
Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. OS and Mitochondrial Dysfunction in ALS
3. Genetic Variants and OS
3.1. SOD1 Mutations
3.2. TARDBP Mutation
3.3. FUS Mutation
3.4. C9orf72 Mutation Associated with OS
3.5. Other Mutations Associated with OS
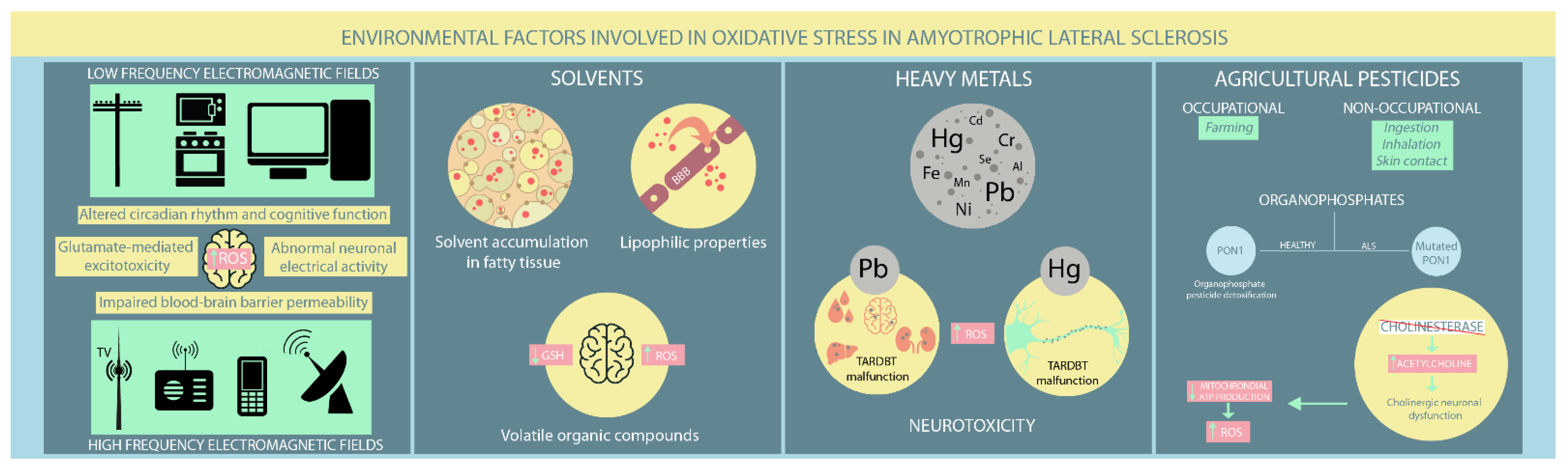
4. Environmental Factors Associated with OS in ALS
4.1. EMFs
4.2. Solvents
4.3. Heavy Metals
4.4. Agricultural Pesticides
5. ALS Therapies Effects on OS Pathways
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar]
- Greco, V.; Longone, P.; Spalloni, A.; Pieroni, L.; Urbani, A. Crosstalk Between Oxidative Stress and Mitochondrial Damage: Focus on Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2019, 1158, 71–82. [Google Scholar] [PubMed]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef]
- Valko, K.; Ciesla, L. Amyotrophic lateral sclerosis. Prog. Med. Chem. 2019, 58, 63–117. [Google Scholar] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox. Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell Longev. 2020, 15, 5021694. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Agar, J.; Durham, H. Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph. Lateral. Scler. Other Motor. Neuron. Disord. 2003, 4, 232–242. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef]
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 2007, 207, 107–117. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free. Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 2019, 138, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137 Pt. 8, 2329–2345. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Vehviläinen, P.; Koistinaho, J.; Gundars, G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell Neurosci. 2014, 8, 126. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Sasaki, S.; Warita, H.; Murakami, T.; Shibata, N.; Komori, T.; Abe, K.; Kobayashi, M.; Iwata, M. Ultrastructural study of aggregates in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005, 109, 247–255. [Google Scholar] [CrossRef]
- Higgins, C.M.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef]
- Hitchler, M.J.; Domann, F.E. Regulation of CuZnSOD and its redox signaling potential: Implications for amyotrophic lateral sclerosis. Antioxid. Redox. Signal. 2014, 20, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Outten, C.E.; Proescher, J.B.; Rosenfeld, L.; Watson, W.H.; Whitson, L.J.; Hart, P.J.; Jensen, L.T.; Cizewski Culotta, V. The effects of glutaredoxin and copper activation pathways on the disulfide and stability of Cu,Zn superoxide dismutase. J. Biol. Chem. 2006, 281, 28648–28656. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Feneberg, E.; Gordon, D.; Thompson, A.G.; Finelli, M.J.; Dafinca, R.; Candalija, A.; Charles, P.D.; Mäger, I.; Wood, M.J.; Fischer, R.; et al. An ALS-linked mutation in TDP-43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol. Dis. 2020, 144, 105050. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Aulas, A.; Vande Velde, C. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Koppers, M.; Groen, E.J.N.; van den Heuvel, D.M.A.; Dini Modigliani, S.; Anink, J.J.; Fumoto, K.; van Diggelen, F.; Snelting, A.; Sodaar, P.; et al. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta. Neuropathol. 2016, 132, 175–196. [Google Scholar] [CrossRef]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Park, S.K.; Park, S.; Liebman, S.W. Respiration Enhances TDP-43 Toxicity, but TDP-43 Retains Some Toxicity in the Absence of Respiration. J. Mol. Biol. 2019, 431, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Özdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta. Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Tian, Y.P.; Che, F.Y.; Su, Q.P.; Lu, Y.C.; You, C.P.; Huang, L.M.; Wang, S.G.; Wang, L.; Yu, J.X. Effects of mutant TDP-43 on the Nrf2/ARE pathway and protein expression of MafK and JDP2 in NSC-34 cells. Genet. Mol. Res. 2017, 16, gmr16029638. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.D.; Ichida, J.K. C9ORF72 protein function and immune dysregulation in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 713, 134523. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Vatsavayai, S.C.; Nana, A.L.; Yokoyama, J.S.; Seeley, W.W. C9orf72-FTD/ALS pathogenesis: Evidence from human neuropathological studies. Acta Neuropathol. 2019, 137, 1–26. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef]
- Emara, M.M.; Ivanov, P.; Hickman, T.; Dawra, N.; Tisdale, S.; Kedersha, N.; Hu, G.F.; Anderson, P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J. Biol. Chem. 2010, 285, 10959–10968. [Google Scholar] [CrossRef]
- Hoang, T.T.; Johnson, D.A.; Raines, R.T.; Johnson, J.A. Angiogenin activates the astrocytic Nrf2/antioxidant-response element pathway and thereby protects murine neurons from oxidative stress. J. Biol. Chem. 2019, 294, 15095–15103. [Google Scholar] [CrossRef]
- Hoang, T.T.; Smith, T.P.; Raines, R.T. A Boronic Acid Conjugate of Angiogenin that Shows ROS-Responsive Neuroprotective Activity. Angew. Chem. Int. Ed. Engl. 2017, 56, 2619–2622. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; LeClerc, A.L.; Keagle, P.J.; Glass, J.D.; Wills, A.M.; van Blitterswijk, M.; Bosco, D.A.; Rodriguez-Leyva, I.; Gellera, C.; Ratti, A.; et al. Paraoxonase gene mutations in amyotrophic lateral sclerosis. Ann. Neurol. 2010, 68, 102–107. [Google Scholar] [CrossRef]
- Wills, A.M.; Cronin, S.; Slowik, A.; Kasperaviciute, D.; Van Es, M.A.; Morahan, J.M.; Valdmanis, P.N.; Meininger, V.; Melki, J.; Shaw, C.E.; et al. A large-scale international meta-analysis of paraoxonase gene polymorphisms in sporadic ALS. Neurology 2009, 73, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; Blokhuis, A.; van Es, M.A.; van Vught, P.W.; Rowicka, P.A.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. Rare and common paraoxonase gene variants in amyotrophic lateral sclerosis patients. Neurobiol. Aging 2012, 33, 1845.e1. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Luna, J.; Leleu, J.P.; Preux, P.M.; Corcia, P.; Couratier, P.; Marin, B.; Boumediene, F.; Fralim Consortium. Residential exposure to ultra high frequency electromagnetic fields emitted by Global System for Mobile (GSM) antennas and amyotrophic lateral sclerosis incidence: A geo-epidemiological population-based study. Environ. Res. 2019, 176, 108525. [Google Scholar] [CrossRef]
- Gunnarsson, L.G.; Bodin, L. Amyotrophic Lateral Sclerosis and Occupational Exposures: A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public. Health. 2018, 15, 2371. [Google Scholar] [CrossRef]
- Gunnarsson, L.G.; Bodin, L. Occupational Exposures and Neurodegenerative Diseases-A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public. Health. 2019, 16, 337. [Google Scholar] [CrossRef]
- Koeman, T.; Slottje, P.; Schouten, L.J.; Peters, S.; Huss, A.; Veldink, J.H.; Kromhout, H.; van den Brandt, P.A.; Vermeulen, R. Occupational exposure and amyotrophic lateral sclerosis in a prospective cohort. Occup. Environ. Med. 2017, 74, 578–585. [Google Scholar] [CrossRef]
- Filippini, T.; Tesauro, M.; Fiore, M.; Malagoli, C.; Consonni, M.; Violi, F.; Iacuzio, L.; Arcolin, E.; Oliveri Conti, G.; Cristaldi, A.; et al. Environmental and Occupational Risk Factors of Amyotrophic Lateral Sclerosis: A Population-Based Case-Control Study. Int. J. Environ. Res. Public. Health. 2020, 17, 2882. [Google Scholar] [CrossRef]
- Seelen, M.; Vermeulen, R.C.; van Dillen, L.S.; van der Kooi, A.J.; Huss, A.; de Visser, M.; van den Berg, L.H.; Veldink, J.H. Residential exposure to extremely low frequency electromagnetic fields and the risk of ALS. Neurology 2014, 83, 1767–1769. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Sanchez de la Torre, J.R.; Paz-Fajardo, L.; Limia, C.; Santurtun, A.; Cifra, M.; Kourtidis, K.; Fdez-Arroyabe, P. The role of magnetic fields in neurodegenerative diseases. Int. J. Biometeorol. 2021, 65, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Capozzella, A.; Sacco, C.; Chighine, A.; Loreti, B.; Scala, B.; Casale, T.; Sinibaldi, F.; Tomei, G.; Giubilati, R.; Tomei, F.; et al. Work related etiology of amyotrophic lateral sclerosis (ALS): A meta-analysis. Ann. Ig. 2014, 26, 456–472. [Google Scholar] [PubMed]
- Consales, C.; Merla, C.; Marino, C.; Benassi, B. Electromagnetic fields, oxidative stress, and neurodegeneration. Int. J. Cell Biol. 2012, 2012, 683897. [Google Scholar] [CrossRef]
- Poulletier de Gannes, F.; Ruffié, G.; Taxile, M.; Ladevèze, E.; Hurtier, A.; Haro, E.; Duleu, S.; Charlet de Sauvage, R.; Billaudel, B.; Geffard, M.; et al. Amyotrophic lateral sclerosis (ALS) and extremely-low frequency (ELF) magnetic fields: A study in the SOD-1 transgenic mouse model. Amyotroph. Lateral. Scler. 2009, 10, 370–373. [Google Scholar] [CrossRef]
- Filippini, T.; Hatch, E.E.; Vinceti, M. Residential exposure to electromagnetic fields and risk of amyotrophic lateral sclerosis: A dose-response meta-analysis. Sci. Rep. 2021, 11, 11939. [Google Scholar] [CrossRef]
- Falone, S.; Mirabilio, A.; Carbone, M.C.; Zimmitti, V.; Di Loreto, S.; Mariggiò, M.A.; Mancinelli, R.; Di Ilio, C.; Amicarelli, F. Chronic exposure to 50 Hz magnetic fields causes a significant weakening of antioxidant defence systems in aged rat brain. Int. J. Biochem. Cell Biol. 2008, 40, 2762–2770. [Google Scholar] [CrossRef]
- Jelenković, A.; Janać, B.; Pesić, V.; Jovanović, D.M.; Vasiljević, I.; Prolić, Z. Effects of extremely low-frequency magnetic field in the brain of rats. Brain. Res. Bull. 2006, 68, 355–360. [Google Scholar] [CrossRef]
- Oshiro, S.; Morioka, M.S.; Kikuchi, M. Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv. Pharmacol. Sci. 2011, 2011, 378278. [Google Scholar] [CrossRef]
- Liebl, M.P.; Windschmitt, J.; Besemer, A.S.; Schäfer, A.K.; Reber, H.; Behl, C.; Clement, A.M. Low-frequency magnetic fields do not aggravate disease in mouse models of Alzheimer’s disease and amyotrophic lateral sclerosis. Sci. Rep. 2015, 5, 8585. [Google Scholar] [CrossRef]
- Ratner, M.H.; Jabre, J.F.; Ewing, W.M.; Abou-Donia, M.; Oliver, L.C. Amyotrophic lateral sclerosis—A case report and mechanistic review of the association with toluene and other volatile organic compounds. Am. J. Ind. Med. 2018, 61, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, A.S.; Hansen, J.; Thompson, S.; Gredal, O.; Weisskopf, M.G. A mixtures approach to solvent exposures and amyotrophic lateral sclerosis: A population-based study in Denmark. Eur. J. Epidemiol. 2020, 35, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.L.; Kamel, F.; Lundholm, C.; Feychting, M.; Weibull, C.E.; Sandler, D.P.; Wiebert, P.; Sparén, P.; Ye, W.; Fang, F. Occupational exposures and the risk of amyotrophic lateral sclerosis. Occup. Environ. Med. 2017, 74, 87–92. [Google Scholar] [CrossRef]
- Fang, F.; Quinlan, P.; Ye, W.; Barber, M.K.; Umbach, D.M.; Sandler, D.P.; Kamel, F. Workplace exposures and the risk of amyotrophic lateral sclerosis. Environ. Health. Perspect. 2009, 117, 1387–1392. [Google Scholar] [CrossRef]
- Roberts, A.L.; Johnson, N.J.; Cudkowicz, M.E.; Eum, K.D.; Weisskopf, M.G. Job-related formaldehyde exposure and ALS mortality in the USA. J. Neurol. Neurosurg. Psychiatry 2016, 87, 786–788. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Morozova, N.; O’Reilly, E.J.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2009, 80, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.M.; Barchowsky, A.; Bowser, R.; Youk, A.; Talbott, E.O. Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: A meta-analysis of epidemiological studies: Pesticide exposure as a risk factor for ALS. Environ. Res. 2012, 117, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; Caller, T.A.; Tandan, R.; Duell, E.J.; Henegan, P.L.; Field, N.C.; Bradley, W.G.; Stommel, E.W. Environmental and Occupational Exposures and Amyotrophic Lateral Sclerosis in New England. Neurodegener. Dis. 2017, 17, 110–116. [Google Scholar] [CrossRef]
- Malek, A.M.; Barchowsky, A.; Bowser, R.; Heiman-Patterson, T.; Lacomis, D.; Rana, S.; Youk, A.; Talbott, E.O. Exposure to hazardous air pollutants and the risk of amyotrophic lateral sclerosis. Environ. Pollut. 2015, 197, 181–186. [Google Scholar] [CrossRef]
- Georgieva, T.; Michailova, A.; Panev, T.; Popov, T. Possibilities to control the health risk of petrochemical workers. Int. Arch. Occup. Environ. Health 2002, 75, S21–S26. [Google Scholar] [CrossRef]
- Cheong, I.; Marjańska, M.; Deelchand, D.K.; Eberly, L.E.; Walk, D.; Öz, G. Ultra-High Field Proton MR Spectroscopy in Early-Stage Amyotrophic Lateral Sclerosis. Neurochem. Res. 2017, 42, 1833–1844, Erratum in 2017, 42, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Mohamed, M.A.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E.L. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA. Neurol. 2013, 70, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Myhre, O.; Fonnum, F. The effect of aliphatic, naphthenic, and aromatic hydrocarbons on production of reactive oxygen species and reactive nitrogen species in rat brain synaptosome fraction: The involvement of calcium, nitric oxide synthase, mitochondria, and phospholipase A. Biochem. Pharmacol. 2001, 62, 119–128. [Google Scholar] [CrossRef]
- Mills, K.R.; Nithi, K.A. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve 1997, 20, 1137–1141. [Google Scholar] [CrossRef]
- Kikuchi, H.; Doh-ura, K.; Kawashima, T.; Kira, J.; Iwaki, T. Immunohistochemical analysis of spinal cord lesions in amyotrophic lateral sclerosis using microtubule-associated protein 2 (MAP2) antibodies. Acta Neuropathol. 1999, 97, 13–21. [Google Scholar] [CrossRef]
- Farah, C.A.; Nguyen, M.D.; Julien, J.P.; Leclerc, N. Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2003, 84, 77–86. [Google Scholar] [CrossRef]
- Gotohda, T.; Tokunaga, I.; Kitamura, O.; Kubo, S. Toluene inhalation induced neuronal damage in the spinal cord and changes of neurotrophic factors in rat. Leg. Med. 2007, 9, 123–127. [Google Scholar] [CrossRef]
- Oggiano, R.; Solinas, G.; Forte, G.; Bocca, B.; Farace, C.; Pisano, A.; Sotgiu, M.A.; Clemente, S.; Malaguarnera, M.; Fois, A.G.; et al. Trace elements in ALS patients and their relationships with clinical severity. Chemosphere 2018, 197, 457–466. [Google Scholar] [CrossRef]
- Peters, S.; Broberg, K.; Gallo, V.; Levi, M.; Kippler, M.; Vineis, P.; Veldink, J.; van den Berg, L.; Middleton, L.; Travis, R.C.; et al. Blood Metal Levels and Amyotrophic Lateral Sclerosis Risk: A Prospective Cohort. Ann. Neurol. 2021, 89, 125–133. [Google Scholar] [CrossRef]
- Parkin Kullmann, J.A.; Pamphlett, R. A Comparison of Mercury Exposure from Seafood Consumption and Dental Amalgam Fillings in People with and without Amyotrophic Lateral Sclerosis (ALS): An International Online Case-Control Study. Int. J. Environ. Res. Public Health 2018, 15, 2874. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; O’Brien, K.M.; Jackson, B.P.; Sandler, D.P.; Kaye, W.E.; Wagner, L.; Stommel, E.W.; Horton, D.K.; Mehta, P.; Weinberg, C.R. Keratinous biomarker of mercury exposure associated with amyotrophic lateral sclerosis risk in a nationwide U.S. study. Amyotroph. Lateral. Scler. Frontotemporal. Degener. 2020, 21, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Takayanagi, T.; Ishitani, A.; Hirota, T. Mercury in hair of patients with ALS. Rinsho Shinkeigaku 1989, 29, 844–848. [Google Scholar] [PubMed]
- Bocca, B.; Forte, G.; Oggiano, R.; Clemente, S.; Asara, Y.; Peruzzu, A.; Farace, C.; Pala, S.; Fois, A.G.; Pirina, P.; et al. Level of neurotoxic metals in amyotrophic lateral sclerosis: A population-based case-control study. J. Neurol. Sci. 2015, 359, 11–17. [Google Scholar] [CrossRef]
- Moriwaka, F.; Satoh, H.; Ejima, A.; Watanabe, C.; Tashiro, K.; Hamada, T.; Matsumoto, A.; Shima, K.; Yanagihara, T.; Fukazawa, T. Mercury and selenium contents in amyotrophic lateral sclerosis in Hokkaido, the northernmost island of Japan. J. Neurol. Sci. 1993, 118, 38–42. [Google Scholar] [CrossRef]
- Dickerson, A.S.; Hansen, J.; Gredal, O.; Weisskopf, M.G. Study of Occupational Chromium, Iron, and Nickel Exposure and Amyotrophic Lateral Sclerosis in Denmark. Int. J. Environ. Res. Public Health 2020, 17, 8086. [Google Scholar] [CrossRef]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef]
- Farace, C.; Fenu, G.; Lintas, S.; Oggiano, R.; Pisano, A.; Sabalic, A.; Solinas, G.; Bocca, B.; Forte, G.; Madeddu, R. Amyotrophic lateral sclerosis and lead: A systematic update. Neurotoxicology 2020, 81, 80–88. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Mandrioli, J.; Violi, F.; Bargellini, A.; Weuve, J.; Fini, N.; Grill, P.; Michalke, B. Lead, cadmium and mercury in cerebrospinal fluid and risk of amyotrophic lateral sclerosis: A case-control study. J. Trace. Elem. Med. Biol. 2017, 43, 121–125. [Google Scholar] [CrossRef]
- Kamel, F.; Umbach, D.M.; Stallone, L.; Richards, M.; Hu, H.; Sandler, D.P. Association of lead exposure with survival in amyotrophic lateral sclerosis. Environ. Health Perspect. 2008, 116, 943–947. [Google Scholar] [CrossRef]
- Barbeito, A.G.; Martinez-Palma, L.; Vargas, M.R.; Pehar, M.; Mañay, N.; Beckman, J.S.; Barbeito, L.; Cassina, P. Lead exposure stimulates VEGF expression in the spinal cord and extends survival in a mouse model of ALS. Neurobiol. Dis. 2010, 37, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.O.; Atchison, W.D. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology 2009, 30, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Dhawan, U.; Boudeau, S.; Lei, S.; Carlomagno, Y.; Knobel, M.; Al Mohanna, L.F.A.; Boomhower, S.R.; Newland, M.C.; Sherr, D.H.; et al. Heavy Metal Neurotoxicants Induce ALS-Linked TDP-43 Pathology. Toxicol. Sci. 2019, 167, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Gajowiak, A.; Styś, A.; Starzyński, R.R.; Staroń, R.; Lipiński, P. Misregulation of iron homeostasis in amyotrophic lateral sclerosis. Postepy. Hig. Med. Dosw. 2016, 70, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Patti, F.; Fiore, M.; Chisari, C.G.; D’Amico, E.; Lo Fermo, S.; Toscano, S.; Copat, C.; Ferrante, M.; Zappia, M. CSF neurotoxic metals/metalloids levels in amyotrophic lateral sclerosis patients: Comparison between bulbar and spinal onset. Environ. Res. 2020, 188, 109820. [Google Scholar] [CrossRef]
- Tesauro, M.; Bruschi, M.; Filippini, T.; D’Alfonso, S.; Mazzini, L.; Corrado, L.; Consonni, M.; Vinceti, M.; Fusi, P.; Urani, C. Metal(loid)s role in the pathogenesis of amyotrophic lateral sclerosis: Environmental, epidemiological, and genetic data. Environ. Res. 2021, 192, 110292. [Google Scholar] [CrossRef]
- Gil-Bea, F.J.; Aldanondo, G.; Lasa-Fernández, H.; López de Munain, A.; Vallejo-Illarramendi, A. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert. Rev. Mol. Med. 2017, 19, e7. [Google Scholar] [CrossRef]
- Pitts, M.W.; Byrns, C.N.; Ogawa-Wong, A.N.; Kremer, P.; Berry, M.J. Selenoproteins in nervous system development and function. Biol. Trace. Elem. Res. 2014, 161, 231–245. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Sies, H. Selenium homeostasis and antioxidant selenoproteins in brain: Implications for disorders in the central nervous system. Arch. Biochem. Biophys. 2013, 536, 152–157. [Google Scholar] [CrossRef]
- Estevez, A.O.; Mueller, C.L.; Morgan, K.L.; Szewczyk, N.J.; Teece, L.; Miranda-Vizuete, A.; Estevez, M. Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans. Neurotoxicology 2012, 33, 1021–1032. [Google Scholar] [CrossRef]
- Van Campenhout, K.; Infante, H.G.; Adams, F.; Blust, R. Induction and binding of Cd, Cu, and Zn to metallothionein in carp (Cyprinus carpio) using HPLC-ICP-TOFMS. Toxicol. Sci. 2004, 80, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Oggiano, R.; Pisano, A.; Sabalic, A.; Farace, C.; Fenu, G.; Lintas, S.; Forte, G.; Bocca, B.; Madeddu, R. An overview on amyotrophic lateral sclerosis and cadmium. Neurol. Sci. 2021, 42, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Maya, S.; Prakash, T.; Madhu, K.D.; Goli, D. Multifaceted effects of aluminium in neurodegenerative diseases: A review. Biomed. Pharmacother. 2016, 83, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Kihira, T.; Mukoyama, M.; Ando, K.; Yase, Y.; Yasui, M. Determination of manganese concentrations in the spinal cords from amyotrophic lateral sclerosis patients by inductively coupled plasma emission spectroscopy. J. Neurol. Sci. 1990, 98, 251–258. [Google Scholar] [CrossRef]
- Roos, E.; Wärmländer, S.K.T.S.; Meyer, J.; Sholts, S.B.; Jarvet, J.; Gräslund, A.; Roos, P.M. Amyotrophic Lateral Sclerosis After Exposure to Manganese from Traditional Medicine Procedures in Kenya. Biol. Trace. Elem. Res. 2021, 199, 3618–3624. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Violi, F.; Rothman, K.J.; Costanzini, S.; Malagoli, C.; Wise, L.A.; Odone, A.; Signorelli, C.; Iacuzio, L.; et al. Pesticide exposure assessed through agricultural crop proximity and risk of amyotrophic lateral sclerosis. Environ. Health 2017, 16, 91. [Google Scholar] [CrossRef]
- Tosi, L.; Righetti, C.; Adami, L.; Zanette, G. October 1942: A strange epidemic paralysis in Saval, Verona, Italy. Revision and diagnosis 50 years later of tri-ortho-cresyl phosphate poisoning. J. Neurol. Neurosurg. Psychiatry 1994, 57, 810–813. [Google Scholar] [CrossRef]
- Burns, C.J.; Beard, K.K.; Cartmill, J.B. Mortality in chemical workers potentially exposed to 2,4-dichlorophenoxyacetic acid (2,4-D) 1945–94: An update. Occup. Environ. Med. 2001, 58, 24–30. [Google Scholar] [CrossRef]
- Vinceti, M.; Violi, F.; Tzatzarakis, M.; Mandrioli, J.; Malagoli, C.; Hatch, E.E.; Fini, N.; Fasano, A.; Rakitskii, V.N.; Kalantzi, O.I.; et al. Pesticides, polychlorinated biphenyls and polycyclic aromatic hydrocarbons in cerebrospinal fluid of amyotrophic lateral sclerosis patients: A case-control study. Environ. Res. 2017, 155, 261–267. [Google Scholar] [CrossRef]
- Mostafalou, S.; Abdollahi, M. The link of organophosphorus pesticides with neurodegenerative and neurodevelopmental diseases based on evidence and mechanisms. Toxicology 2018, 409, 44–52. [Google Scholar] [CrossRef]
- Del Pino, J.; Moyano, P.; Anadon, M.J.; García, J.M.; Díaz, M.J.; García, J.; Frejo, M.T. Acute and long-term exposure to chlorpyrifos induces cell death of basal forebrain cholinergic neurons through AChE variants alteration. Toxicology 2015, 336, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karami-Mohajeri, S.; Ahmadipour, A.; Rahimi, H.R.; Abdollahi, M. Adverse effects of organophosphorus pesticides on the liver: A brief summary of four decades of research. Arh. Hig. Rada. Toksikol. 2017, 68, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2007, 1, CD001447. [Google Scholar]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Writing Group on Behalf of the Edaravone (MCI-186) ALS 17 Study Group. Exploratory double-blind, parallel-group, placebo-controlled extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 2017, 18 (Suppl. 1), 20–31. [Google Scholar] [CrossRef]
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Takahashi, F.; Takei, K.; Tsuda, K.; Palumbo, J. Post-hoc analysis of MCI186–17, the extension study to MCI186–16, the confirmatory double-blind, parallel-group, placebo-controlled study of edaravone in amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. Frontotemporal Degener. 2017, 18 (Suppl. 1), 32–39. [Google Scholar] [CrossRef]
- Jami, M.S.; Salehi-Najafabadi, Z.; Ahmadinejad, F.; Hoedt, E.; Chaleshtori, M.H.; Ghatrehsamani, M.; Neubert, T.A.; Larsen, J.P.; Møller, S.G. Edaravone leads to proteome changes indicative of neuronal cell protection in response to oxidative stress. Neurochem. Int. 2015, 90, 134–141. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.-S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants. 2017, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Pérez-González, A.; Galano, A. OH radical scavenging activity of Edaravone: Mechanism and kinetics. J. Phys. Chem. B 2011, 115, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Matic, I.; Strobbe, D.; Frison, M.; Campanella, M. Controlled and Impaired Mitochondrial Quality in Neurons: Molecular Physiology and Prospective Pharmacology. Pharmacol. Res. 2015, 99, 410–424. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Rauchová, H. Coenzyme Q10 effects in neurological diseases. Physiol. Res. 2021, 70 (Suppl. 4), S683–S714. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef]
- Lum, J.S.; Brown, M.L.; Farrawell, N.E.; McAlary, L.; Ly, D.; Chisholm, C.G.; Snow, J.; Vine, K.L.; Karl, T.; Kreilaus, F.; et al. CuATSM improves motor function and extends survival but is not tolerated at a high dose in SOD1G93A mice with a C57BL/6 background. Sci. Rep. 2021, 11, 19392. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motataianu, A.; Serban, G.; Barcutean, L.; Balasa, R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int. J. Mol. Sci. 2022, 23, 9339. https://doi.org/10.3390/ijms23169339
Motataianu A, Serban G, Barcutean L, Balasa R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. International Journal of Molecular Sciences. 2022; 23(16):9339. https://doi.org/10.3390/ijms23169339
Chicago/Turabian StyleMotataianu, Anca, Georgiana Serban, Laura Barcutean, and Rodica Balasa. 2022. "Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors" International Journal of Molecular Sciences 23, no. 16: 9339. https://doi.org/10.3390/ijms23169339