
Sepsis and Acute Kidney Injury: A Review Focusing on the Bidirectional Interplay


Abstract
:1. Introduction
2. Epidemiology
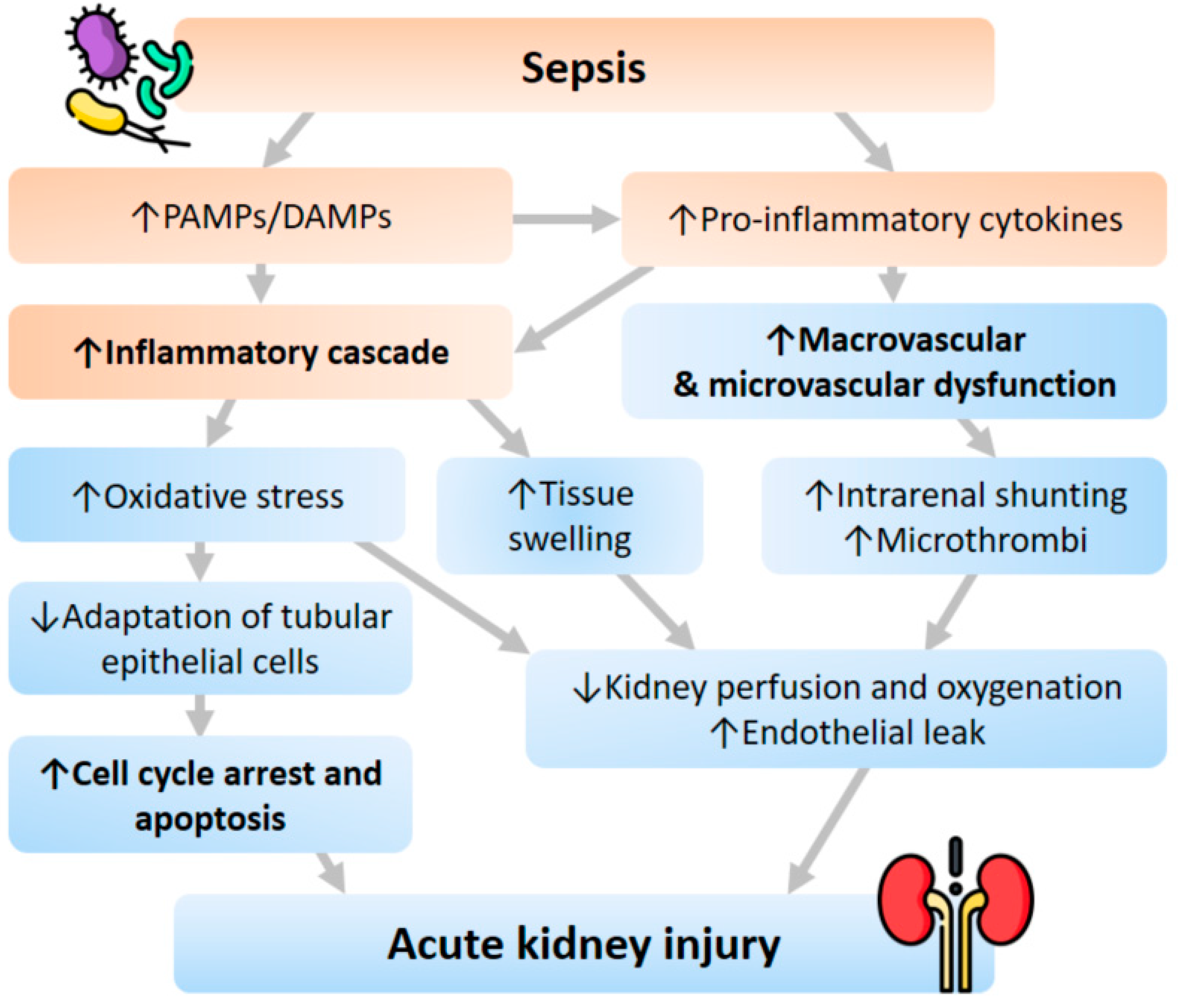
3. Pathophysiology of SA-AKI
3.1. Inflammatory Cascade
3.2. Macrovascular and Microvascular Dysfunction
3.3. Cell Cycle Arrest and Apoptosis
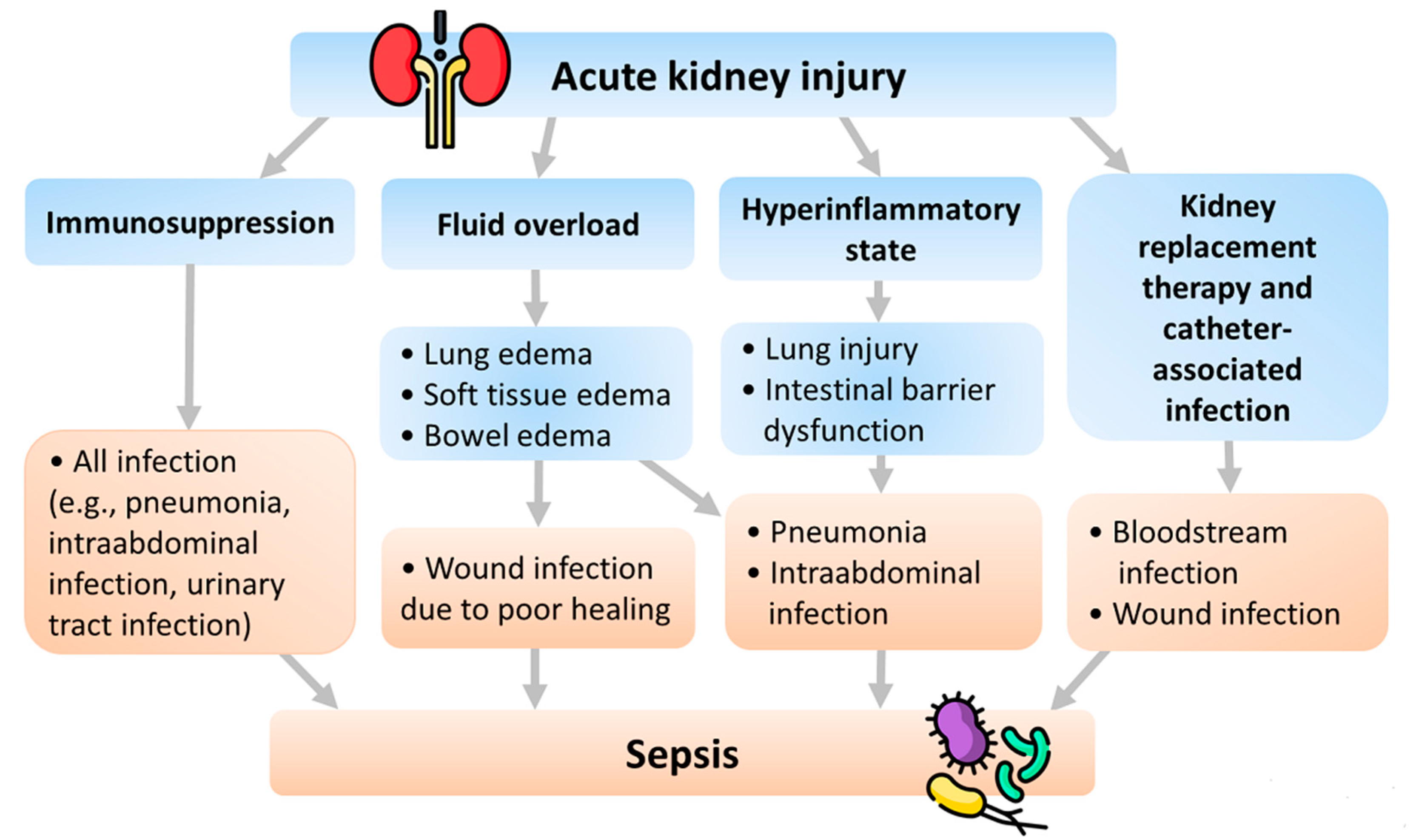
4. Pathophysiology of Sepsis Following AKI
4.1. Fluid Overload
4.2. Hyperinflammatory State
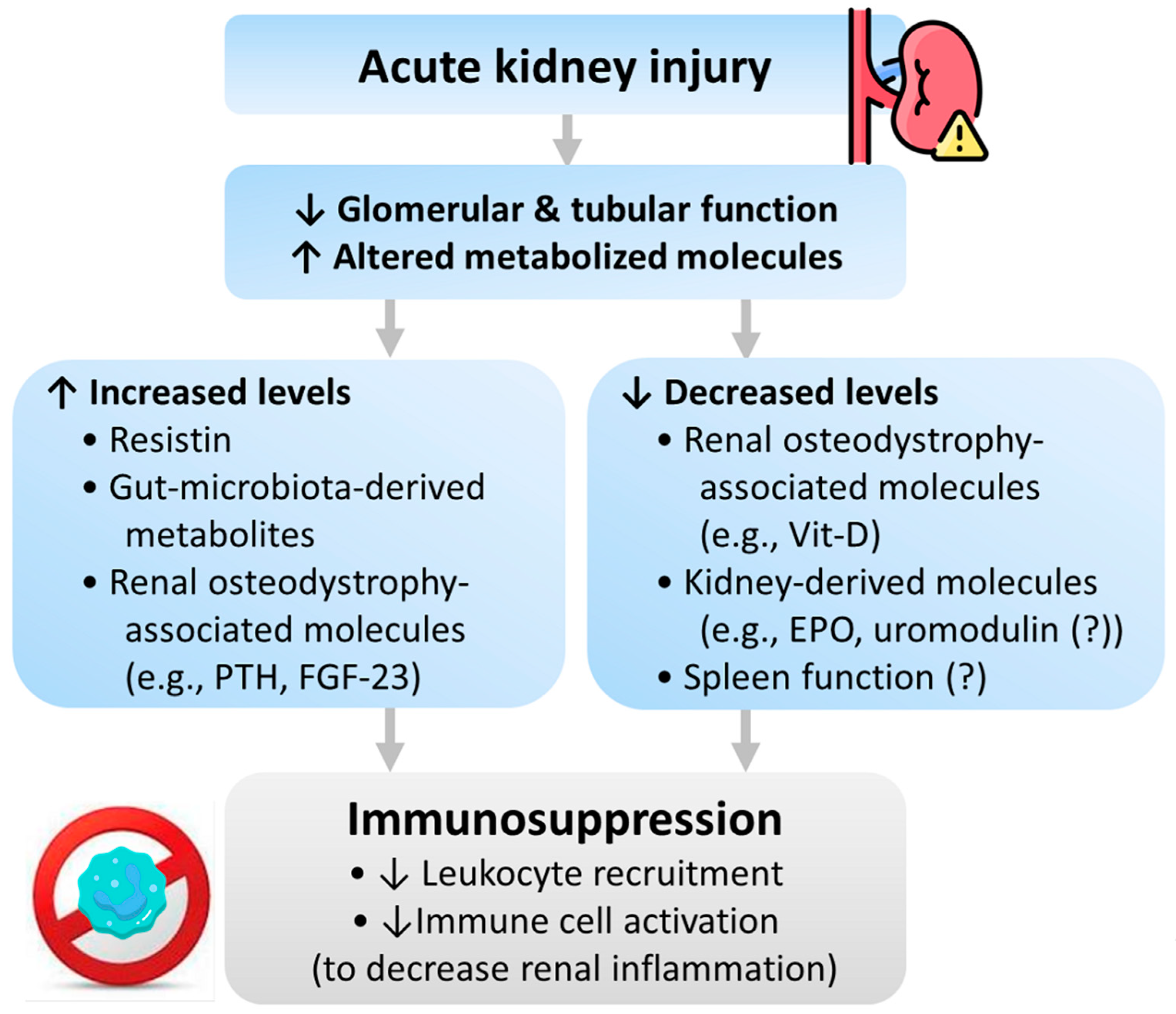
4.3. Immunosuppression
4.3.1. Resistin
4.3.2. Gut Microbiota-Derived Metabolites
4.3.3. Renal Osteodystrophy-Associated Molecules
4.3.4. Kidney-Derived Molecules
4.3.5. Kidney and Spleen Interactions
4.4. KRT and Catheter-Associated Infection
5. Potentially Preventive Strategies
6. Limitations and Further Prospects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pannu, N.; James, M.; Hemmelgarn, B.; Klarenbach, S. Association between AKI, recovery of renal function, and long-term outcomes after hospital discharge. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.C.; Huang, T.M.; Lai, C.F.; Shiao, C.C.; Lin, Y.F.; Chu, T.S.; Wu, P.C.; Chao, C.T.; Wang, J.Y.; Kao, T.W.; et al. Acute-on-chronic kidney injury at hospital discharge is associated with long-term dialysis and mortality. Kidney Int. 2011, 80, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaghbeer, M.; Dealmeida, D.; Bilderback, A.; Ambrosino, R.; Kellum, J.A. Clinical Decision Support for In-Hospital AKI. J. Am. Soc. Nephrol. 2018, 29, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pr. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Yang, S.Y.; Chiou, T.T.; Shiao, C.C.; Lin, H.Y.; Chan, M.J.; Wu, C.H.; Sun, C.Y.; Wang, W.J.; Huang, Y.T.; Wu, V.C.; et al. Nomenclature and diagnostic criteria for acute kidney injury—2020 consensus of the Taiwan AKI-task force. J. Med. Assoc. 2022, 121, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Rabb, H. The distant organ effects of acute kidney injury. Kidney Int. 2012, 81, 942–948. [Google Scholar] [CrossRef]
- Druml, W. Systemic consequences of acute kidney injury. Curr. Opin. Crit. Care 2014, 20, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Siew, E.D. Biomarkers for the Early Detection and Prognosis of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 149–173. [Google Scholar] [CrossRef]
- Charlton, J.R.; Portilla, D.; Okusa, M.D. A basic science view of acute kidney injury biomarkers. Nephrol. Dial. Transplant. 2014, 29, 1301–1311. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gomez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.S.; Wang, C.Y.; Pan, S.C.; Huang, T.M.; Lin, M.C.; Lai, C.F.; Wu, C.H.; Wu, V.C.; Chien, K.L.; National Taiwan University Hospital Study Group on Acute Renal Failure. Risk of developing severe sepsis after acute kidney injury: A population-based cohort study. Crit. Care 2013, 17, R231. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Caballero, C.L.; Del Rio-Pertuz, G.; Gomez, H. Sepsis-Associated Acute Kidney Injury. Crit. Care Clin. 2021, 37, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Su, L.; Han, G.; Yan, P.; Xie, L. Prognostic Value of Procalcitonin in Adult Patients with Sepsis: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0129450. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Lai, M.Y.; Kan, W.C.; Shiao, C.C. Independent Predictive Ability of Procalcitonin of Acute Kidney Injury among Critically Ill Patients. J. Clin. Med. 2020, 9, 1939. [Google Scholar] [CrossRef]
- Vincent, J.L.; Sakr, Y.; Sprung, C.L.; Ranieri, V.M.; Reinhart, K.; Gerlach, H.; Moreno, R.; Carlet, J.; Le Gall, J.R.; Payen, D.; et al. Sepsis in European intensive care units: Results of the SOAP study. Crit. Care Med. 2006, 34, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nie, S.; Liu, Z.; Chen, C.; Xu, G.; Zha, Y.; Qian, J.; Liu, B.; Han, S.; Xu, A.; et al. Epidemiology and Clinical Correlates of AKI in Chinese Hospitalized Adults. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1510–1518. [Google Scholar] [CrossRef]
- Kellum, J.A.; Chawla, L.S.; Keener, C.; Singbartl, K.; Palevsky, P.M.; Pike, F.L.; Yealy, D.M.; Huang, D.T.; Angus, D.C.; ProCESS; et al. The Effects of Alternative Resuscitation Strategies on Acute Kidney Injury in Patients with Septic Shock. Am. J. Respir. Crit. Care Med. 2016, 193, 281–287. [Google Scholar] [CrossRef]
- Selby, N.M.; Kolhe, N.V.; McIntyre, C.W.; Monaghan, J.; Lawson, N.; Elliott, D.; Packington, R.; Fluck, R.J. Defining the cause of death in hospitalised patients with acute kidney injury. PLoS ONE 2012, 7, e48580. [Google Scholar] [CrossRef] [PubMed]
- Thakar, C.V.; Yared, J.P.; Worley, S.; Cotman, K.; Paganini, E.P. Renal dysfunction and serious infections after open-heart surgery. Kidney Int. 2003, 64, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.R.; Teixeira, J.P.; Ambruso, S.; Bronsert, M.; Pal, J.D.; Cleveland, J.C.; Reece, T.B.; Fullerton, D.A.; Faubel, S.; Aftab, M. Stage 1 acute kidney injury is independently associated with infection following cardiac surgery. J. Thorac. Cardiovasc. Surg. 2021, 161, 1346–1355.e1343. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.R.; You, Z.; Holmen, J.; SooHoo, M.; Gist, K.M.; Colbert, J.F.; Chonchol, M.; Faubel, S.; Jovanovich, A. Incident infection following acute kidney injury with recovery to baseline creatinine: A propensity score matched analysis. PLoS ONE 2019, 14, e0217935. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.C.; Wang, C.Y.; Shiao, C.C.; Chang, C.H.; Huang, H.Y.; Huang, T.M.; Lai, C.F.; Lin, M.C.; Ko, W.J.; Wu, K.D.; et al. Increased risk of active tuberculosis following acute kidney injury: A nationwide, population-based study. PLoS ONE 2013, 8, e69556. [Google Scholar] [CrossRef] [PubMed]
- Reynvoet, E.; Vandijck, D.M.; Blot, S.I.; Dhondt, A.W.; De Waele, J.J.; Claus, S.; Buyle, F.M.; Vanholder, R.C.; Hoste, E.A. Epidemiology of infection in critically ill patients with acute renal failure. Crit. Care Med. 2009, 37, 2203–2209. [Google Scholar] [CrossRef]
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700. [Google Scholar] [CrossRef]
- Lankadeva, Y.R.; Kosaka, J.; Iguchi, N.; Evans, R.G.; Booth, L.C.; Bellomo, R.; May, C.N. Effects of Fluid Bolus Therapy on Renal Perfusion, Oxygenation, and Function in Early Experimental Septic Kidney Injury. Crit. Care Med. 2019, 47, e36–e43. [Google Scholar] [CrossRef]
- Kosaka, J.; Lankadeva, Y.R.; May, C.N.; Bellomo, R. Histopathology of Septic Acute Kidney Injury: A Systematic Review of Experimental Data. Crit. Care Med. 2016, 44, e897–e903. [Google Scholar] [CrossRef]
- Kalakeche, R.; Hato, T.; Rhodes, G.; Dunn, K.W.; El-Achkar, T.M.; Plotkin, Z.; Sandoval, R.M.; Dagher, P.C. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J. Am. Soc. Nephrol. 2011, 22, 1505–1516. [Google Scholar] [CrossRef]
- Dellepiane, S.; Marengo, M.; Cantaluppi, V. Detrimental cross-talk between sepsis and acute kidney injury: New pathogenic mechanisms, early biomarkers and targeted therapies. Crit. Care 2016, 20, 61. [Google Scholar] [CrossRef]
- Calzavacca, P.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Variable responses of regional renal oxygenation and perfusion to vasoactive agents in awake sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R1226–R1233. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Post, E.H.; Kellum, J.A.; Bellomo, R.; Vincent, J.L. Renal perfusion in sepsis: From macro- to microcirculation. Kidney Int. 2017, 91, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Chelazzi, C.; Villa, G.; Mancinelli, P.; De Gaudio, A.R.; Adembri, C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit. Care 2015, 19, 26. [Google Scholar] [CrossRef]
- Guerci, P.; Ergin, B.; Ince, C. The macro- and microcirculation of the kidney. Best Pr. Res. Clin. Anaesthesiol. 2017, 31, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed]
- Escobar, D.A.; Botero-Quintero, A.M.; Kautza, B.C.; Luciano, J.; Loughran, P.; Darwiche, S.; Rosengart, M.R.; Zuckerbraun, B.S.; Gomez, H. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J. Surg. Res. 2015, 194, 262–272. [Google Scholar] [CrossRef]
- Opal, S.M.; Ellis, J.L.; Suri, V.; Freudenberg, J.M.; Vlasuk, G.P.; Li, Y.; Chahin, A.B.; Palardy, J.E.; Parejo, N.; Yamamoto, M.; et al. Pharmacological Sirt1 Activation Improves Mortality and Markedly Alters Transcriptional Profiles That Accompany Experimental Sepsis. Shock 2016, 45, 411–418. [Google Scholar] [CrossRef]
- Brandstrup, B.; Tonnesen, H.; Beier-Holgersen, R.; Hjortso, E.; Ording, H.; Lindorff-Larsen, K.; Rasmussen, M.S.; Lanng, C.; Wallin, L.; Iversen, L.H.; et al. Effects of intravenous fluid restriction on postoperative complications: Comparison of two perioperative fluid regimens: A randomized assessor-blinded multicenter trial. Ann. Surg. 2003, 238, 641–648. [Google Scholar] [CrossRef]
- He, J.; Yang, B. Aquaporins in Renal Diseases. Int. J. Mol. Sci. 2019, 20, 366. [Google Scholar] [CrossRef]
- Hales, C.A.; Du, H.K.; Volokhov, A.; Mourfarrej, R.; Quinn, D.A. Aquaporin channels may modulate ventilator-induced lung injury. Respir. Physiol. 2001, 124, 159–166. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Dursun, B.; Altmann, C.; Ahuja, N.; He, Z.; Bhargava, R.; Edelstein, C.E.; Jani, A.; Hoke, T.S.; Klein, C.; et al. Cytokine production increases and cytokine clearance decreases in mice with bilateral nephrectomy. Nephrol. Dial. Transplant. 2012, 27, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Edelstein, C.L. Mechanisms and mediators of lung injury after acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 48–60. [Google Scholar] [CrossRef]
- Hoke, T.S.; Douglas, I.S.; Klein, C.L.; He, Z.; Fang, W.; Thurman, J.M.; Tao, Y.; Dursun, B.; Voelkel, N.F.; Edelstein, C.L.; et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J. Am. Soc. Nephrol. 2007, 18, 155–164. [Google Scholar] [CrossRef]
- Bijuklic, K.; Jennings, P.; Kountchev, J.; Hasslacher, J.; Aydin, S.; Sturn, D.; Pfaller, W.; Patsch, J.R.; Joannidis, M. Migration of leukocytes across an endothelium-epithelium bilayer as a model of renal interstitial inflammation. Am. J. Physiol. Cell Physiol. 2007, 293, C486–C492. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Le, P.; Klenzak, J.; Freedman, S.; McMenamin, M.E.; Ikizler, T.A.; Group, P. Impaired monocyte cytokine production in critically ill patients with acute renal failure. Kidney Int. 2004, 66, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Wen, X.; Keener, C.; Pike, F.; Palevsky, P.M.; Unruh, M.; Finkel, K.; Vijayan, A.; Elder, M.; Chen, Y.F.; et al. Associations between Intensity of RRT, Inflammatory Mediators, and Outcomes. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Loftus, T.J.; Hawkins, R.B.; Raymond, S.L.; Stortz, J.A.; Hollen, M.K.; Weiss, B.P.; Miller, E.S.; Bihorac, A.; Larson, S.D.; et al. Innate Immunity in the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome and Its Implications for Therapy. Front. Immunol. 2018, 9, 595. [Google Scholar] [CrossRef]
- Murugan, R.; Karajala-Subramanyam, V.; Lee, M.; Yende, S.; Kong, L.; Carter, M.; Angus, D.C.; Kellum, J.A.; Genetic and Inflammatory Markers of Sepsis (GenIMS) Investigators. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010, 77, 527–535. [Google Scholar] [CrossRef]
- Ahuja, N.; Andres-Hernando, A.; Altmann, C.; Bhargava, R.; Bacalja, J.; Webb, R.G.; He, Z.; Edelstein, C.L.; Faubel, S. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am. J. Physiol. Ren. Physiol. 2012, 303, F864–F872. [Google Scholar] [CrossRef]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef]
- Lie, M.L.; White, L.E.; Santora, R.J.; Park, J.M.; Rabb, H.; Hassoun, H.T. Lung T lymphocyte trafficking and activation during ischemic acute kidney injury. J. Immunol. 2012, 189, 2843–2851. [Google Scholar] [CrossRef]
- Altmann, C.; Andres-Hernando, A.; McMahan, R.H.; Ahuja, N.; He, Z.; Rivard, C.J.; Edelstein, C.L.; Barthel, L.; Janssen, W.J.; Faubel, S. Macrophages mediate lung inflammation in a mouse model of ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F421–F432. [Google Scholar] [CrossRef] [PubMed]
- White, L.E.; Cui, Y.; Shelak, C.M.; Lie, M.L.; Hassoun, H.T. Lung endothelial cell apoptosis during ischemic acute kidney injury. Shock 2012, 38, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Bishop, J.V.; Wen, X.; Murugan, R.; Chandra, S.; Filippi, M.D.; Kellum, J.A. Differential effects of kidney-lung cross-talk during acute kidney injury and bacterial pneumonia. Kidney Int. 2011, 80, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Miller, L.; Ruiz-Velasco, V.; Kellum, J.A. Reversal of Acute Kidney Injury-Induced Neutrophil Dysfunction: A Critical Role for Resistin. Crit. Care Med. 2016, 44, e492–e501. [Google Scholar] [CrossRef]
- Rossaint, J.; Spelten, O.; Kassens, N.; Mueller, H.; Van Aken, H.K.; Singbartl, K.; Zarbock, A. Acute loss of renal function attenuates slow leukocyte rolling and transmigration by interfering with intracellular signaling. Kidney Int. 2011, 80, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.P.; Stone, S.F.; Neil, C.L.; van Eeden, P.E.; Fatovich, D.M.; Arendts, G.; Brown, S.G. Sustained elevation of resistin, NGAL and IL-8 are associated with severe sepsis/septic shock in the emergency department. PLoS ONE 2014, 9, e110678. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Gressner, O.A.; Sanson, E.; Tacke, F.; Trautwein, C. Serum resistin levels in critically ill patients are associated with inflammation, organ dysfunction and metabolism and may predict survival of non-septic patients. Crit. Care 2009, 13, R95. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Ilic, D.; Raupachova, J.; Horl, W.H. Resistin inhibits essential functions of polymorphonuclear leukocytes. J. Immunol. 2008, 181, 3761–3768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ankawi, G.; Sun, J.; Digvijay, K.; Yin, Y.; Rosner, M.H.; Ronco, C. Gut-kidney crosstalk in septic acute kidney injury. Crit. Care 2018, 22, 117. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.T.; Kan, W.C.; Shiao, C.C. Acute Kidney Injury and Gut Dysbiosis: A Narrative Review Focus on Pathophysiology and Treatment. Int. J. Mol. Sci. 2022, 23, 3658. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, C.J.; Go, Y.S.; Lee, H.Y.; Kim, M.G.; Oh, S.W.; Cho, W.Y.; Im, S.H.; Jo, S.K. Intestinal microbiota control acute kidney injury severity by immune modulation. Kidney Int. 2020, 98, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.K. Kidney-Gut Crosstalk in AKI. Kidney360 2021, 2, 886–889. [Google Scholar] [CrossRef]
- Park, S.W.; Chen, S.W.; Kim, M.; Brown, K.M.; Kolls, J.K.; D’Agati, V.D.; Lee, H.T. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab. Invest. 2011, 91, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Kim, J.Y.; Ham, A.; Brown, K.M.; Mori-Akiyama, Y.; Ouellette, A.J.; D’Agati, V.D.; Lee, H.T. Paneth cell-mediated multiorgan dysfunction after acute kidney injury. J. Immunol. 2012, 189, 5421–5433. [Google Scholar] [CrossRef] [PubMed]
- Jeng, L.; Yamshchikov, A.V.; Judd, S.E.; Blumberg, H.M.; Martin, G.S.; Ziegler, T.R.; Tangpricha, V. Alterations in vitamin D status and anti-microbial peptide levels in patients in the intensive care unit with sepsis. J. Transl. Med. 2009, 7, 28. [Google Scholar] [CrossRef]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef]
- de Haan, K.; Groeneveld, A.B.; de Geus, H.R.; Egal, M.; Struijs, A. Vitamin D deficiency as a risk factor for infection, sepsis and mortality in the critically ill: Systematic review and meta-analysis. Crit. Care 2014, 18, 660. [Google Scholar] [CrossRef]
- Castellanos, M.; Jung, E.; Park, S.Y.; Schuller-Levis, G.; Odaimi, M.; Elsayegh, S.; Kleiner, M.; Elsoueidi, R.; Shtaynberg, N.; Park, E. Effect of parathyroid hormone and teriparatide on immune function of human adherent and non-adherent leukocytes. Clin. Nephrol. 2010, 74, 83–90. [Google Scholar] [CrossRef]
- Alexiewicz, J.M.; Smogorzewski, M.; Fadda, G.Z.; Massry, S.G. Impaired phagocytosis in dialysis patients: Studies on mechanisms. Am. J. Nephrol. 1991, 11, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Christov, M. Fibroblast growth factor 23 in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Invest. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, E.A.; Han, X.; Xiao, Z.; Quarles, L.D. Role of Fibroblast Growth Factor-23 in Innate Immune Responses. Front. Endocrinol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Chonchol, M.; Greene, T.; Zhang, Y.; Hoofnagle, A.N.; Cheung, A.K. Low Vitamin D and High Fibroblast Growth Factor 23 Serum Levels Associate with Infectious and Cardiac Deaths in the HEMO Study. J. Am. Soc. Nephrol. 2016, 27, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Bartz, T.M.; Dalrymple, L.; de Boer, I.H.; Kestenbaum, B.; Shlipak, M.G.; Garimella, P.S.; Ix, J.H.; Chonchol, M. Fibroblast Growth Factor 23 and the Risk of Infection-Related Hospitalization in Older Adults. J. Am. Soc. Nephrol. 2017, 28, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Noiri, E.; Hamasaki, Y.; Matsubara, T.; Ishii, T.; Yahagi, N.; Nangaku, M.; Doi, K. Erythropoietin concentration in acute kidney injury is associated with insulin-like growth factor-binding protein-1. Nephrology 2016, 21, 693–699. [Google Scholar] [CrossRef]
- Micanovic, R.; LaFavers, K.; Garimella, P.S.; Wu, X.R.; El-Achkar, T.M. Uromodulin (Tamm-Horsfall protein): Guardian of urinary and systemic homeostasis. Nephrol. Dial. Transplant. 2020, 35, 33–43. [Google Scholar] [CrossRef]
- Micanovic, R.; Khan, S.; Janosevic, D.; Lee, M.E.; Hato, T.; Srour, E.F.; Winfree, S.; Ghosh, J.; Tong, Y.; Rice, S.E.; et al. Tamm-Horsfall Protein Regulates Mononuclear Phagocytes in the Kidney. J. Am. Soc. Nephrol. 2018, 29, 841–856. [Google Scholar] [CrossRef]
- Bates, J.M.; Raffi, H.M.; Prasadan, K.; Mascarenhas, R.; Laszik, Z.; Maeda, N.; Hultgren, S.J.; Kumar, S. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: Rapid communication. Kidney Int. 2004, 65, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Raffi, H.S.; Bates, J.M., Jr.; Laszik, Z.; Kumar, S. Tamm-horsfall protein protects against urinary tract infection by proteus mirabilis. J. Urol. 2009, 181, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; McCracken, R.; Liu, Y.; Heitmeier, M.R.; Bourgeois, S.; Ryerse, J.; Wu, X.R. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 304, F1066–F1075. [Google Scholar] [CrossRef]
- LaFavers, K.A.; Macedo, E.; Garimella, P.S.; Lima, C.; Khan, S.; Myslinski, J.; McClintick, J.; Witzmann, F.A.; Winfree, S.; Phillips, C.L.; et al. Circulating uromodulin inhibits systemic oxidative stress by inactivating the TRPM2 channel. Sci. Transl. Med. 2019, 11, eaaw3639. [Google Scholar] [CrossRef] [PubMed]
- Delgado, G.E.; Kleber, M.E.; Scharnagl, H.; Kramer, B.K.; Marz, W.; Scherberich, J.E. Serum Uromodulin and Mortality Risk in Patients Undergoing Coronary Angiography. J. Am. Soc. Nephrol. 2017, 28, 2201–2210. [Google Scholar] [CrossRef]
- Garimella, P.S.; Katz, R.; Ix, J.H.; Fried, L.F.; Kritchevsky, S.B.; Devarajan, P.; Bennett, M.R.; Parikh, C.R.; Shlipak, M.G.; Harris, T.B.; et al. Association of urinary uromodulin with kidney function decline and mortality: The health ABC study. Clin. Nephrol. 2017, 87, 278–286. [Google Scholar] [CrossRef]
- Leiherer, A.; Muendlein, A.; Saely, C.H.; Ebner, J.; Brandtner, E.M.; Fraunberger, P.; Drexel, H. Serum uromodulin is a predictive biomarker for cardiovascular events and overall mortality in coronary patients. Int. J. Cardiol. 2017, 231, 6–12. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Altmann, C.; Ahuja, N.; Lanaspa, M.A.; Nemenoff, R.; He, Z.; Ishimoto, T.; Simpson, P.A.; Weiser-Evans, M.C.; Bacalja, J.; et al. Splenectomy exacerbates lung injury after ischemic acute kidney injury in mice. Am. J. Physiol. Ren. Physiol. 2011, 301, F907–F916. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Hu, X.; Yuen, P.S.; Leelahavanichkul, A.; Yasuda, H.; Kim, S.M.; Schnermann, J.; Jonassen, T.E.; Frokiaer, J.; Nielsen, S.; et al. AP214, an analogue of alpha-melanocyte-stimulating hormone, ameliorates sepsis-induced acute kidney injury and mortality. Kidney Int. 2008, 73, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Blot, S.I.; Lameire, N.H.; Vanholder, R.C.; De Bacquer, D.; Colardyn, F.A. Effect of nosocomial bloodstream infection on the outcome of critically ill patients with acute renal failure treated with renal replacement therapy. J. Am. Soc. Nephrol. 2004, 15, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.J.; Lopez-Herce, J.; Vierge, E.; Castillo, A.; Bustinza, A.; Bellon, J.M.; Sanchez, A.; Fernandez, S. Infection in critically ill pediatric patients on continuous renal replacement therapy. Int. J. Artif. Organs. 2017, 40, 224–229. [Google Scholar] [CrossRef]
- Self, W.H.; Semler, M.W.; Wanderer, J.P.; Wang, L.; Byrne, D.W.; Collins, S.P.; Slovis, C.M.; Lindsell, C.J.; Ehrenfeld, J.M.; Siew, E.D.; et al. Balanced Crystalloids versus Saline in Noncritically Ill Adults. N. Engl. J. Med. 2018, 378, 819–828. [Google Scholar] [CrossRef]
- Semler, M.W.; Self, W.H.; Wanderer, J.P.; Ehrenfeld, J.M.; Wang, L.; Byrne, D.W.; Stollings, J.L.; Kumar, A.B.; Hughes, C.G.; Hernandez, A.; et al. Balanced Crystalloids versus Saline in Critically Ill Adults. N. Engl. J. Med. 2018, 378, 829–839. [Google Scholar] [CrossRef]
- Honore, P.M.; Jacobs, R.; Hendrickx, I.; Bagshaw, S.M.; Joannes-Boyau, O.; Boer, W.; De Waele, E.; Van Gorp, V.; Spapen, H.D. Prevention and treatment of sepsis-induced acute kidney injury: An update. Ann. Intensive Care 2015, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.W.; Roberts, D.J.; De Waele, J.; Jaeschke, R.; Malbrain, M.L.; De Keulenaer, B.; Duchesne, J.; Bjorck, M.; Leppaniemi, A.; Ejike, J.C.; et al. Intra-abdominal hypertension and the abdominal compartment syndrome: Updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med. 2013, 39, 1190–1206. [Google Scholar] [CrossRef]
- Fujii, H.; Yonekura, Y.; Yamashita, Y.; Kono, K.; Nakai, K.; Goto, S.; Sugano, M.; Goto, S.; Fujieda, A.; Ito, Y.; et al. Anti-oxidative effect of AST-120 on kidney injury after myocardial infarction. Br. J. Pharm. 2016, 173, 1302–1313. [Google Scholar] [CrossRef]
- Nguyen, H.B.; Eshete, B.; Lau, K.H.; Sai, A.; Villarin, M.; Baylink, D. Serum 1,25-dihydroxyvitamin, D. An outcome prognosticator in human sepsis. PLoS ONE 2013, 8, e64348. [Google Scholar] [CrossRef]
- Graidis, S.; Papavramidis, T.S.; Papaioannou, M. Vitamin D and Acute Kidney Injury: A Two-Way Causality Relation and a Predictive, Prognostic, and Therapeutic Role of Vitamin D. Front. Nutr. 2020, 7, 630951. [Google Scholar] [CrossRef]
- Doi, K.; Ishizu, T.; Tsukamoto-Sumida, M.; Hiruma, T.; Yamashita, T.; Ogasawara, E.; Hamasaki, Y.; Yahagi, N.; Nangaku, M.; Noiri, E. The high-mobility group protein B1-Toll-like receptor 4 pathway contributes to the acute lung injury induced by bilateral nephrectomy. Kidney Int. 2014, 86, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, A.; Miller, L.; Kellum, J.A.; Singbartl, K. Hemoadsorption corrects hyperresistinemia and restores anti-bacterial neutrophil function. Intensive Care Med. Exp. 2017, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Alpers, C.E.; Azeloglu, E.U.; Balis, U.G.J.; Barasch, J.M.; Barisoni, L.; Blank, K.N.; Bomback, A.S.; Brown, K.; Dagher, P.C.; et al. Rationale and design of the Kidney Precision Medicine Project. Kidney Int. 2021, 99, 498–510. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Strategies | Remarks |
|---|---|
| Prompt and proper antibiotics administration | Treatment of infection and sepsis |
| Vasopressor keeping a mean arterial blood pressure > 65 mmHg (norepinephrine preferred) | Maintain renal perfusion and autoregulation |
| Balanced crystalloid fluid administration | Avoidance of chloride overload. With benefit on major kidney adverse events |
| Avoidance of nephrotoxic agents | e.g., some antibiotics and contrast media |
| Application of the KDIGO bundle (?) | Effects under evaluation |
| High awareness of abdominal compartment syndrome | High intra- abdominal pressure is a deteriorating factor of SA-AKI |
| Strategies | Remarks |
|---|---|
| Probiotics and short chain fatty acid supplementation | Decrease gut-microbiota-derived metabolites and their immunosuppressive effects |
| AST-120 administration | |
| Vitamin D supplementation | Immunomodulation effect |
| Avoidance of fluid overload | Decrease tissue edema and infection risk |
| Avoidance of unnecessary catheter cannulation | Decrease bloodstream infection risk |
| KRT strategies | Hemofiltration and hemoadsorption for removing some cytokines and molecules that are harmful to immunity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-M.; Chou, Y.-T.; Kan, W.-C.; Shiao, C.-C. Sepsis and Acute Kidney Injury: A Review Focusing on the Bidirectional Interplay. Int. J. Mol. Sci. 2022, 23, 9159. https://doi.org/10.3390/ijms23169159
Chang Y-M, Chou Y-T, Kan W-C, Shiao C-C. Sepsis and Acute Kidney Injury: A Review Focusing on the Bidirectional Interplay. International Journal of Molecular Sciences. 2022; 23(16):9159. https://doi.org/10.3390/ijms23169159
Chicago/Turabian StyleChang, Yu-Ming, Yu-Ting Chou, Wei-Chih Kan, and Chih-Chung Shiao. 2022. "Sepsis and Acute Kidney Injury: A Review Focusing on the Bidirectional Interplay" International Journal of Molecular Sciences 23, no. 16: 9159. https://doi.org/10.3390/ijms23169159