
Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s Disease

Abstract
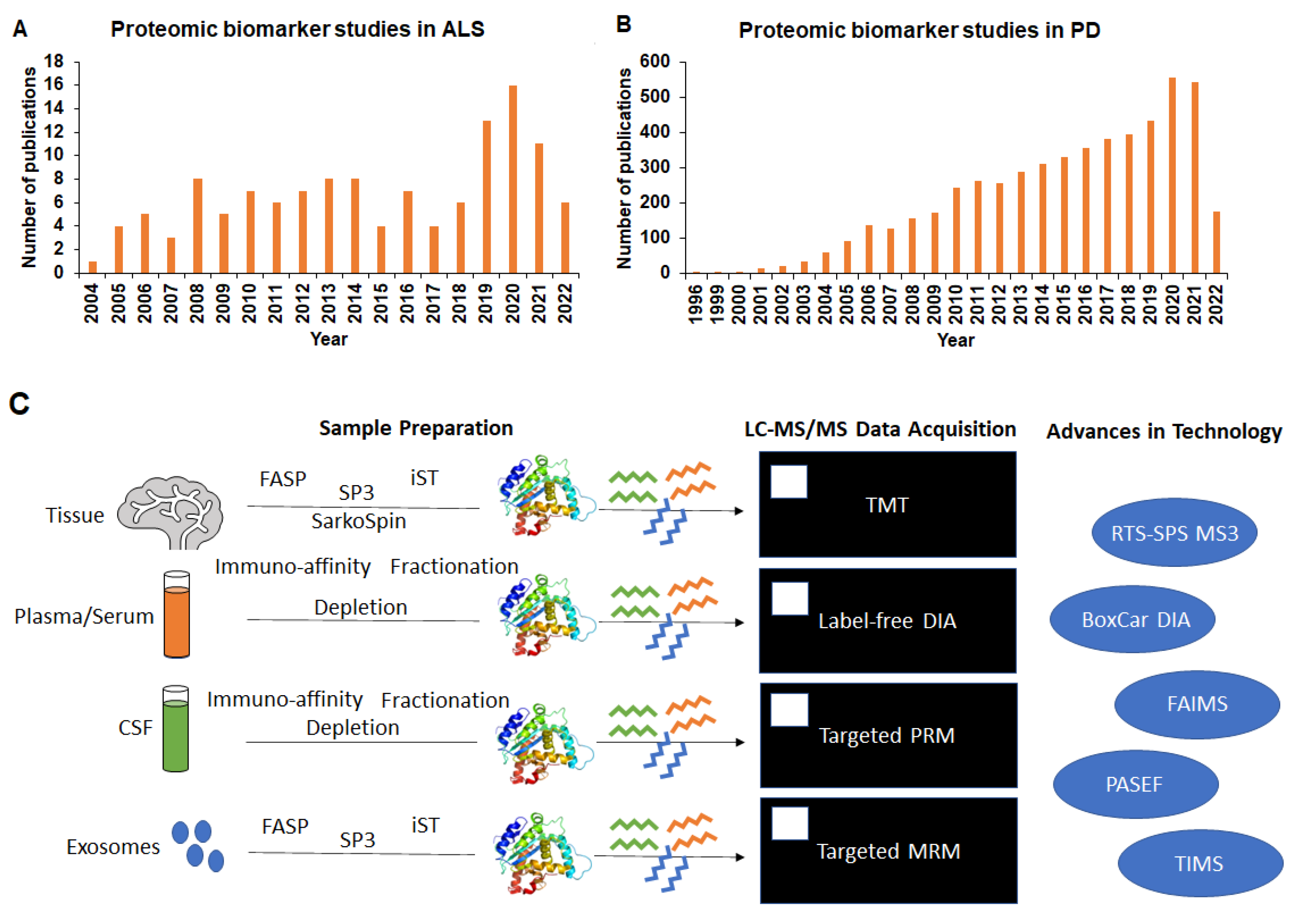
:1. Introduction
2. Overview of Proteomics Technological Advancement for Biomarker Discovery
3. Proteomics for Biomarker Discovery in ALS
3.1. Differential Expression of Proteins and Interactome from Post-Mortem Human Tissue as Biomarkers in ALS
3.2. Post-Translational Modifications in TDP43 from Tissue Proteomics Studies in ALS
3.3. Plasma and Serum as Sources for Proteomic Biomarkers in ALS
3.4. Cerebrospinal Fluid (CSF) Proteomic Biomarker Identification
3.5. Exosomes Proteomics in Biomarker Identification
4. Proteomics for Biomarker Discovery in PD
4.1. Differential Expression of Proteins from Post-Mortem Human Tissue as Biomarkers in PD
4.2. Post-Translational Modifications in Key Proteins from Tissue Proteomics Studies in PD
4.3. Plasma and Serum as Sources for Proteomic Biomarkers in PD
4.4. Cerebrospinal Fluid (CSF) as a Source for Proteomic Biomarkers in PD
4.5. Exosomes Proteomics in Biomarker Identification
5. Clinical Trials Using Proteomics for Biomarker Discovery in ALS and PD
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef]
- Edgar, S.; Ellis, M.; Abdul-Aziz, N.A.; Goh, K.J.; Shahrizaila, N.; Kennerson, M.L.; Ahmad-Annuar, A. Mutation analysis of SOD1, C9orf72, TARDBP and FUS genes in ethnically-diverse Malaysian patients with amyotrophic lateral sclerosis (ALS). Neurobiol. Aging 2021, 108, 200–206. [Google Scholar] [CrossRef]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013, 21, 102–108. [Google Scholar] [CrossRef]
- Andersen, P.M. Mutation in C9orf72 changes the boundaries of ALS and FTD. Lancet Neurol. 2012, 11, 205–207. [Google Scholar] [CrossRef]
- Yao, L.; He, X.; Cui, B.; Zhao, F.; Zhou, C. NEK1 mutations and the risk of amyotrophic lateral sclerosis (ALS): A meta-analysis. Neurol. Sci. 2021, 42, 1277–1285. [Google Scholar] [CrossRef]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Yoshimura, J.; Doi, K.; Morishita, S.; Goto, J.; Toda, T.; et al. Loss-of-function variants in NEK1 are associated with an increased risk of sporadic ALS in the Japanese population. J. Hum. Genet. 2021, 66, 237–241. [Google Scholar] [CrossRef]
- Riley, J.F.; Fioramonti, P.J.; Rusnock, A.K.; Hehnly, H.; Castaneda, C.A. ALS-linked mutations impair UBQLN2 stress-induced biomolecular condensate assembly in cells. J. Neurochem. 2021, 159, 145–155. [Google Scholar] [CrossRef]
- Lin, B.C.; Phung, T.H.; Higgins, N.R.; Greenslade, J.E.; Prado, M.A.; Finley, D.; Karbowski, M.; Polster, B.M.; Monteiro, M.J. ALS/FTD mutations in UBQLN2 are linked to mitochondrial dysfunction through loss-of-function in mitochondrial protein import. Hum. Mol. Genet. 2021, 30, 1230–1246. [Google Scholar] [CrossRef]
- Baron, D.M.; Fenton, A.R.; Saez-Atienzar, S.; Giampetruzzi, A.; Sreeram, A.; Shankaracharya; Keagle, P.J.; Doocy, V.R.; Smith, N.J.; Danielson, E.W.; et al. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 2022, 39, 110598. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef]
- Basu, S.; Rajendra, K.C.; Alagar, S.; Bahadur, R.P. Impaired nuclear transport induced by juvenile ALS causing P525L mutation in NLS domain of FUS: A molecular mechanistic study. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140766. [Google Scholar] [CrossRef]
- Robertson, J.; Bilbao, J.; Zinman, L.; Hazrati, L.N.; Tokuhiro, S.; Sato, C.; Moreno, D.; Strome, R.; Mackenzie, I.R.; Rogaeva, E. A novel double mutation in FUS gene causing sporadic ALS. Neurobiol. Aging 2011, 32, 553.e27–553.e30. [Google Scholar] [CrossRef]
- Chio, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef]
- Zhou, Y.; Luo, X.; Li, F.; Tian, X.; Zhu, L.; Yang, Y.; Ren, Y.; Pang, H. Association of Parkinson’s disease with six single nucleotide polymorphisms located in four PARK genes in the northern Han Chinese population. J. Clin. Neurosci. 2012, 19, 1011–1015. [Google Scholar] [CrossRef]
- Chung, S.J.; Armasu, S.M.; Biernacka, J.M.; Lesnick, T.G.; Rider, D.N.; Lincoln, S.J.; Ortolaza, A.I.; Farrer, M.J.; Cunningham, J.M.; Rocca, W.A.; et al. Common variants in PARK loci and related genes and Parkinson’s disease. Mov. Disord. 2011, 26, 280–288. [Google Scholar] [CrossRef]
- Klein, C.; Schneider, S.A.; Lang, A.E. Hereditary parkinsonism: Parkinson disease look-alikes—An algorithm for clinicians to “PARK” genes and beyond. Mov. Disord. 2009, 24, 2042–2058. [Google Scholar] [CrossRef]
- Domingo, A.; Klein, C. Genetics of Parkinson disease. Handb. Clin. Neurol. 2018, 147, 211–227. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Brzhozovskiy, A.; Kononikhin, A.; Bugrova, A.E.; Kovalev, G.I.; Schmit, P.O.; Kruppa, G.; Nikolaev, E.N.; Borchers, C.H. The Parallel Reaction Monitoring-Parallel Accumulation-Serial Fragmentation (prm-PASEF) Approach for Multiplexed Absolute Quantitation of Proteins in Human Plasma. Anal. Chem. 2022, 94, 2016–2022. [Google Scholar] [CrossRef]
- Lesur, A.; Dittmar, G. The clinical potential of prm-PASEF mass spectrometry. Expert Rev. Proteom. 2021, 18, 75–82. [Google Scholar] [CrossRef]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef]
- Garabedian, A.; Benigni, P.; Ramirez, C.E.; Baker, E.S.; Liu, T.; Smith, R.D.; Fernandez-Lima, F. Towards Discovery and Targeted Peptide Biomarker Detection Using nanoESI-TIMS-TOF MS. J. Am. Soc. Mass Spectrom. 2018, 29, 817–826. [Google Scholar] [CrossRef]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Sinitcyn, P.; Hamzeiy, H.; Salinas Soto, F.; Itzhak, D.; McCarthy, F.; Wichmann, C.; Steger, M.; Ohmayer, U.; Distler, U.; Kaspar-Schoenefeld, S.; et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021, 39, 1563–1573. [Google Scholar] [CrossRef]
- Muntel, J.; Gandhi, T.; Verbeke, L.; Bernhardt, O.M.; Treiber, T.; Bruderer, R.; Reiter, L. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics 2019, 15, 348–360. [Google Scholar] [CrossRef]
- Zhou, Y.; Tan, Z.; Xue, P.; Wang, Y.; Li, X.; Guan, F. High-throughput, in-depth and estimated absolute quantification of plasma proteome using data-independent acquisition/mass spectrometry (“HIAP-DIA”). Proteomics 2021, 21, e2000264. [Google Scholar] [CrossRef]
- Mehta, D.; Scandola, S.; Uhrig, R.G. BoxCar and Library-Free Data-Independent Acquisition Substantially Improve the Depth, Range, and Completeness of Label-Free Quantitative Proteomics. Anal. Chem. 2022, 94, 793–802. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Salovska, B.; Li, W.; Di, Y.; Liu, Y. BoxCarmax: A High-Selectivity Data-Independent Acquisition Mass Spectrometry Method for the Analysis of Protein Turnover and Complex Samples. Anal. Chem. 2021, 93, 3103–3111. [Google Scholar] [CrossRef]
- Gaun, A.; Lewis Hardell, K.N.; Olsson, N.; O’Brien, J.J.; Gollapudi, S.; Smith, M.; McAlister, G.; Huguet, R.; Keyser, R.; Buffenstein, R.; et al. Automated 16-Plex Plasma Proteomics with Real-Time Search and Ion Mobility Mass Spectrometry Enables Large-Scale Profiling in Naked Mole-Rats and Mice. J. Proteome Res. 2021, 20, 1280–1295. [Google Scholar] [CrossRef]
- Hebert, A.S.; Prasad, S.; Belford, M.W.; Bailey, D.J.; McAlister, G.C.; Abbatiello, S.E.; Huguet, R.; Wouters, E.R.; Dunyach, J.J.; Brademan, D.R.; et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 9529–9537. [Google Scholar] [CrossRef]
- He, B.; Huang, Z.; Huang, C.; Nice, E.C. Clinical applications of plasma proteomics and peptidomics: Towards precision medicine. Proteom. Clin. Appl. 2022, e2100097. [Google Scholar] [CrossRef]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Muller, J.B.; Meier, F.; et al. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell Proteom. 2018, 17, 2284–2296. [Google Scholar] [CrossRef]
- Kumar, D.; Hassan, M.I. Ultra-sensitive techniques for detecting neurological biomarkers: Prospects for early diagnosis. Biochem Biophys. Res. Commun. 2021, 584, 15–18. [Google Scholar] [CrossRef]
- Cole, K.H.; Luptak, A. High-throughput methods in aptamer discovery and analysis. Methods Enzymol. 2019, 621, 329–346. [Google Scholar] [CrossRef]
- Chen, L.C.; Tzeng, S.C.; Peck, K. Aptamer microarray as a novel bioassay for protein-protein interaction discovery and analysis. Biosens. Bioelectron. 2013, 42, 248–255. [Google Scholar] [CrossRef]
- Huang, J.; Chen, X.; Fu, X.; Li, Z.; Huang, Y.; Liang, C. Advances in Aptamer-Based Biomarker Discovery. Front. Cell Dev. Biol. 2021, 9, 659760. [Google Scholar] [CrossRef]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef]
- Raghunathan, R.; Sethi, M.K.; Zaia, J. On-slide tissue digestion for mass spectrometry based glycomic and proteomic profiling. MethodsX 2019, 6, 2329–2347. [Google Scholar] [CrossRef]
- Scicchitano, M.S.; Dalmas, D.A.; Boyce, R.W.; Thomas, H.C.; Frazier, K.S. Protein extraction of formalin-fixed, paraffin-embedded tissue enables robust proteomic profiles by mass spectrometry. J. Histochem. Cytochem. 2009, 57, 849–860. [Google Scholar] [CrossRef]
- Mitsa, G.; Guo, Q.; Goncalves, C.; Preston, S.E.J.; Lacasse, V.; Aguilar-Mahecha, A.; Benlimame, N.; Basik, M.; Spatz, A.; Batist, G.; et al. A Non-Hazardous Deparaffinization Protocol Enables Quantitative Proteomics of Core Needle Biopsy-Sized Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue Specimens. Int. J. Mol. Sci. 2022, 23, 4443. [Google Scholar] [CrossRef]
- Reimel, B.A.; Pan, S.; May, D.H.; Shaffer, S.A.; Goodlett, D.R.; McIntosh, M.W.; Yerian, L.M.; Bronner, M.P.; Chen, R.; Brentnall, T.A. Proteomics on Fixed Tissue Specimens-A Review. Curr. Proteom. 2009, 6, 63–69. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, X.; Huang, P.; Tian, R. Spatial proteomics for understanding the tissue microenvironment. Analyst 2021, 146, 3777–3798. [Google Scholar] [CrossRef]
- Shin, J.J.H.; Crook, O.M.; Borgeaud, A.C.; Cattin-Ortola, J.; Peak-Chew, S.Y.; Breckels, L.M.; Gillingham, A.K.; Chadwick, J.; Lilley, K.S.; Munro, S. Spatial proteomics defines the content of trafficking vesicles captured by golgin tethers. Nat. Commun. 2020, 11, 5987. [Google Scholar] [CrossRef]
- Navarro-Romero, A.; Montpeyo, M.; Martinez-Vicente, M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef]
- Engelen-Lee, J.; Blokhuis, A.M.; Spliet, W.G.M.; Pasterkamp, R.J.; Aronica, E.; Demmers, J.A.A.; Broekhuizen, R.; Nardo, G.; Bovenschen, N.; Van Den Berg, L.H. Proteomic profiling of the spinal cord in ALS: Decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 210–220. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Liu, F.; Morderer, D.; Wren, M.C.; Vettleson-Trutza, S.A.; Wang, Y.; Rabichow, B.E.; Salemi, M.R.; Phinney, B.S.; Oskarsson, B.; Dickson, D.W.; et al. Proximity proteomics of C9orf72 dipeptide repeat proteins identifies molecular chaperones as modifiers of poly-GA aggregation. Acta Neuropathol. Commun. 2022, 10, 22. [Google Scholar] [CrossRef]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef]
- Feneberg, E.; Charles, P.D.; Finelli, M.J.; Scott, C.; Kessler, B.M.; Fischer, R.; Ansorge, O.; Gray, E.; Talbot, K.; Turner, M.R. Detection and quantification of novel C-terminal TDP-43 fragments in ALS-TDP. Brain Pathol. 2021, 31, e12923. [Google Scholar]
- Bereman, M.S.; Beri, J.; Enders, J.R.; Nash, T. Machine Learning Reveals Protein Signatures in CSF and Plasma Fluids of Clinical Value for ALS. Sci. Rep. 2018, 8, 16334. [Google Scholar] [CrossRef]
- Xu, Z.; Lee, A.; Nouwens, A.; Henderson, R.D.; McCombe, P.A. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 362–376. [Google Scholar] [CrossRef]
- Mohanty, L.; Henderson, R.D.; McCombe, P.A.; Lee, A. Levels of clusterin, CD5L, ficolin-3, and gelsolin in ALS patients and controls. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 21, 631–634. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Thezenas, M.L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Kessler, B.M.; Turner, M.R. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 258–268. [Google Scholar] [CrossRef]
- Barschke, P.; Oeckl, P.; Steinacker, P.; Al Shweiki, M.R.; Weishaupt, J.H.; Landwehrmeyer, G.B.; Anderl-Straub, S.; Weydt, P.; Diehl-Schmid, J.; Danek, A.; et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J. Neurol. Neurosurg. Psychiatry 2020, 91, 503–511. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Mager, I.; Thezenas, M.L.; Charles, P.D.; Talbot, K.; Fischer, R.; Kessler, B.M.; Wood, M.; Turner, M.R. CSF extracellular vesicle proteomics demonstrates altered protein homeostasis in amyotrophic lateral sclerosis. Clin. Proteom. 2020, 17, 31. [Google Scholar] [CrossRef]
- Hayashi, N.; Doi, H.; Kurata, Y.; Kagawa, H.; Atobe, Y.; Funakoshi, K.; Tada, M.; Katsumoto, A.; Tanaka, K.; Kunii, M.; et al. Proteomic analysis of exosome-enriched fractions derived from cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurosci. Res. 2020, 160, 43–49. [Google Scholar] [CrossRef]
- Chen, P.C.; Wu, D.; Hu, C.J.; Chen, H.Y.; Hsieh, Y.C.; Huang, C.C. Exosomal TAR DNA-binding protein-43 and neurofilaments in plasma of amyotrophic lateral sclerosis patients: A longitudinal follow-up study. J. Neurol. Sci. 2020, 418, 117070. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef]
- Didonna, A.; Benetti, F. Post-translational modifications in neurodegeneration. AIMS Biophys. 2016, 3, 27–49. [Google Scholar]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Kametani, F.; Obi, T.; Shishido, T.; Akatsu, H.; Murayama, S.; Saito, Y.; Yoshida, M.; Hasegawa, M. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 2016, 6, 23281. [Google Scholar] [CrossRef]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Cao, X.; Sandberg, A.; Araujo, J.E.; Cvetkovski, F.; Berglund, E.; Eriksson, L.E.; Pernemalm, M. Evaluation of Spin Columns for Human Plasma Depletion to Facilitate MS-Based Proteomics Analysis of Plasma. J. Proteome Res. 2021, 20, 4610–4620. [Google Scholar] [CrossRef]
- Lee, P.Y.; Osman, J.; Low, T.Y.; Jamal, R. Plasma/serum proteomics: Depletion strategies for reducing high-abundance proteins for biomarker discovery. Bioanalysis 2019, 11, 1799–1812. [Google Scholar] [CrossRef]
- El Rassi, Z.; Puangpila, C. Liquid-phase based separation systems for depletion, prefractionation, and enrichment of proteins in biological fluids and matrices for in-depth proteomics analysis-An update covering the period 2014–2016. Electrophoresis 2017, 38, 150–161. [Google Scholar] [CrossRef]
- Keshishian, H.; Burgess, M.W.; Specht, H.; Wallace, L.; Clauser, K.R.; Gillette, M.A.; Carr, S.A. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 2017, 12, 1683–1701. [Google Scholar] [CrossRef]
- Zubiri, I.; Lombardi, V.; Bremang, M.; Mitra, V.; Nardo, G.; Adiutori, R.; Lu, C.H.; Leoni, E.; Yip, P.; Yildiz, O.; et al. Tissue-enhanced plasma proteomic analysis for disease stratification in amyotrophic lateral sclerosis. Mol. Neurodegener. 2018, 13, 60. [Google Scholar] [CrossRef]
- Leoni, E.; Bremang, M.; Mitra, V.; Zubiri, I.; Jung, S.; Lu, C.H.; Adiutori, R.; Lombardi, V.; Russell, C.; Koncarevic, S.; et al. Author Correction: Combined Tissue-Fluid Proteomics to Unravel Phenotypic Variability in Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 18603. [Google Scholar] [CrossRef]
- Huang, J.; Khademi, M.; Lindhe, O.; Jonsson, G.; Piehl, F.; Olsson, T.; Kockum, I. Assessing the Preanalytical Variability of Plasma and Cerebrospinal Fluid Processing and Its Effects on Inflammation-Related Protein Biomarkers. Mol. Cell Proteom. 2021, 20, 100157. [Google Scholar] [CrossRef]
- Macron, C.; Nunez Galindo, A.; Cominetti, O.; Dayon, L. A Versatile Workflow for Cerebrospinal Fluid Proteomic Analysis with Mass Spectrometry: A Matter of Choice between Deep Coverage and Sample Throughput. Methods Mol. Biol. 2019, 2044, 129–154. [Google Scholar] [CrossRef]
- Varghese, A.M.; Sharma, A.; Mishra, P.; Vijayalakshmi, K.; Harsha, H.C.; Sathyaprabha, T.N.; Bharath, S.M.; Nalini, A.; Alladi, P.A.; Raju, T.R. Chitotriosidase-a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin. Proteom. 2013, 10, 19. [Google Scholar] [CrossRef]
- Pinnell, J.R.; Cui, M.; Tieu, K. Exosomes in Parkinson disease. J. Neurochem. 2021, 157, 413–428. [Google Scholar] [CrossRef]
- Zhang, N.; Gu, D.; Meng, M.; Gordon, M.L. TDP-43 Is Elevated in Plasma Neuronal-Derived Exosomes of Patients With Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 166. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of Alzheimer’s disease. FASEB J. 2018, 32, 888–893. [Google Scholar] [CrossRef]
- Licker, V.; Turck, N.; Kövari, E.; Burkhardt, K.; Côte, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.C.; Burkhard, P.R. Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 2014, 14, 784–794. [Google Scholar] [CrossRef]
- Petyuk, V.A.; Yu, L.; Olson, H.M.; Yu, F.; Clair, G.; Qian, W.J.; Shulman, J.M.; Bennett, D.A. Proteomic Profiling of the Substantia Nigra to Identify Determinants of Lewy Body Pathology and Dopaminergic Neuronal Loss. J. Proteome Res. 2021, 20, 2266–2282. [Google Scholar] [CrossRef]
- Chelliah, S.S.; Bhuvanendran, S.; Magalingam, K.B.; Kamarudin, M.N.A.; Radhakrishnan, A.K. Identification of blood-based biomarkers for diagnosis and prognosis of Parkinson’s disease: A systematic review of proteomics studies. Ageing Res. Rev. 2022, 73, 101514. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, X.; Yu, H.; Liu, X.; Yang, F.; Yao, J.; Jin, H.; Yang, P. Quantitative proteomic analysis of serum proteins in patients with Parkinson’s disease using an isobaric tag for relative and absolute quantification labeling, two-dimensional liquid chromatography, and tandem mass spectrometry. Analyst 2012, 137, 490–495. [Google Scholar] [CrossRef]
- Hu, L.; Dong, M.X.; Huang, Y.L.; Lu, C.Q.; Qian, Q.; Zhang, C.C.; Xu, X.M.; Liu, Y.; Chen, G.H.; Wei, Y.D. Integrated Metabolomics and Proteomics Analysis Reveals Plasma Lipid Metabolic Disturbance in Patients With Parkinson’s Disease. Front. Mol. Neurosci. 2020, 13, 80. [Google Scholar] [CrossRef]
- Yang, L.; Stewart, T.; Shi, M.; Pottiez, G.; Dator, R.; Wu, R.; Aro, P.; Schuster, R.J.; Ginghina, C.; Pan, C.; et al. An alpha-synuclein MRM assay with diagnostic potential for Parkinson’s disease and monitoring disease progression. Proteom. Clin. Appl. 2017, 11, 1700045. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, M.; Chung, K.A.; Zabetian, C.P.; Leverenz, J.B.; Berg, D.; Srulijes, K.; Trojanowski, J.Q.; Lee, V.M.; Siderowf, A.D.; et al. Phosphorylated alpha-synuclein in Parkinson’s disease. Sci. Transl. Med. 2012, 4, 121ra120. [Google Scholar] [CrossRef]
- Rotunno, M.S.; Lane, M.; Zhang, W.; Wolf, P.; Oliva, P.; Viel, C.; Wills, A.M.; Alcalay, R.N.; Scherzer, C.R.; Shihabuddin, L.S.; et al. Cerebrospinal fluid proteomics implicates the granin family in Parkinson’s disease. Sci. Rep. 2020, 10, 2479. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef]
- Kitamura, Y.; Kojima, M.; Kurosawa, T.; Sasaki, R.; Ichihara, S.; Hiraku, Y.; Tomimoto, H.; Murata, M.; Oikawa, S. Proteomic Profiling of Exosomal Proteins for Blood-based Biomarkers in Parkinson’s Disease. Neuroscience 2018, 392, 121–128. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Wei, L.; Yu, F.; Yu, B.; Xu, A. Long Noncoding RNA POU3F3 and alpha-Synuclein in Plasma L1CAM Exosomes Combined with beta-Glucocerebrosidase Activity: Potential Predictors of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1104–1119. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of alpha-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Foulds, P.; Mann, D.M.; Allsop, D. Phosphorylated alpha-synuclein as a potential biomarker for Parkinson’s disease and related disorders. Expert Rev. Mol. Diagn. 2012, 12, 115–117. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated i.in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Kragh, C.L.; Lund, L.B.; Febbraro, F.; Hansen, H.D.; Gai, W.P.; El-Agnaf, O.; Richter-Landsberg, C.; Jensen, P.H. Alpha-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J. Biol. Chem. 2009, 284, 10211–10222. [Google Scholar] [CrossRef]
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of alpha-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109. [Google Scholar] [CrossRef]
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of alpha-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of alpha-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675. [Google Scholar] [CrossRef]
- Na, C.H.; Sathe, G.; Rosenthal, L.S.; Moghekar, A.R.; Dawson, V.L.; Dawson, T.M.; Pandey, A. Development of a novel method for the quantification of tyrosine 39 phosphorylated alpha- and beta-synuclein in human cerebrospinal fluid. Clin. Proteom. 2020, 17, 13. [Google Scholar] [CrossRef]
- Ohrfelt, A.; Zetterberg, H.; Andersson, K.; Persson, R.; Secic, D.; Brinkmalm, G.; Wallin, A.; Mulugeta, E.; Francis, P.T.; Vanmechelen, E.; et al. Identification of novel alpha-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 2011, 36, 2029–2042. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G.; et al. Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204. [Google Scholar] [CrossRef]
- Raghunathan, R.; Sethi, M.K.; Klein, J.A.; Zaia, J. Proteomics, Glycomics, and Glycoproteomics of Matrisome Molecules. Mol. Cell Proteom. 2019, 18, 2138–2148. [Google Scholar] [CrossRef]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef]
- Downs, M.; Sethi, M.K.; Raghunathan, R.; Layne, M.D.; Zaia, J. Matrisome changes in Parkinson’s disease. Anal. Bioanal. Chem. 2022, 414, 3005–3015. [Google Scholar] [CrossRef]
- Posavi, M.; Diaz-Ortiz, M.; Liu, B.; Swanson, C.R.; Skrinak, R.T.; Hernandez-Con, P.; Amado, D.A.; Fullard, M.; Rick, J.; Siderowf, A.; et al. Characterization of Parkinson’s disease using blood-based biomarkers: A multicohort proteomic analysis. PLoS Med. 2019, 16, e1002931. [Google Scholar] [CrossRef]
- Cerri, S.; Ghezzi, C.; Sampieri, M.; Siani, F.; Avenali, M.; Dornini, G.; Zangaglia, R.; Minafra, B.; Blandini, F. The Exosomal/Total alpha-Synuclein Ratio in Plasma Is Associated With Glucocerebrosidase Activity and Correlates With Measures of Disease Severity in PD Patients. Front. Cell Neurosci. 2018, 12, 125. [Google Scholar] [CrossRef]
- Zheng, H.; Xie, Z.; Zhang, X.; Mao, J.; Wang, M.; Wei, S.; Fu, Y.; Zheng, H.; He, Y.; Chen, H.; et al. Investigation of alpha-Synuclein Species in Plasma Exosomes and the Oligomeric and Phosphorylated alpha-Synuclein as Potential Peripheral Biomarker of Parkinson’s Disease. Neuroscience 2021, 469, 79–90. [Google Scholar] [CrossRef]
- He, T. Implementation of Proteomics in Clinical Trials. Proteom. Clin. Appl. 2019, 13, e1800198. [Google Scholar] [CrossRef]
- Gomez Ravetti, M.; Moscato, P. Identification of a 5-protein biomarker molecular signature for predicting Alzheimer’s disease. PLoS ONE 2008, 3, e3111. [Google Scholar] [CrossRef]
- Carr, S.A.; Abbatiello, S.E.; Ackermann, B.L.; Borchers, C.; Domon, B.; Deutsch, E.W.; Grant, R.P.; Hoofnagle, A.N.; Huttenhain, R.; Koomen, J.M.; et al. Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell Proteom. 2014, 13, 907–917. [Google Scholar] [CrossRef]
- Krieger, J.R.; Wybenga-Groot, L.E.; Tong, J.; Bache, N.; Tsao, M.S.; Moran, M.F. Evosep One Enables Robust Deep Proteome Coverage Using Tandem Mass Tags while Significantly Reducing Instrument Time. J. Proteome Res. 2019, 18, 2346–2353. [Google Scholar] [CrossRef]
- Fu, Q.; Liu, Z.; Bhawal, R.; Anderson, E.T.; Sherwood, R.W.; Yang, Y.; Thannhauser, T.; Schroyen, M.; Tang, X.; Zhang, H.; et al. Comparison of MS(2), synchronous precursor selection MS(3), and real-time search MS(3) methodologies for lung proteomes of hydrogen sulfide treated swine. Anal. Bioanal. Chem. 2021, 413, 419–429. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages |
|---|---|---|
| Sample Preparation Methods | ||
| Filter-aided sample preparation (FASP) | Unbiased filter-based approach which removes detergents | Molecular weight cutoffs and can be challenging for aggregated proteins |
| Single-pot solid-phase-enhanced sample preparation (SP3) | Bead-based approach with low sample loss Unbiased robust recovery and can be automated using magnetic beads | Beads have a limited capacity and should not be overloaded to cause inconsistencies |
| In-StageTip (iST) | Peptide can be fractionated on the tip to gain depth | Tips must be compatible with solubilization reagents used |
| SarkoSpin | Can isolate insoluble pathological protein aggregated | Utilize detergents that require sample clean up prior to mass spectrometry |
| Depletion | Gain depth | Low throughput, induce variability |
| Immunoenrichment | Gain depth | Low throughput, induce variability |
| Offline fractionation | Gain depth | Low throughput |
| Mass Spectrometry Quantitation Strategies | ||
| Isobaric TMT tags | Relative quantitation in MS2/MS3 dimension with multiplexing to save time | Require (SPS) MS3 for accurate quantitation to overcome challenges with ratio compression |
| Label-free DIA | Relative quantitation by area under the peak, enabling the acquisition of complete data in large cohorts | Requires expertise in MS method design for window strategy and data interpretation |
| MRM/PRM-targeted quantitation | Absolute quantitation with standard curves and the ability to monitor disease progression with time | Limited in the number of targets that can be analyzed |
| Advances in Instrumentation and Technology | ||
| BOXCAR data-independent acquisition | Improves data completeness | Requires good method design |
| Real-time search RTS-SPS-MS3 | Improves quantitation | Special feature in an instrument |
| High-field asymmetric ion mobility spectrometry FAIMS | Improves sensitivity and selectivity | In front end and not true ion mobility to separate isomers |
| Modified LC- Evosep | Improves throughput and robustness | Has defined methods and is not customizable |
| Automation | Improves reproducibility | Tedious to implement changes in workflows |
| Trapped ion mobility TIMS | PTM and isoform identification | Needs expertise to achieve good separation |
| Parallel acquisition serial fragmentation PASEF | Improves scan speed and sensitivity | Needs optimization based on gradient |
| Multiplexed immunoassay O-link | Improves dynamic range based on antibody specificity | Limited in the panel of targets based on the availability of antibodies |
| Aptamer-based assay | Improves dynamic range based on aptamer specificity | Limited in the panel of targets based on the availability of aptamers |
| Disease | Marker | Quantitation | Tissue | Summary | Reference |
|---|---|---|---|---|---|
| Tissue-based proteomic markers in ALS | |||||
| ALS | TDP43 | PRM absolute quantitation | Prefrontal/motor cortex and spinal cord | An increase in C: N-terminal TDP43 peptide ratio > 1.5, new truncation site-specific trend observed in ALS-TDP | [51,55] |
| ALS (sporadic) | Calmodulin | Label-free | Spinal cord | Downregulated in ALS | [51] |
| ALS (sporadic) | ATP5D | Label-free | Spinal cord | Downregulated in ALS | [51] |
| ALS | UCHL1 | Label-free and MRM | Spinal cord | Upregulated in ALS and correlated with CSF | [52] |
| ALS | MAP2 | Label-free and MRM | Spinal cord | Upregulated in ALS and correlated with CSF | [52] |
| ALS | GPNMB | Label-free and MRM | Spinal cord | Upregulated in ALS and correlated with CSF | [52] |
| Plasma/Serum proteomics biomarkers in ALS | |||||
| ALS | Gelsolin | LFQ and MRM | Plasma | Differentially expressed in ALS | [56,57,58] |
| ALS | Clusterin | MRM | Plasma | Downregulated in ALS | [57,58] |
| ALS | CD5L | MRM | Plasma | Differentially expressed in ALS | [57,58] |
| ALS | Ficolin 3 | MRM | Plasma | Upregulated in ALS | [57,58] |
| CSF proteomic biomarkers in ALS | |||||
| ALS | α-1-antichymotrypsin | LFQ | CSF | In CSF, 118 proteins were significantly altered in ALS compared to controls | [56] |
| ALS | Amyloid beta A4 protein | LFQ | CSF | In CSF, 118 proteins were significantly altered in ALS compared to controls | [56] |
| ALS | Gelsolin | LFQ | CSF | In CSF, 118 proteins were significantly altered in ALS compared to controls | [56] |
| ALS | Chitinase-3-like protein 1 (CHI3L1) | LFQ | CSF | Upregulated | [59] |
| ALS | Chitinase-3-like protein 2 (CHI3L2) | LFQ, TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [59,60] |
| ALS | Chitotriosidase-1 (CHIT-1) | LFQ, TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [59,60] |
| ALS | Ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1) | LFQ, TMT, MRM | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [52,59,60] |
| ALS | MAP2 | MRM | CSF | Upregulated | [52] |
| ALS | CAPG | MRM | CSF | Upregulated | [52] |
| ALS | GPNMB | MRM | CSF | Upregulated | [52] |
| ALS | CRYAB | TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [60] |
| ALS | PFN1 | TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [60] |
| ALS | TFRC | TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [60] |
| ALS | TREM2 | TMT | CSF | Upregulated in C9orf72 variant-associated symptomatic ALS compared to asymptomatic controls with C9orf72 variants | [60] |
| ALS | TXNDC17 | TMT | CSF | Upregulated in mutated C9orf72 variant-associated symptomatic ALS compared to asymptomatic controls with C9orf72 variants | [60] |
| ALS | NEFM | TMT | CSF | Upregulated in mutated C9orf72 symptomatic ALS compared to asymptomatic controls with C9orf72 mutations | [60] |
| Exosomal biomarkers in ALS | |||||
| ALS | Gelsolin | LFQ | CSF exosomes | Upregulated in C9orf mutated ALS cases | [61] |
| ALS | Clusterin | LFQ | CSF exosomes | Upregulated | [61] |
| ALS | UBA1 | LFQ | CSF exosomes | Upregulated in C9orf mutated ALS cases | [61] |
| ALS | NIR | LFQ | CSF exosomes | Upregulated in sporadic ALS | [62] |
| ALS | TDP43 | LFQ | Plasma exosomes | Levels correlated with longitudinal progression | [63] |
| Disease | Marker | Quantitation | Tissue | Summary | Reference |
|---|---|---|---|---|---|
| Tissue-based proteomic markers in PD | |||||
| PD | Mitochondrial dysfunction, oxidative stress, cytoskeleton impairment-related proteins | TMT | Substantia nigra | Significant changes in expression levels of 204 nigral proteins in human PD samples | [85] |
| PD | RGS6 | LFQ | Substantia nigra (Lewy body pathology) | Changes in proteins related to (1) Arp2/3 complex-mediated actin nucleation; (2) synaptic function; (3) poly(A) RNA binding; (4) basement membrane and endothelium; and (5) hydrogen peroxide metabolic processes | [86] |
| PD | GANAB | LFQ | Substantia nigra (Lewy body pathology) | Changes in proteins related to (1) Arp2/3 complex-mediated actin nucleation; (2) synaptic function; (3) poly(A) RNA binding; (4) basement membrane and endothelium; and (5) hydrogen peroxide metabolic processes | [86] |
| PD | CD59 | LFQ | Substantia nigra (Lewy body pathology) | Changes in proteins related to (1) Arp2/3 complex-mediated actin nucleation; (2) synaptic function; (3) poly(A) RNA binding; (4) basement membrane and endothelium; and (5) hydrogen peroxide metabolic processes | [86] |
| Plasma/serum proteomic biomarkers in PD | |||||
| PD | Apolipoprotein A1 | iTRAQ | Plasma/serum | Downregulated in PD | [87,88] |
| PD | Apolipoprotein A-IV | LFQ | Plasma/serum | Downregulated in PD | [87,88] |
| PD | Apolipoprotein B | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Apolipoprotein CI | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Apolipoprotein CIII | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Apolipoprotein C4 | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Apolipoprotein C4 | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Apolipoprotein M | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Inter-alpha-trypsin inhibitor heavy | LFQ | Plasma/serum | Downregulated in PD | [87] |
| PD | Complement C4A | LFQ | Plasma/serum | Downregulated in PD | [87] |
| PD | Complement C4B | iTRAQ | Plasma/serum | Downregulated in PD | [87,88] |
| PD | Complement C3 | LFQ | Plasma/serum | Downregulated in PD | [87] |
| PD | Haptoglobin | LFQ | Plasma | Downregulated in PD | [89] |
| PD | Clusterin | LFQ | Plasma/serum | Upregulated in PD | [87] |
| PD | Transthyretin | LFQ | Plasma/serum | Upregulated in PD | [87] |
| PD | Zinc α-2 glycoprotein | LFQ | Plasma/serum | Upregulated in PD | [87] |
| PD | Vitamin D binding protein | LFQ | Plasma/serum | Upregulated in PD | [87] |
| PD | Afamin | LFQ | Plasma/serum | Upregulated in PD | [87] |
| CSF proteomic biomarkers in PD | |||||
| PD | α-synuclein peptide (81–96) | MRM | CSF | α-Synuclein peptide altered in PD | [90] |
| PD | α-synuclein pS129 | MRM | CSF | α-Synuclein pS129 correlates with disease severity | [91] |
| PD | Granins | DIA | CSF | Granins are downregulated in PD | [92] |
| Exosomal biomarkers in PD | |||||
| PD | α-synuclein | LFQ | Serum neuronal(L1CAM+) exosomes | Upregulated in prodromal and clinical PD compared to controls and other neurodegenerative diseases | [93] |
| PD | Clusterin | LFQ | Serum neuronal (L1CAM+) exosomes, plasma exosomes | Upregulated in FTD but not PD, served as a combined marker with α-synuclein and is downregulated in PD in plasma exosomes | [93,94] |
| PD | α-synuclein | SRM | Plasma neuronal (L1CAM+) exosomes | α-synuclein is upregulated in PD | [95,96] |
| PD | Complement C1r | LFQ | Plasma exosomes | Downregulated in PD | [94] |
| PD | Apolipoprotein A1 | LFQ | Plasma exosomes | Downregulated in PD | [94] |
| Disease | Clinical Trial | Summary | Reference |
|---|---|---|---|
| ALS | NCT01948102 | An observational study for the identification of prognostic and diagnostic markers in skin and adipose samples using proteomics to measure changes in abundance and/or post-translational modifications of proteins in the trial | [6] |
| PD | NCT00315250 | An interventional study with the aim of developing imaging, clinical, and biochemical biomarkers for PD uses proteomics in combination with metabolomics and gene expression to categorize Parkinson’s syndrome vs. non-Parkinson’s syndrome | [7] |
| PD | NCT02263235 | A study in Alzheimer’s, PD, and other neurological disorders without cognitive decline uses targeted quantitative proteomics by MRM in CSF, blood, urine, and saliva for diagnostic purposes after administering stable isotope-labelled leucine for the diagnosis of neurological disorders | [4] |
| PD | NCT02524405 | An investigational study in Alzheimer’s and PD (called the brain–eye amyloid memory study (BEAM)), MRI, and amyloid PET were used for primary and secondary outcomes, genetic analysis for ApoE4 status, and proteomics and lipidomics analyses | [2] |
| PD | NCT02387281 | An observational study in PD studying freezing of gait (FOG) proteomics on CSF is used in combination with analysis of catecholamines along with MRI and other cognitive tests to assess types of FOG and if there is a connection with cognitive differences and gait patterns presented in PD | [3] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghunathan, R.; Turajane, K.; Wong, L.C. Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 9299. https://doi.org/10.3390/ijms23169299
Raghunathan R, Turajane K, Wong LC. Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(16):9299. https://doi.org/10.3390/ijms23169299
Chicago/Turabian StyleRaghunathan, Rekha, Kathleen Turajane, and Li Chin Wong. 2022. "Biomarkers in Neurodegenerative Diseases: Proteomics Spotlight on ALS and Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 16: 9299. https://doi.org/10.3390/ijms23169299