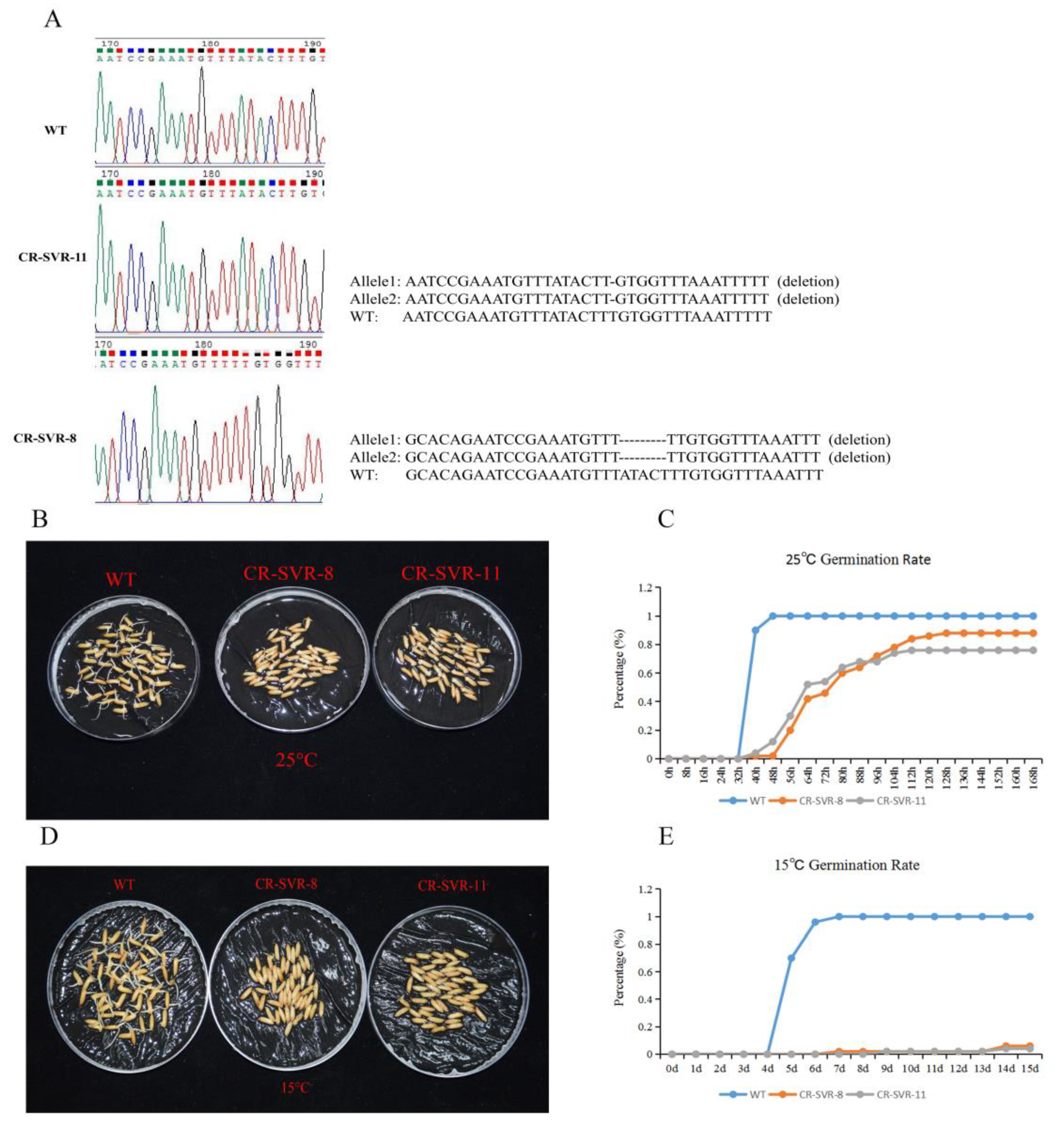
2.7. CRISPR/Cas9-Engineered Mutations in SVR Cause Delay of Germination
3. Discussion
3.1. lncRNAs Differ from mRNAs in Structure and Expression Pattern
3.2. lncRNAs May Work Cooperatively with mRNAs to Regulate Seed Vigor through the Auxin-Activated Pathway
4. Materials and Methods
4.1. Seed Germination and Sampling
4.2. RNA Extraction, Library Construction, and Strand-Specific Sequencing
4.3. Identification of lncRNAs
4.4. Prediction of Target Genes
4.5. Analysis of Differential Expression Patterns and Enrichment Analysis
4.6. Network Construction
4.7. qRT–PCR
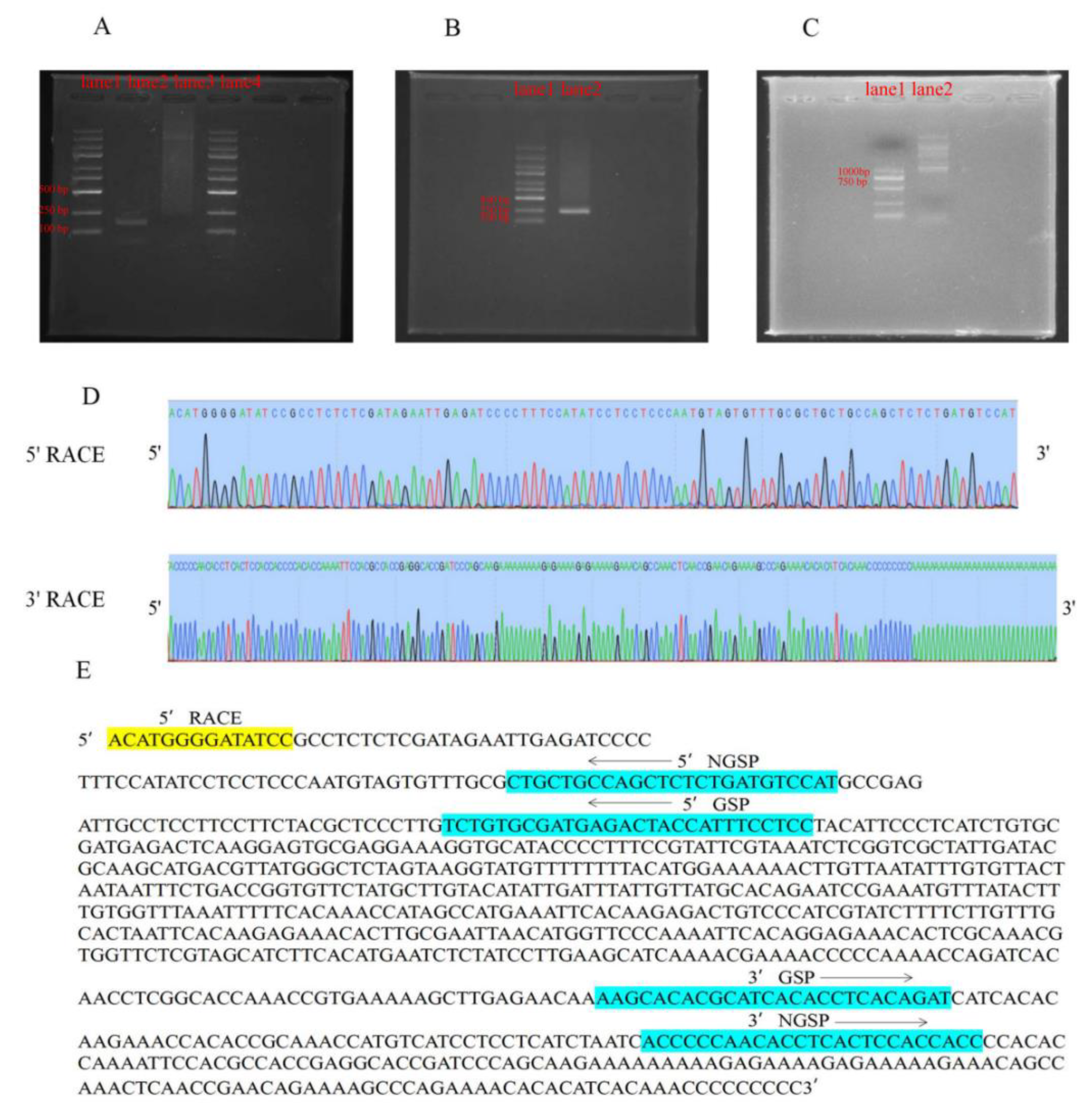
4.8. RACE Analysis
4.9. Construction of CRISPR/Cas9 Vector and Transgenic Plants
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farooq, M.; Siddique, K.H.M.; Rehman, H.; Aziz, T.; Lee, D.J.; Wahid, A. Rice direct seeding: Experiences, challenges and opportunities. Soil Tillage Res. 2011, 111, 87–98. [Google Scholar] [CrossRef]
- Xie, L.; Tan, Z.; Zhou, Y.; Xu, R.; Feng, L.; Xing, Y.; Qi, X. Identification and fine mapping of quantitative trait loci for seed vigor in germination and seedling establishment in rice. J. Integr. Plant Biol. 2014, 56, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Fujino, K.; Sekiguchi, H.; Matsuda, Y.; Sugimoto, K.; Ono, K.; Yano, M. Molecular identification of a major quantitative trait locus, qLTG3-1, controlling low-temperature germinability in rice. Proc. Natl. Acad. Sci. USA 2008, 105, 12623–12628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long non-coding RNA in plants in the era of reference sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, J.; Gan, Y. The new insight of auxin functions: Transition from seed dormancy to germination and floral opening in plants. Plant Growth Regul. 2020, 91, 169–174. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Zhang, Y.-C.; Sun, Y.-M.; Yu, Y.; Lei, M.-Q.; Yang, Y.-W.; Lian, J.-P.; Feng, Y.-Z.; Zhang, Z.; Yang, L.; et al. The parent-of-origin lncRNA MISSEN regulates rice endosperm development. Nat. Commun. 2021, 12, 6525. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Yang, J.; Mathioni, S.M.; Yu, J.; Shen, J.; Yang, X.; Wang, L.; Zhang, Q.; Cai, Z.; Xu, C.; et al. PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15144–15149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the timing of flowering in plants. Annu. Rev. Cell Dev. 2009, 25, 277–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lucia, F.; Crevillen, P.; Jones, A.M.E.; Greb, T.; Dean, C. A PHD-Polycomb Repressive Complex 2 triggers the epigenetic silencing of FLC during vernalization. Proc. Natl. Acad. Sci. USA 2008, 105, 16831–16836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, M.R.; Amasino, R.M. A Single Amino Acid Change in the Enhancer of Zeste Ortholog CURLY LEAF Results in Vernalization-Independent, Rapid Flowering in Arabidopsis. Plant Physiol. 2009, 151, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, D.; Primavesi, L.; Bishopp, A.; Roberts, G.; Doonan, J.; Jenuwein, T.; Goodrich, J. Silencing by plant Polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J. 2006, 25, 4638–4649. [Google Scholar] [CrossRef]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Spalding, E.P.; Gray, W.M. Rapid Auxin-Mediated Cell Expansion. Annu. Rev. Plant Biol. 2020, 71, 379–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef] [PubMed]
- McClure, B.A.; Guilfoyle, T. Characterization of a class of small auxin-inducible soybean polyadenylated RNAs. Plant Mol. Biol. 1987, 9, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family in rice (Oryza sativa). Genomics 2006, 88, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hao, X.; Cao, J. Small auxin upregulated RNA (SAUR) gene family in maize: Identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J. Integr. Plant Biol. 2014, 56, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Xiao, M.Z.; Liu, Y.; Fu, J.L.; He, Y.; Jiang, D.A. The small auxin-up RNA OsSAUR45 affects auxin synthesis and transport in rice. Plant Mol. Biol. 2017, 94, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.M.; Zhu, T.; Rothstein, S.J. SAUR39, a Small Auxin-Up RNA Gene, Acts as a Negative Regulator of Auxin Synthesis and Transport in Rice. Plant Physiol. 2009, 151, 691–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, W.; Sun, S.; Peng, L.; Huang, Z.; He, Y.; Wang, Z. The Rice Small Auxin-Up RNA Gene OsSAUR33 Regulates Seed Vigor via Sugar Pathway during Early Seed Germination. Int. J. Mol. Sci. 2021, 22, 1562. [Google Scholar] [CrossRef]
- Ren, H.; Gray, W.M. SAUR Proteins as Effectors of Hormonal and Environmental Signals in Plant Growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamm, P.; Kumar, P.P. Auxin and gibberellin responsive Arabidopsis SMALL AUXIN UP RNA36 regulates hypocotyl elongation in the light. Plant Cell Rep. 2013, 32, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, Y.; Gao, C.; She, W.; Lin, W.; Chen, Y.; Han, N.; Bian, H.; Zhu, M.; Wang, J. Tissue-specific expression of SMALL AUXIN UP RNA41 differentially regulates cell expansion and root meristem patterning in Arabidopsis. Plant Cell Physiol. 2013, 54, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inzé, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, K.; Isaacs, C.G.; Reeves, P.H.; Maloney, G.S.; Muday, G.K.; Nagpal, P.; Reed, J.W. Arabidopsis SMALL AUXIN UP RNA63 promotes hypocotyl and stamen filament elongation. Plant J. 2012, 71, 684–697. [Google Scholar] [CrossRef]
- Spartz, A.K.; Ren, H.; Park, M.Y.; Grandt, K.N.; Lee, S.H.; Murphy, A.S.; Sussman, M.R.; Overvoorde, P.J.; Gray, W.M. SAUR Inhibition of PP2C-D Phosphatases Activates Plasma Membrane H+-ATPases to Promote Cell Expansion in Arabidopsis. Plant Cell 2014, 26, 2129–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, S.A.; Ahmad, S.M.; Mumtaz, P.T.; Malik, A.A.; Dar, M.A.; Urwat, U.; Shah, R.A.; Ganai, N.A. Long non-coding RNAs: Mechanism of action and functional utility. Non-Coding RNA Res. 2016, 1, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Osak, M.; Bogu, G.K.; Stanton, L.W.; Johnson, R.; Lipovich, L. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 2010, 16, 1478–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Guo, X.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Wang, X.; Chu, M. Transcriptome Analysis Reveals Differentially Expressed Genes and Long Non-coding RNAs Associated with Fecundity in Sheep Hypothalamus with Different FecB Genotypes. Front. Cell Dev. Biol. 2021, 9, 633747. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Liang, Q.; Li, C.; Fu, S.; Kundu, J.K.; Zhou, X.; Wu, J. Transcriptome Analysis of Rice Reveals the lncRNA-mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses 2020, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Li, Q.; Jin, J.; Chen, H.; Zou, Y.; Huang, X.; Ding, Y. Genome-Wide Identification of lncRNAs Involved in Fertility Transition in the Photo-Thermosensitive Genic Male Sterile Rice Line Wuxiang, S. Front. Plant Sci. 2021, 11, 580050. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapa ssp. chinensis). Sci. Rep. 2019, 9, 5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Gan, L.; Wang, C.; Zhao, X.; Zhang, M.; Du, J.; Zhou, S.; Zhu, C. Genome-Wide Identification and Characterization of Potato Long Non-coding RNAs Associated with Phytophthora infestans Resistance. Front. Plant Sci. 2021, 12, 619062. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, K.; Han, Z.; Wang, P.; Fang, Y. Genome-wide identification of Arabidopsis long noncoding RNAs in response to the blue light. Sci. Rep. 2020, 10, 6229. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N. Genome-Wide Analysis Uncovers Regulation of Long Intergenic Noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yamada, M.; Han, X.; Ohler, U.; Benfey, P.N. High-Resolution Expression Map of the Arabidopsis Root Reveals Alternative Splicing and lincRNA Regulation. Dev. Cell 2016, 39, 508–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shahid, M.Q.; Wen, M.; Chen, S.; Yu, H.; Jiao, Y.; Lu, Z.; Li, Y.; Liu, X. Global identification and analysis revealed differentially expressed lncRNAs associated with meiosis and low fertility in autotetraploid rice. BMC Plant Biol. 2020, 20, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ajadi, A.A.; Wang, Y.; Tong, X.; Wang, H.; Tang, L.; Li, Z.; Shu, Y.; Liu, X.; Li, S.; et al. Genome-Wide Identification of lncRNAs During Rice Seed Development. Genes 2020, 11, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zhang, H.; Wang, Q.; Guo, R.; Wei, L.; Song, H.; Kuang, W.; Liao, J.; Huang, Y.; Wang, Z. Genome-wide identification and characterization of long non-coding RNAs involved in flag leaf senescence of rice. Plant Mol. Biol. 2021, 105, 655–684. [Google Scholar] [CrossRef] [PubMed]
- Kindgren, P.; Ard, R.; Ivanov, M.; Marquardt, S. Transcriptional read-through of the long non-coding RNA SVALKA governs plant cold acclimation. Nat. Commun. 2018, 9, 4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.; Zhang, X.; Ma, X.; Zhao, J. Spatio-temporal transcriptional dynamics of maize long non-coding RNAs responsive to drought stress. Genes 2019, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Tie, W.; Fu, L.; Yan, Y.; Liu, G.; Yan, W.; Li, Y.; Wu, C.; Zhang, J.; Hu, W. Strand-specific RNA-seq based identification and functional prediction of drought-responsive lncRNAs in cassava. BMC Genom. 2019, 20, 214. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Yang, P. Studies on the molecular mechanisms of seed germination. Proteomics 2015, 15, 1671–1679. [Google Scholar] [CrossRef]
- Finch Savage, B. Seeds: Physiology of Development, Germination and Dormancy. Seed Science Research; Bewley, J.D., Bradford, K.J., Hilhorst, H.W.M., Nonogaki, H., Eds.; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2013; Volume 23, p. 289. [Google Scholar]
- Bewley, J.D. Seed Germination and Dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- He, M.; He, C.Q.; Ding, N.Z. Abiotic Stresses: General Defenses of Land Plants and Chances for Engineering Multistress Tolerance. Front. Plant Sci. 2018, 9, 1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, U.C.; Nayyar, H.; Jha, R.; Khurshid, M.; Zhou, M.; Mantri, N.; Siddique, K.H.M. Long non-coding RNAs: Emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 2020, 20, 466. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Shi, Y.; Yang, S. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019, 222, 1690–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanneste, S.; Friml, J. Auxin: A trigger for change in plant development. Cell 2009, 136, 1005–10016. [Google Scholar] [CrossRef]
- Chapman, E.J.; Estelle, M. Mechanism of auxin-regulated gene expression in plants. Annu. Rev. Genet. 2009, 43, 265–285. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, H.; Zhao, Y.; Feng, Z.; Li, Q.; Yang, H.Q.; Luan, S.; Li, J.; He, Z.H. Auxin controls seed dormancy through stimulation of abscisic acid signaling by inducing ARF-mediated ABI3 activation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2013, 110, 15485–15490. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.M.; Sarkar, S.F.; Bonetta, D.; McCourt, P. The ABSCISIC ACID INSENSITIVE 3 (ABI3) gene is modulated by farnesylation and is involved in auxin signaling and lateral root development in Arabidopsis. Plant J. 2003, 34, 67–75. [Google Scholar] [CrossRef]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef]
- Ramaih, S.; Guedira, M.; Paulsen, G.M. Relationship of indoleacetic acid and tryptophan to dormancy and preharvest sprouting of wheat. Funct. Plant Biol. 2003, 30, 939–945. [Google Scholar] [CrossRef]
- Shuai, H.; Meng, Y.; Luo, X.; Chen, F.; Zhou, W.; Dai, Y.; Qi, Y.; Du, J.; Yang, F.; Liu, J.; et al. Exogenous auxin represses soybean seed germination through decreasing the gibberellin/abscisic acid (GA/ABA) ratio. Sci. Rep. 2017, 7, 12620. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Lin, X.; Zhu, Y.; Chen, W.; Lin, L.; Chen, D.; Huang, J.; Pan, K.; Lin, Y.; Wu, B.; Dai, Y.; et al. Integrated analysis of long non-coding RNAs and mRNA expression profiles reveals the potential role of lncRNAs in gastric cancer pathogenesis. Int. J. Oncol. 2014, 45, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liu, H.; Ding, Y.; Zhou, Y.; Jin, W.; Xie, K.; Chen, L.L. CRISPR-P 2.0: An improved CRISPR-Cas9 tool for genome editing in plants. Mol. Plant 2017, 10, 530–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Q.; Liu, J.; Weng, H.; Yuan, X.; Xiao, W.; Wang, H. A Long Noncoding RNA Derived from lncRNA–mRNA Networks Modulates Seed Vigor. Int. J. Mol. Sci. 2022, 23, 9472. https://doi.org/10.3390/ijms23169472
Gao Q, Liu J, Weng H, Yuan X, Xiao W, Wang H. A Long Noncoding RNA Derived from lncRNA–mRNA Networks Modulates Seed Vigor. International Journal of Molecular Sciences. 2022; 23(16):9472. https://doi.org/10.3390/ijms23169472
Chicago/Turabian StyleGao, Qiaoli, Jinzhao Liu, Huibin Weng, Xi Yuan, Wuming Xiao, and Hui Wang. 2022. "A Long Noncoding RNA Derived from lncRNA–mRNA Networks Modulates Seed Vigor" International Journal of Molecular Sciences 23, no. 16: 9472. https://doi.org/10.3390/ijms23169472