
Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment

 , ,
, ,
Abstract
:1. Introduction
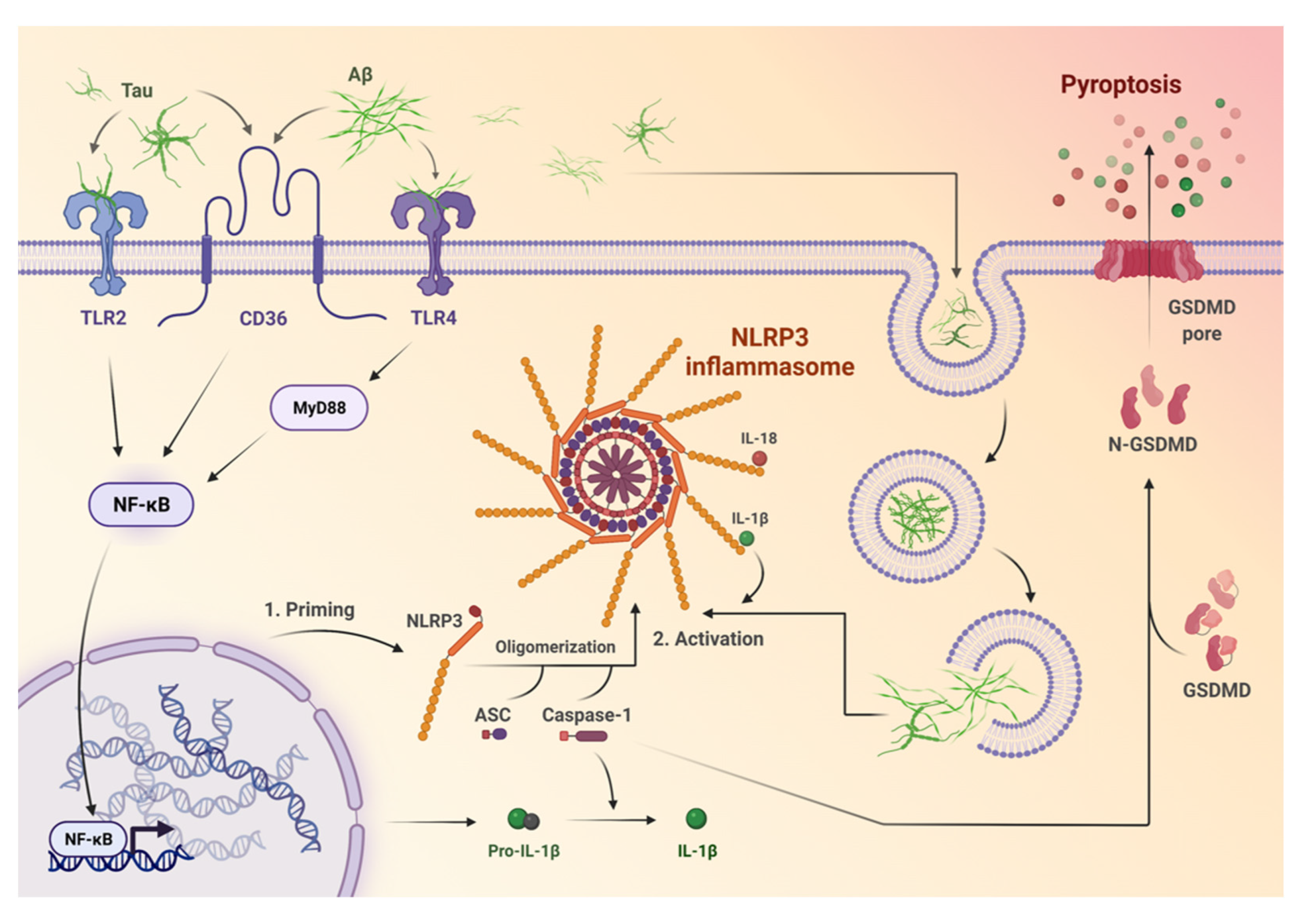
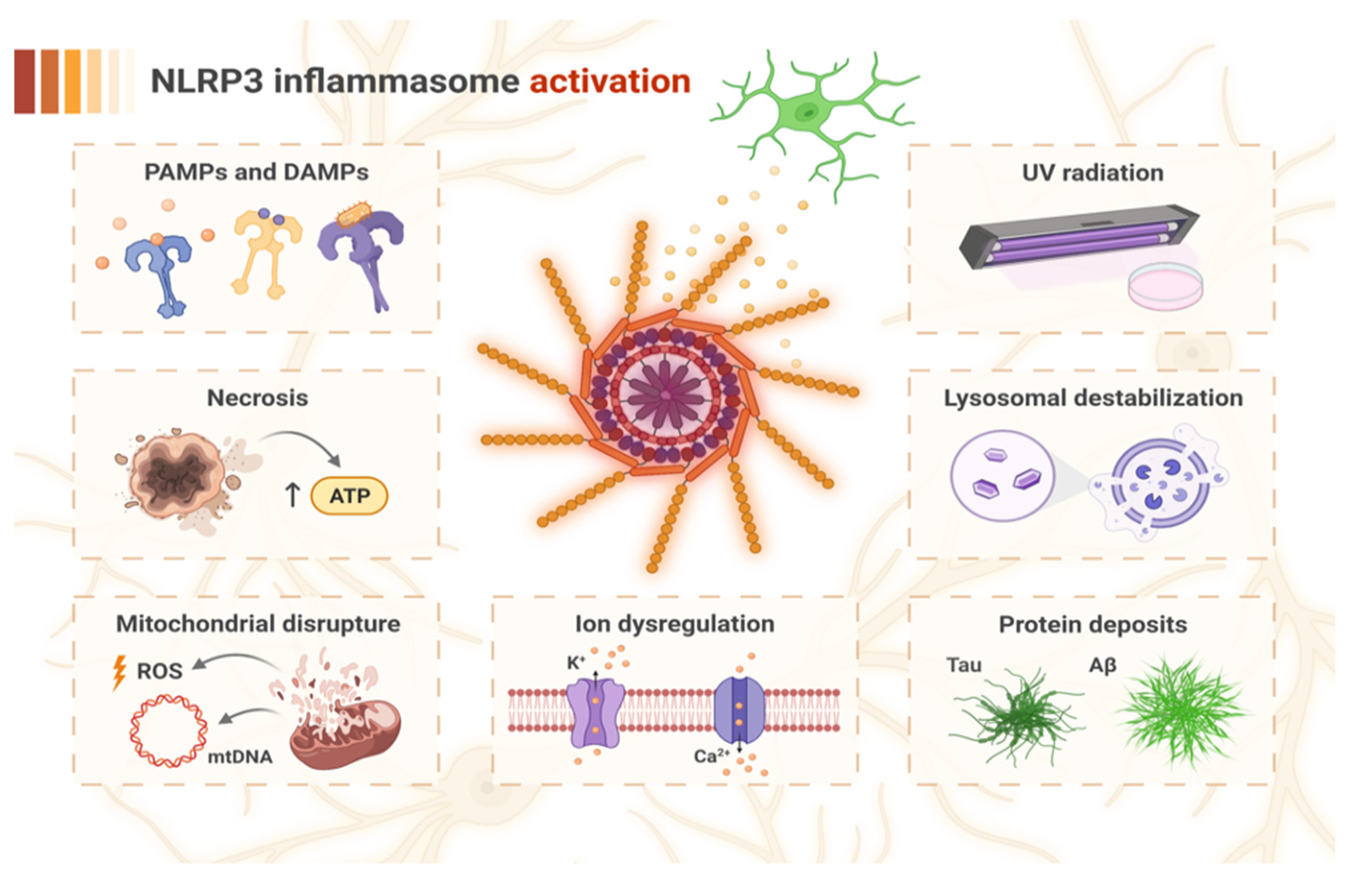
2. NLRP3 Inflammasome Activation and Stimulation
3. NLRP3 Inflammasome in AD
4. Neuroinflammation in the Pathogenesis and Progression of AD
5. NLRP3 Inhibitors
5.1. MCC950
5.2. JC124
5.3. VX-765
5.4. OLT1177
5.5. NSAIDs
5.6. Colchicine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Poplawski, T.; Walczak, A.; Gradzik, K.; Wawrzynkiewicz, A.; Wojtczak, R.; Mucha, B.; Diehl, J.A.; Majsterek, I. Inhibition of the PERK-dependent unfolded protein response signaling pathway involved in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Markiewicz, L.; Diehl, J.A.; Pytel, D.; Majsterek, I. Unfolded Protein Response and PERK Kinase as a New Therapeutic Target in the Pathogenesis of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 3169–3184. [Google Scholar] [CrossRef]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fandrich, M. Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Schmidt, M.; Sachse, C.; Richter, W.; Xu, C.; Fandrich, M.; Grigorieff, N. Comparison of Alzheimer Abeta(1–40) and Abeta(1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. USA 2009, 106, 19813–19818. [Google Scholar] [CrossRef]
- Rozpedek-Kaminska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef]
- Lee, H.G.; Won, S.M.; Gwag, B.J.; Lee, Y.B. Microglial P2X(7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011, 43, 7–14. [Google Scholar] [CrossRef]
- Rinne, J.O.; Nagren, K. Positron emission tomography in at risk patients and in the progression of mild cognitive impairment to Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984. [Google Scholar] [CrossRef] [PubMed]
- Milner, M.T.; Maddugoda, M.; Gotz, J.; Burgener, S.S.; Schroder, K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Freeman, L.C.; Ting, J.P. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 2016, 136, 29–38. [Google Scholar] [CrossRef]
- Feng, Y.S.; Tan, Z.X.; Wu, L.Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef] [PubMed]
- Fulp, J.; He, L.; Toldo, S.; Jiang, Y.; Boice, A.; Guo, C.; Li, X.; Rolfe, A.; Sun, D.; Abbate, A.; et al. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis and Biological Characterization. J. Med. Chem. 2018, 61, 5412–5423. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2014, 76, 57–78. [Google Scholar] [CrossRef]
- Jha, S.; Ting, J.P. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J. Immunol. 2009, 183, 7623–7629. [Google Scholar] [CrossRef]
- Duncan, J.A.; Ting, J.P. Rebuilding host-pathogen interaction from the ground up: In vitro reconstitution of the inflammasome. Cell Host Microbe 2007, 1, 7–9. [Google Scholar] [CrossRef]
- Meng, G.; Grabiec, A.; Vallon, M.; Ebe, B.; Hampel, S.; Bessler, W.; Wagner, H.; Kirschning, C.J. Cellular recognition of tri-/di-palmitoylated peptides is independent from a domain encompassing the N-terminal seven leucine-rich repeat (LRR)/LRR-like motifs of TLR2. J. Biol. Chem. 2003, 278, 39822–39829. [Google Scholar] [CrossRef]
- O’Connor, W., Jr.; Harton, J.A.; Zhu, X.; Linhoff, M.W.; Ting, J.P. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J. Immunol. 2003, 171, 6329–6333. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Scott, P.; Mueller, J.L.; Misaghi, A.; Stevens, S.; Yancopoulos, G.D.; Murphy, A.; Valenzuela, D.M.; Liu-Bryan, R. Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum. 2010, 62, 2170–2179. [Google Scholar] [CrossRef]
- Dowds, T.A.; Masumoto, J.; Zhu, L.; Inohara, N.; Nunez, G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 2004, 279, 21924–21928. [Google Scholar] [CrossRef]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.J.; Kim, J.U.; Yook, T.H.; Kim, K.H.; Lee, J.Y.; Yang, G. A Novel Treatment Strategy by Natural Products in NLRP3 Inflammasome-Mediated Neuroinflammation in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 1324. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Demento, S.L.; Eisenbarth, S.C.; Foellmer, H.G.; Platt, C.; Caplan, M.J.; Mark Saltzman, W.; Mellman, I.; Ledizet, M.; Fikrig, E.; Flavell, R.A.; et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine 2009, 27, 3013–3021. [Google Scholar] [CrossRef]
- Duncan, J.A.; Gao, X.; Huang, M.T.; O’Connor, B.P.; Thomas, C.E.; Willingham, S.B.; Bergstralh, D.T.; Jarvis, G.A.; Sparling, P.F.; Ting, J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009, 182, 6460–6469. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Yang, L.; Kouadir, M.; Yang, Y.; Wang, J.; Zhou, X.; Yin, X.; Zhao, D. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J. Neuroinflamm. 2012, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.A.; Ruane, D.; Claass, B.; Creagh, E.; Harris, J.; Malyala, P.; Singh, M.; O’Hagan, D.T.; Petrilli, V.; Tschopp, J.; et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 870–875. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, E.; Piippo, N.; Hytti, M.; Hyttinen, J.M.T.; Kaarniranta, K.; Kauppinen, A. Only IL-1β release is inflammasome-dependent upon ultraviolet B irradiation although IL-18 is also secreted. FASEB J. 2020, 34, 6437–6448. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Regulation of the NLRP3 Inflammasome by Post-Translational Modifications and Small Molecules. Front. Immunol. 2020, 11, 618231. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-Del-Olmo, I.; Nichols, E.M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 565924. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, Q. Activation of NLRP3 Inflammasome and Onset of Alzheimer’s Disease. Front. Immunol. 2021, 12, 701282. [Google Scholar] [CrossRef]
- Apelt, J.; Schliebs, R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001, 894, 21–30. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Harte, M. Investigating Markers of the NLRP3 Inflammasome Pathway in Alzheimer’s Disease: A Human Post-Mortem Study. Genes 2021, 12, 1753. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Heneka, M.T. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019, 33, 13075–13084. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brone, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Huang, N.Q.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav. Brain Res. 2017, 316, 234–244. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.M.; Chen, W.S.; Sheu, J.J.; Chen, Y.H.; Chen, J.H.; Chang, C.C. Autoimmune rheumatic diseases increase dementia risk in middle-aged patients: A nationwide cohort study. PLoS ONE 2018, 13, e0186475. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- De Nazareth, A.M. Type 2 diabetes mellitus in the pathophysiology of Alzheimer’s disease. Dement. Neuropsychol. 2017, 11, 105–113. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef]
- Love, S.; Miners, J.S. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016, 131, 645–658. [Google Scholar] [CrossRef]
- Reitz, C. Dyslipidemia and the risk of Alzheimer’s disease. Curr. Atheroscler. Rep. 2013, 15, 307. [Google Scholar] [CrossRef]
- Eglit, G.M.L.; Weigand, A.J.; Nation, D.A.; Bondi, M.W.; Bangen, K.J. Hypertension and Alzheimer’s disease: Indirect effects through circle of Willis atherosclerosis. Brain Commun. 2020, 2, fcaa114. [Google Scholar] [CrossRef]
- Lennon, M.J.; Makkar, S.R.; Crawford, J.D.; Sachdev, P.S. Midlife Hypertension and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019, 71, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Sadighi Akha, A.A. Aging and the immune system: An overview. J. Immunol. Methods 2018, 463, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef]
- Lu, M.; Sun, X.L.; Qiao, C.; Liu, Y.; Ding, J.H.; Hu, G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol. Aging 2014, 35, 421–430. [Google Scholar] [CrossRef]
- Zendedel, A.; Johann, S.; Mehrabi, S.; Joghataei, M.T.; Hassanzadeh, G.; Kipp, M.; Beyer, C. Activation and Regulation of NLRP3 Inflammasome by Intrathecal Application of SDF-1a in a Spinal Cord Injury Model. Mol. Neurobiol. 2016, 53, 3063–3075. [Google Scholar] [CrossRef]
- Maturana, C.J.; Aguirre, A.; Saez, J.C. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol. 2017, 77, 625–642. [Google Scholar] [CrossRef]
- Von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinson’s Dis. 2018, 4, 24. [Google Scholar] [CrossRef]
- Schaafsma, W.; Zhang, X.; van Zomeren, K.C.; Jacobs, S.; Georgieva, P.B.; Wolf, S.A.; Kettenmann, H.; Janova, H.; Saiepour, N.; Hanisch, U.K.; et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav. Immun. 2015, 48, 205–221. [Google Scholar] [CrossRef]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Hasler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Rhee, S.Y.; Ryu, J.H.; Cheong, J.H.; Kwon, K.; Yang, S.I.; Park, S.H.; Lee, J.; Kim, H.Y.; Han, S.H.; et al. Activation of adenosine A2A receptor up-regulates BDNF expression in rat primary cortical neurons. Neurochem. Res. 2011, 36, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Guo, J.; Cai, H.; Zhao, C.; Wang, H.; Liu, Z.; Ge, Z.M. M2-Like Microglia Polarization Attenuates Neuropathic Pain Associated with Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1255–1265. [Google Scholar] [CrossRef]
- Lajqi, T.; Stojiljkovic, M.; Williams, D.L.; Hudalla, H.; Bauer, M.; Witte, O.W.; Wetzker, R.; Bauer, R.; Schmeer, C. Memory-Like Responses of Brain Microglia Are Controlled by Developmental State and Pathogen Dose. Front. Immunol. 2020, 11, 546415. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kamboh, M.I. A Brief Synopsis on the Genetics of Alzheimer’s Disease. Curr. Genet. Med. Rep. 2018, 6, 133–135. [Google Scholar] [CrossRef]
- Xue, F.; Du, H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells 2021, 10, 321. [Google Scholar] [CrossRef]
- Sheng, L.; Chen, M.; Cai, K.; Song, Y.; Yu, D.; Zhang, H.; Xu, G. Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 2019, 33, 10425–10442. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nistico, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; Halai, R.; Cooper, M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Zhao, G.; Wang, Y.; Ren, P.; Wu, M. MCC950, a Selective Inhibitor of NLRP3 Inflammasome, Reduces the Inflammatory Response and Improves Neurological Outcomes in Mice Model of Spinal Cord Injury. Front. Mol. Biosci. 2020, 7, 37. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Tapia-Abellan, A.; Angosto-Bazarra, D.; Martinez-Banaclocha, H.; de Torre-Minguela, C.; Ceron-Carrasco, J.P.; Perez-Sanchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Vande Walle, L.; Stowe, I.B.; Sacha, P.; Lee, B.L.; Demon, D.; Fossoul, A.; Van Hauwermeiren, F.; Saavedra, P.H.V.; Simon, P.; Subrt, V.; et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- He, X.F.; Xu, J.H.; Li, G.; Li, M.Y.; Li, L.L.; Pei, Z.; Zhang, L.Y.; Hu, X.Q. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhuang, L.; Luo, X.; Liang, J.; Sun, E.; He, Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020, 238, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, L.Y.; Brough, D.; Freeman, S. Inhibiting the NLRP3 Inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Carna, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflammation 2019, 16, 81. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Blevins, H.; Xu, Y.; Sun, X.; Bloom, G.S.; Zhang, S.; Sun, D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2021, 82, 1769–1783. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- Flores, J.; Noel, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- Flores, J.; Noel, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020, 11, 4571. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.Q.; Shi, Y.J.; Ding, S.Q.; Shen, L.; Wang, R.; Wang, Q.Y.; Zha, C.; Ding, H.; Hu, J.G.; et al. VX-765 reduces neuroinflammation after spinal cord injury in mice. Neural Regen. Res. 2021, 16, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hao, J.H.; Yao, D.; Li, R.; Li, X.F.; Yu, Z.Y.; Luo, X.; Liu, X.H.; Wang, M.H.; Wang, W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci. Ther. 2020, 26, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug Development for Alzheimer’s Disease: Microglia Induced Neuroinflammation as a Target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef]
- Noe, F.M.; Polascheck, N.; Frigerio, F.; Bankstahl, M.; Ravizza, T.; Marchini, S.; Beltrame, L.; Bandero, C.R.; Loscher, W.; Vezzani, A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 2013, 59, 183–193. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Fernandez, A.; Skouras, D.B.; Dinarello, C.A.; Lopez-Vales, R. OLT1177 (Dapansutrile), a Selective NLRP3 Inflammasome Inhibitor, Ameliorates Experimental Autoimmune Encephalomyelitis Pathogenesis. Front. Immunol. 2019, 10, 2578. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer’s Disease and Parkinson’s Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals 2010, 3, 1812–1841. [Google Scholar] [CrossRef]
- Dill, J.; Patel, A.R.; Yang, X.L.; Bachoo, R.; Powell, C.M.; Li, S. A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. J. Neurosci. 2010, 30, 963–972. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, Y.; Li, B.; Liu, F.; Ryder, J.W.; Wu, X.; Gonzalez-DeWhitt, P.A.; Gelfanova, V.; Hale, J.E.; May, P.C.; et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science 2003, 302, 1215–1217. [Google Scholar] [CrossRef]
- Fu, Q.; Hue, J.; Li, S. Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J. Neurosci. 2007, 27, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Frautschy, S.A. Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.; Kamal, M.A.; Rizvi, S.M.D. Repurposed Drugs as Potential Therapeutic Candidates for the Management of Alzheimer’s Disease. Curr. Drug Metab. 2017, 18, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin. Investig. Drugs 2009, 18, 1147–1168. [Google Scholar] [CrossRef]
- Benito-Leon, J.; Contador, I.; Vega, S.; Villarejo-Galende, A.; Bermejo-Pareja, F. Non-steroidal anti-inflammatory drugs use in older adults decreases risk of Alzheimer’s disease mortality. PLoS ONE 2019, 14, e0222505. [Google Scholar] [CrossRef]
- Karceski, S.; Karceski, S. Can naproxen slow the progression of Alzheimer disease? Neurology 2019, 92, e2181–e2184. [Google Scholar] [CrossRef]
- Li, X.; Kaida-Yip, F.; Zabel, M. NSAID Use and the Prevention of Alzheimer’s Disease: A Meta-Analysis (P6.184). Neurology 2018, 90, P6.184. [Google Scholar]
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.E.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J.; et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef]
- Laine, L.; Wogen, J.; Yu, H. Gastrointestinal health care resource utilization with chronic use of COX-2-specific inhibitors versus traditional NSAIDs. Gastroenterology 2003, 125, 389–395. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Fiolet, A.T.L.; Opstal, T.S.J.; Dekker, M.; Suquilanda, D.; Zivkovic, M.; Duyvendak, M.; The, S.H.K.; Timmers, L.; Bax, W.A.; et al. Colchicine reduces extracellular vesicle NLRP3 inflammasome protein levels in chronic coronary disease: A LoDoCo2 biomarker substudy. Atherosclerosis 2021, 334, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef]
- Kumar, A.; Seghal, N.; Naidu, P.S.; Padi, S.S.; Goyal, R. Colchicines-induced neurotoxicity as an animal model of sporadic dementia of Alzheimer’s type. Pharmacol. Rep. 2007, 59, 274–283. [Google Scholar]
- Sil, S.; Ghosh, T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s Disease. J. Neuroimmunol. 2016, 291, 115–124. [Google Scholar] [CrossRef]
- Sil, S.; Ghosh, T. Cox-2 Plays a Vital Role in the Impaired Anxiety Like Behavior in Colchicine Induced Rat Model of Alzheimer Disease. Behav. Neurol. 2016, 2016, 1501527. [Google Scholar] [CrossRef]
- Sil, S.; Goswami, A.R.; Dutta, G.; Ghosh, T. Effects of naproxen on immune responses in a colchicine-induced rat model of Alzheimer’s disease. Neuroimmunomodulation 2014, 21, 304–321. [Google Scholar] [CrossRef]
- Tilson, H.A.; Rogers, B.C.; Grimes, L.; Harry, G.J.; Peterson, N.J.; Hong, J.S.; Dyer, R.S. Time-dependent neurobiological effects of colchicine administered directly into the hippocampus of rats. Brain Res. 1987, 408, 163–172. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound Code of Inhibitor | Chemical Name ofInhibitor | Chemical Structure of Inhibitor | Mechanism of Action | Research Models |
|---|---|---|---|---|
| MCC950 (also known as CRID-3 or CP-456773) | 1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl]urea |  | inhibition of NACHT domain of NLRP3 | |
| JC124 | (5-Chloro-2-methoxy-N-(4-(N-methylsulfamoyl) phenethyl)benzamide |  | inhibition of caspase-1 activation, IL-1β secretion, and ASC aggregation | |
| VX-765 (also known as Belnacasan) | N-(4-Amino-3-chlorobenzoyl)-3-methyl-L-valyl-N-[(2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl]-L-prolinamide |  | inhibition of caspase-1 and caspase-4 | |
| OLT1177 (also known as Dapansutrile) | 3-(Methane sulfonyl)propanenitrile |  | inhibition of NLRP-ASC and NLRP3-caspase-1 interaction |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 8979. https://doi.org/10.3390/ijms23168979
Barczuk J, Siwecka N, Lusa W, Rozpędek-Kamińska W, Kucharska E, Majsterek I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. International Journal of Molecular Sciences. 2022; 23(16):8979. https://doi.org/10.3390/ijms23168979
Chicago/Turabian StyleBarczuk, Julia, Natalia Siwecka, Weronika Lusa, Wioletta Rozpędek-Kamińska, Ewa Kucharska, and Ireneusz Majsterek. 2022. "Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment" International Journal of Molecular Sciences 23, no. 16: 8979. https://doi.org/10.3390/ijms23168979