
A Novel Antibody Targeting the Second Extracellular Loop of the Serotonin 5-HT2A Receptor Inhibits Platelet Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
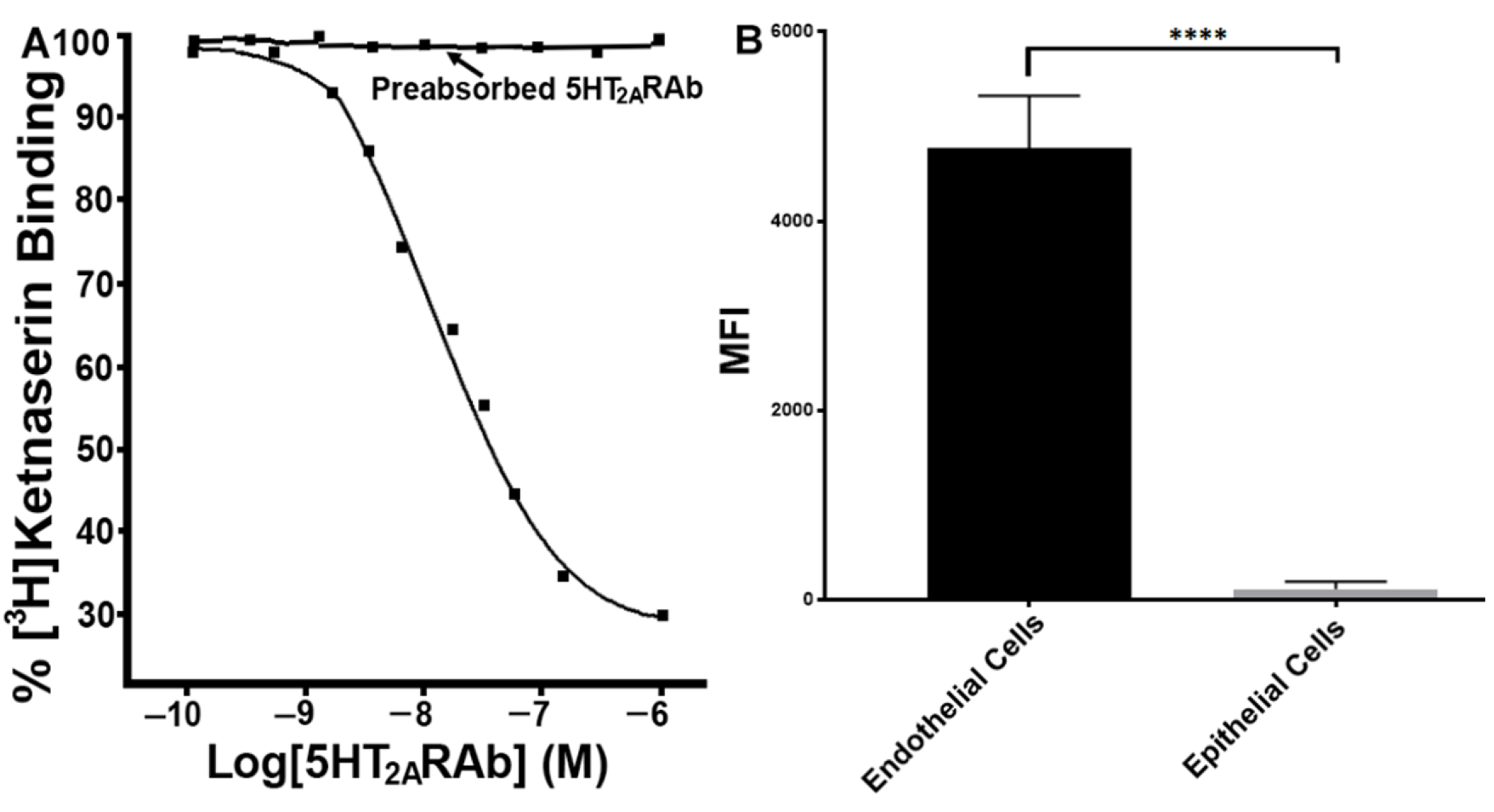
2.1. The 5HT2ARAb Displaces the 5HT2AR Antagonist Ketanserin from Its Binding Sites
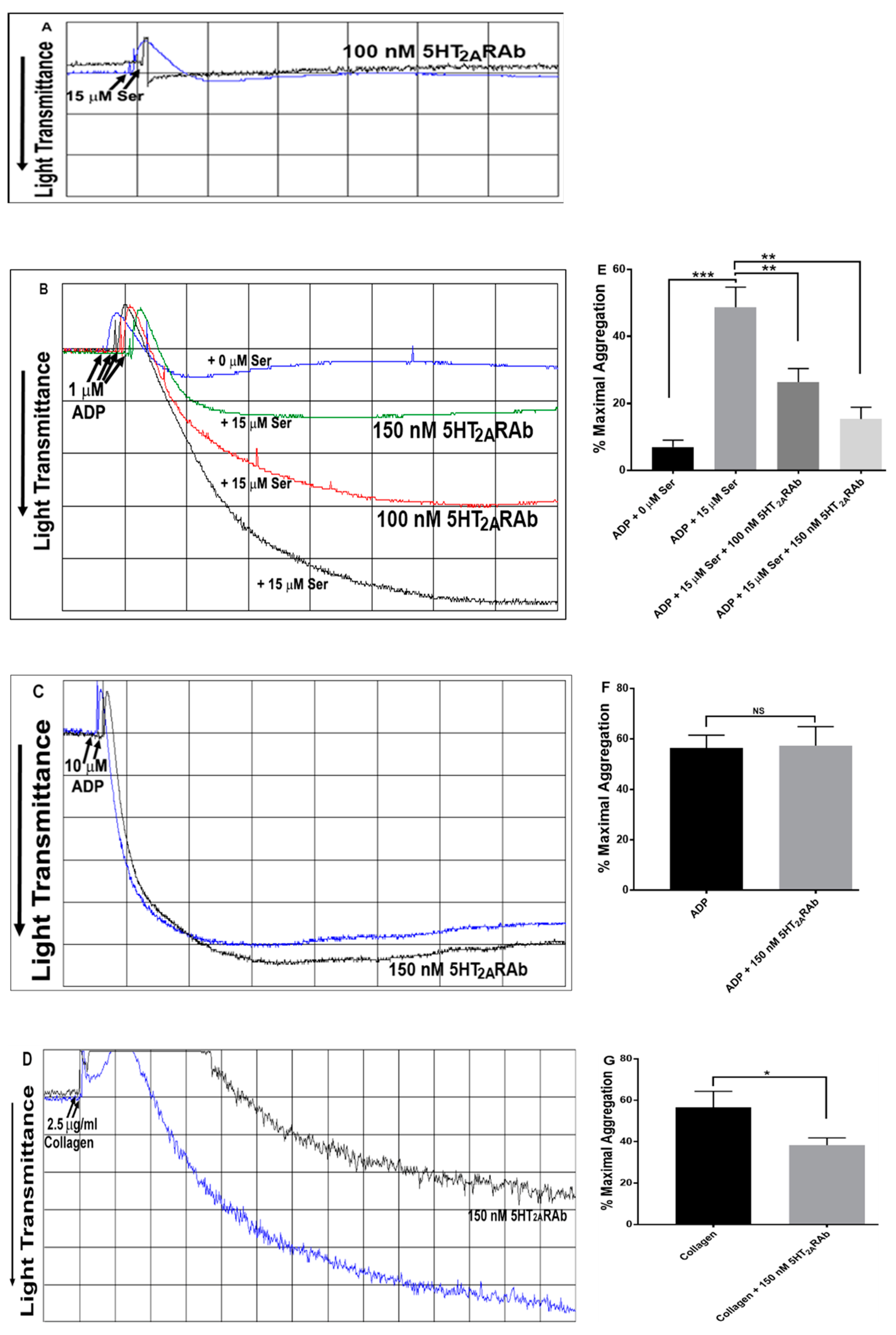
2.2. The 5HT2ARAb Inhibits Serotonin-Enhanced Human Platelet Aggregation In Vitro
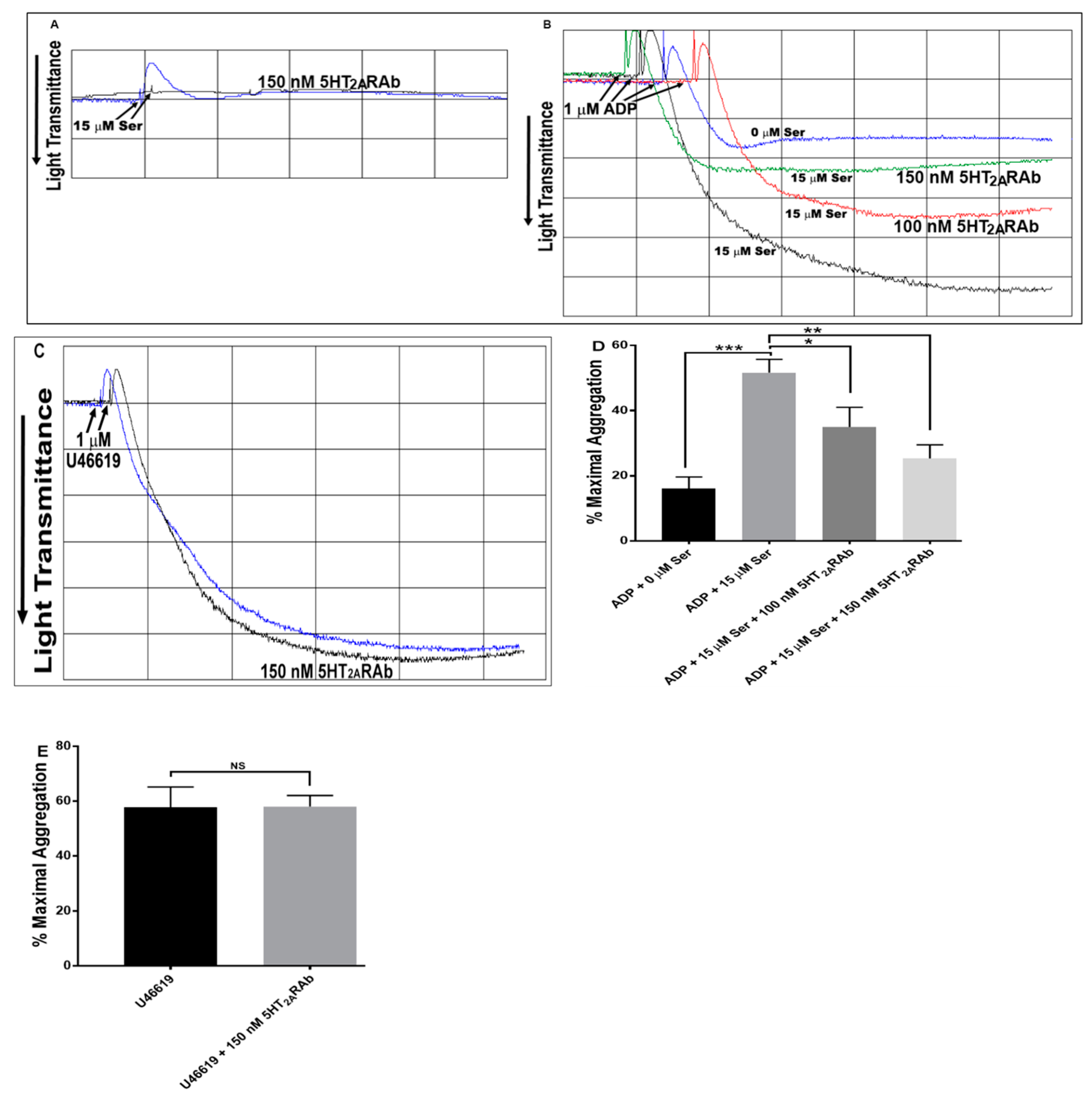
2.3. The 5HT2ARAb Inhibits Serotonin-Enhanced Mouse Platelet Aggregation Ex Vivo
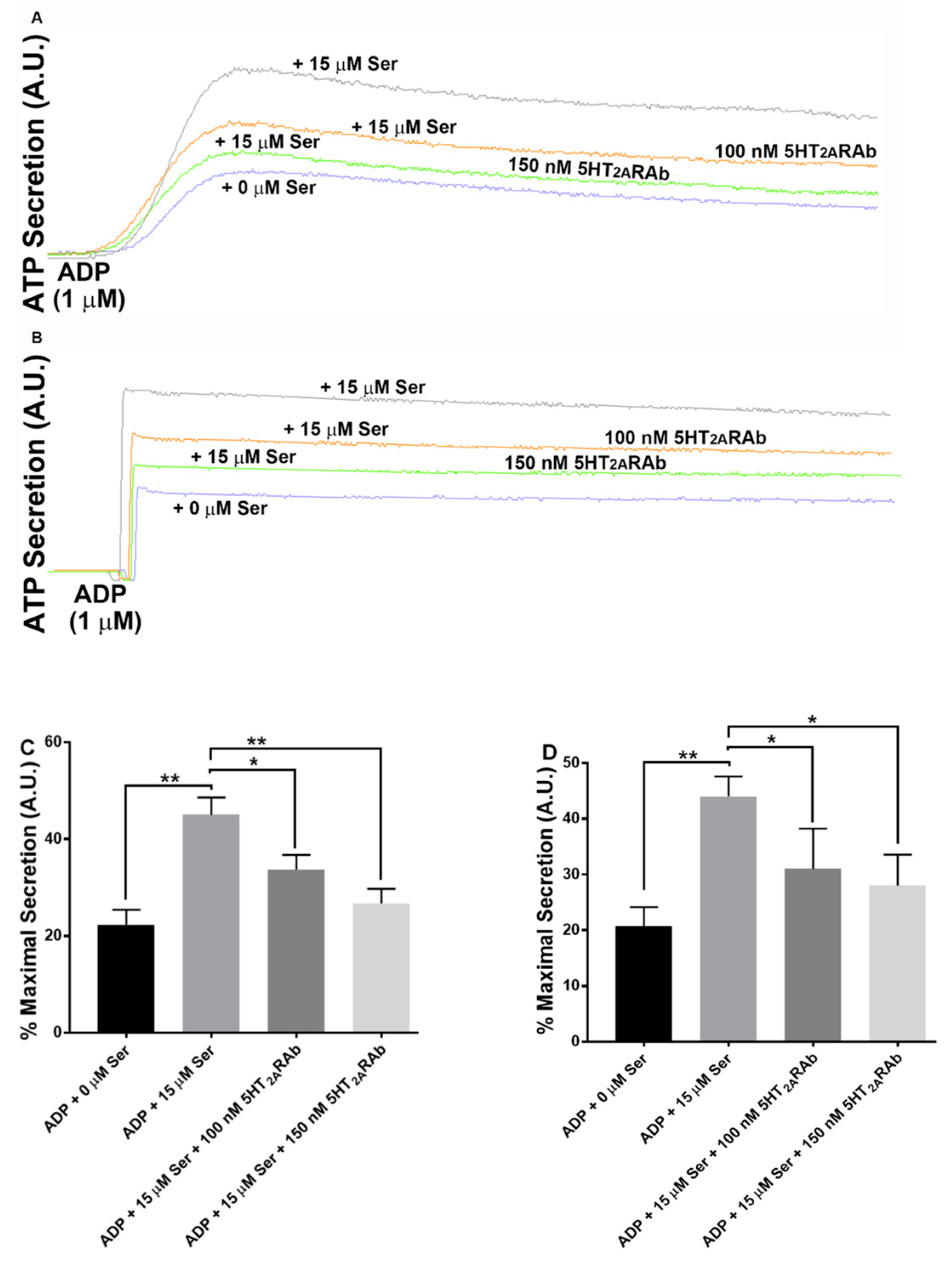
2.4. The 5HT2ARAb Inhibits Serotonin-Enhanced Dense Granule Secretion in Human and Mouse Platelets
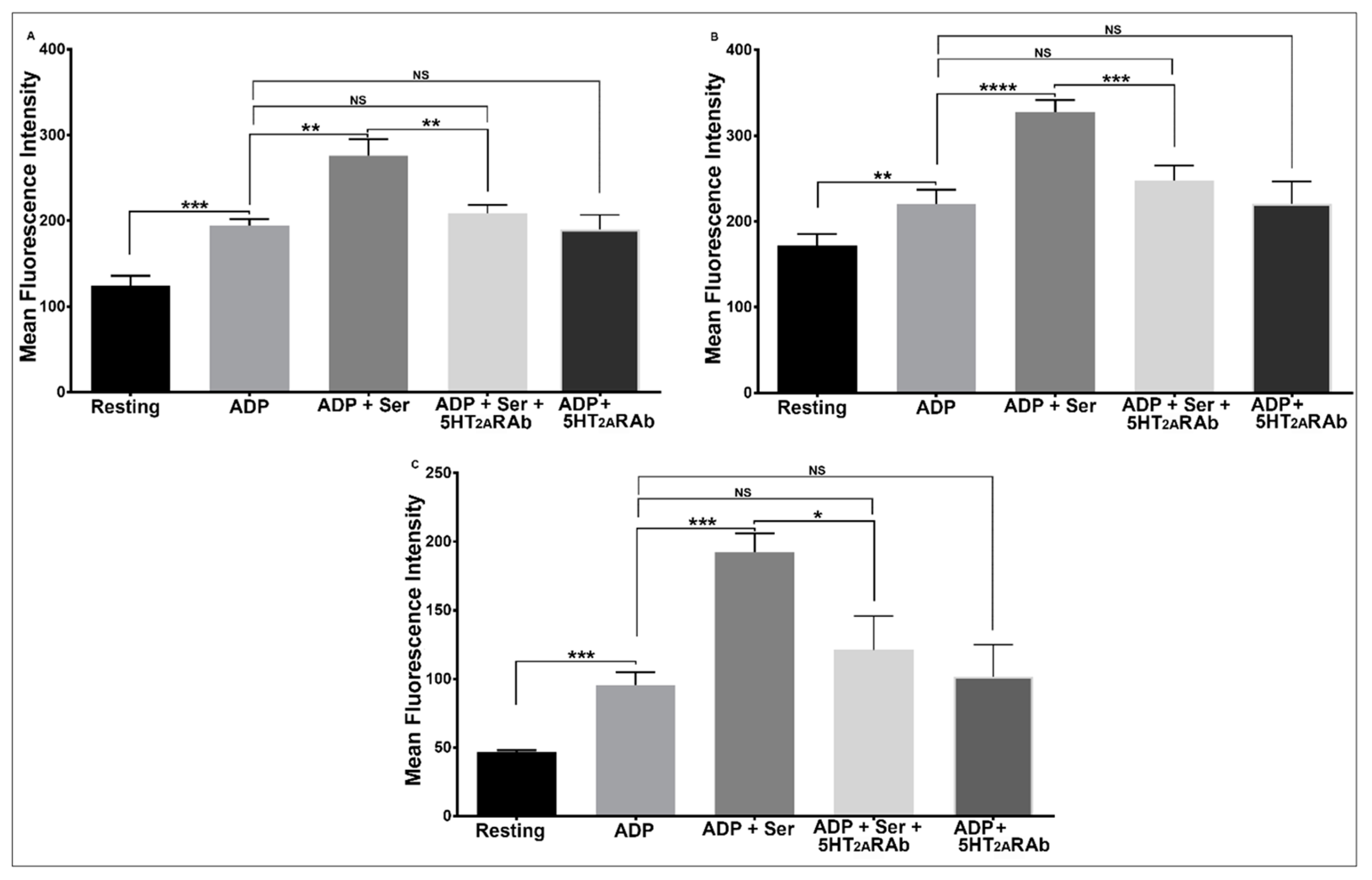
2.5. The 5HT2ARAb Inhibits Serotonin-Enhanced Platelet Secretion, Glycoprotein IIb-IIIa Activation, and Phosphatidylserine Exposure
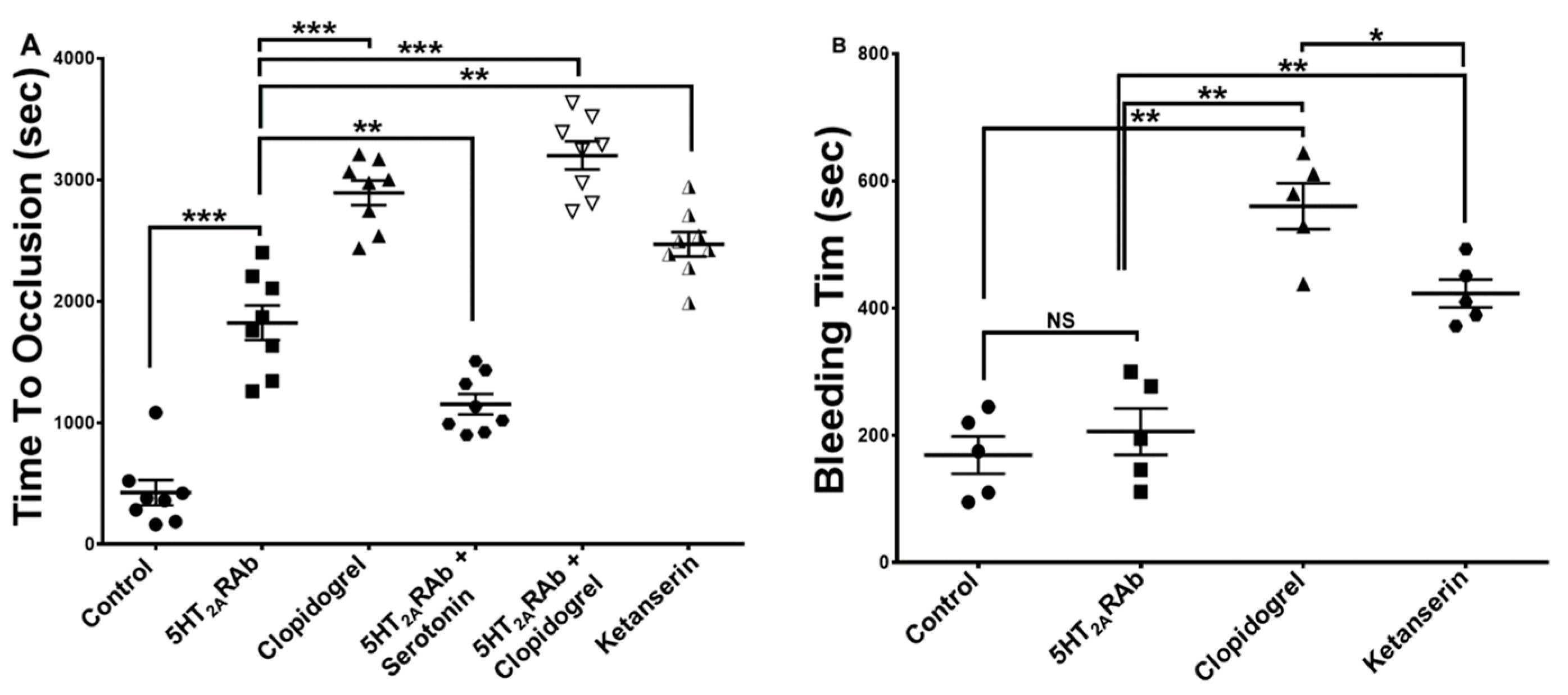
2.6. The 5HT2ARAb Prolongs the Thrombus Occlusion Time but Not the Tail Bleeding Time
2.7. The 5HT2ARAb Does Not Affect Platelet Number in Mice
3. Discussion
4. Methods and Materials
4.1. Reagents and Materials
4.2. Animals
4.3. Human and Murine Platelet Preparation
4.4. In Vitro Platelet Aggregation and ATP Secretion
4.5. Ex Vivo Platelet Aggregation and ATP Secretion
4.6. Flow Cytometric Analysis
4.7. In Vivo Ferric Chloride Carotid Artery Injury–Induced Thrombosis Model
4.8. Tail Bleeding Time
4.9. Radioligand Binding Displacement
4.10. Platelet Count
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at work in primary hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef]
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- McMichael, M. Primary hemostasis. J. Vet. Emerg. Crit. Care 2005, 15, 1–8. [Google Scholar] [CrossRef]
- Nieuwenhuis, H.K.; Sakariassen, K.S.; Houdijk, W.P.; Nievelstein, P.F.; Sixma, J.J. Deficiency of platelet membrane glycoprotein Ia associated with a decreased platelet adhesion to subendothelium: A defect in platelet spreading. Blood 1986, 68, 692–695. [Google Scholar] [CrossRef] [Green Version]
- Staatz, W.D.; Walsh, J.J.; Pexton, T.; Santoro, S.A. The alpha 2 beta 1 integrin cell surface collagen receptor binds to the alpha 1 (I)-CB3 peptide of collagen. J. Biol. Chem. 1990, 265, 4778–4781. [Google Scholar] [CrossRef]
- Charo, I.F.; Feinman, R.D.; Detwiler, T.C. Interrelations of platelet aggregation and secretion. J. Clin. Investig. 1977, 60, 866–873. [Google Scholar] [CrossRef] [Green Version]
- Woulfe, D.S. Platelet G protein-coupled receptors in hemostasis and thrombosis. J. Thromb. Haemost 2005, 3, 2193–2200. [Google Scholar] [CrossRef]
- Dowal, L.; Flaumenhaft, R. Targeting platelet G-protein coupled receptors (GPCRs): Looking beyond conventional GPCR antagonism. Curr. Vasc. Pharmacol. 2010, 8, 140–154. [Google Scholar] [CrossRef]
- Breet, N.; Sluman, M.; van Berkel, M.; Van Werkum, J.; Bouman, H.; Harmsze, A.; Kelder, J.; Zijlstra, F.; Hackeng, C.; Ten Berg, J. Effect of gender difference on platelet reactivity. Neth. Heart J. 2011, 19, 451. [Google Scholar] [CrossRef] [Green Version]
- Ergelen, M.; Gorgulu, S.; Uyarel, H.; Norgaz, T.; Aksu, H.; Ayhan, E.; Gunaydın, Z.Y.; Isık, T.; Tezel, T. The outcome of primary percutaneous coronary intervention for stent thrombosis causing ST-elevation myocardial infarction. Am. Heart J. 2010, 159, 672–676. [Google Scholar] [CrossRef]
- Bembenek, J.P.; Karlinski, M.; Kobayashi, A.; Czlonkowska, A. Deep venous thrombosis in acute stroke patients. Clin. Appl. Thromb. Hemost. 2012, 18, 258–264. [Google Scholar] [CrossRef]
- Coller, B.S. Platelets and thrombolytic therapy. N. Engl. J. Med. 1990, 322, 33–42. [Google Scholar] [CrossRef]
- Weiss, H.J. Antiplatelet drugs-a new pharmacologic approach to the prevention of thrombosis. Am. Heart J. 1976, 92, 86–102. [Google Scholar] [CrossRef]
- Kagaya, A.; Mikuni, M.; Yamamoto, H.; Muraoka, S.; Yamawaki, S.; Takahashi, K. Heterologous supersensitization between serotonin2 and alpha 2-adrenergic receptor-mediated intracellular calcium mobilization in human platelets. J. Neural Transm. Gen. Sect. 1992, 88, 25–36. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.W.; Ramirez, J.; Ortuno, D.; Shi, Y.; Thomsen, W.; Richman, J.G.; Morgan, M.; Dosa, P.; Teegarden, B.R.; Al-Shamma, H.; et al. Anti-thrombotic and vascular effects of AR246686, a novel 5-HT2A receptor antagonist. Eur. J. Pharm. 2008, 586, 234–243. [Google Scholar] [CrossRef]
- De Clerck, F.F.; Herman, A.G. 5-hydroxytryptamine and platelet aggregation. Fed. Proc. 1983, 42, 228–232. [Google Scholar]
- De Clerck, F.F.; Janssen, P.A. Amplification mechanisms in platelet activation and arterial thrombosis. J. Hypertens Suppl. 1990, 8, S87–S93. [Google Scholar]
- Vikenes, K.; Farstad, M.; Nordrehaug, J.E. Serotonin is associated with coronary artery disease and cardiac events. Circulation 1999, 100, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.N.; Lorrain, J.; Lochot, S.; Delahaye, M.; Lale, A.; Savi, P.; Lechaire, I.; Ferrari, P.; Bernat, A.; Schaeffer, P.; et al. Antiplatelet and antithrombotic activity of SL65.0472, a mixed 5-HT1B/5-HT2A receptor antagonist. Thromb. Haemost. 2001, 85, 521–528. [Google Scholar]
- Kihara, H.; Koganei, H.; Hirose, K.; Yamamoto, H.; Yoshimoto, R. Antithrombotic activity of AT-1015, a potent 5-HT(2A) receptor antagonist, in rat arterial thrombosis model and its effect on bleeding time. Eur. J. Pharm. 2001, 433, 157–162. [Google Scholar] [CrossRef]
- Yamada, S.; Akita, H.; Kanazawa, K.; Ishida, T.; Hirata, K.; Ito, K.; Kawashima, S.; Yokoyama, M. T102C polymorphism of the serotonin (5-HT) 2A receptor gene in patients with non-fatal acute myocardial infarction. Atherosclerosis 2000, 150, 143–148. [Google Scholar] [CrossRef]
- Prevention of atherosclerotic complications: Controlled trial of ketanserin. Prevention of Atherosclerotic Complications with Ketanserin Trial Group. BMJ 1989, 298, 424–430. [CrossRef] [Green Version]
- Tanaka, T.; Fujita, M.; Nakae, I.; Tamaki, S.; Hasegawa, K.; Kihara, Y.; Nohara, R.; Sasayama, S. Improvement of exercise capacity by sarpogrelate as a result of augmented collateral circulation in patients with effort angina. J. Am. Coll. Cardiol. 1998, 32, 1982–1986. [Google Scholar] [CrossRef] [Green Version]
- Noble, M.I.; Ford, I.; Cameron, G.; Drake-Holland, A. The Novel Anti-Thrombotic Drug with No-Bleeding Excess. J. Cardiol. Cardiovasc. Ther. 2017, 6, 106–111. [Google Scholar] [CrossRef]
- Cordova-Sintjago, T.; Sakhuja, R.; Kondabolu, K.; Canal, C.E.; Booth, R.G. Molecular Determinants for Ligand Binding at Serotonin 5-HT2A and 5-HT2C GPCRs: Experimental Affinity Results Analyzed by Molecular Modeling and Ligand Docking Studies. Int. J. Quantum Chem. 2012, 112, 3807–3814. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.E.; Nichols, C.D. Serotonin receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef]
- Westkaemper, R.B.; Glennon, R.A. Application of ligand SAR, receptor modeling and receptor mutagenesis to the discovery and development of a new class of 5-HT(2A) ligands. Curr. Top Med. Chem. 2002, 2, 575–598. [Google Scholar] [CrossRef] [Green Version]
- Murad, J.P.; Espinosa, E.V.; Ting, H.J.; Khasawneh, F.T. The C-terminal segment of the second extracellular loop of the thromboxane A2 receptor plays an important role in platelet aggregation. Biochem. Pharm. 2012, 83, 88–96. [Google Scholar] [CrossRef]
- Karim, Z.A.; Alshbool, F.Z.; Vemana, H.P.; Adhami, N.; Dhall, S.; Espinosa, E.V.; Martins-Green, M.; Khasawneh, F.T. Third-hand Smoke: Impact on Hemostasis and Thrombogenesis. J. Cardiovasc. Pharmacol. 2015, 66, 177–182. [Google Scholar] [CrossRef]
- Hensch, N.R.; Karim, Z.A.; Pineda, J.; Mercado, N.; Alshbool, F.Z.; Khasawneh, F.T. P2Y12 antibody inhibits platelet activity and protects against thrombogenesis. Biochem. Biophys. Res. Commun. 2017, 493, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Menys, V.C. Collagen induced human platelet aggregation: Serotonin receptor antagonism retards aggregate growth in vitro. Cardiovasc Res 1993, 27, 1916–1919. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, D.; Pawlak, K.; Chabielska, E.; Malyszko, J.; Takada, A.; Mysliwiec, M.; Buczko, W. A potent 5-hydroxytryptamine receptor (5-HT2A) antagonist, DV-7028, delays arterial thrombosis development in rats. Thromb. Res. 1998, 90, 259–270. [Google Scholar] [CrossRef]
- Czopek, A.; Kubacka, M.; Bucki, A.; Siwek, A.; Filipek, B.; Pawlowski, M.; Kolaczkowski, M. Novel serotonin 5-HT2A receptor antagonists derived from 4-phenylcyclohexane-5-spiro-and 5-methyl-5-phenyl-hydantoin, for use as potential antiplatelet agents. Pharm. Rep. 2021, 73, 1361–1372. [Google Scholar] [CrossRef]
- Marcinkowska, M.; Kubacka, M.; Zagorska, A.; Jaromin, A.; Fajkis-Zajaczkowska, N.; Kolaczkowski, M. Exploring the antiplatelet activity of serotonin 5-HT2A receptor antagonists bearing 6-fluorobenzo[d]isoxazol-3-yl)propyl) motif- as potential therapeutic agents in the prevention of cardiovascular diseases. Biomed. Pharm. 2022, 145, 112424. [Google Scholar] [CrossRef]
- Dawood, B.B.; Wilde, J.; Watson, S.P. Reference curves for aggregation and ATP secretion to aid diagnose of platelet-based bleeding disorders: Effect of inhibition of ADP and thromboxane A2 pathways. Platelets 2007, 18, 329–345. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Dilks, J.R.; Rozenvayn, N.; Monahan-Earley, R.A.; Feng, D.; Dvorak, A.M. The actin cytoskeleton differentially regulates platelet α-granule and dense-granule secretion. Blood 2005, 105, 3879–3887. [Google Scholar] [CrossRef]
- Storrie, B.; Whiteheart, S.W. Editorial: Platelet Secretion. Platelets 2017, 28, 107. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, H.R.; Reyes, N.; Zhang, Q.C.; Okoroh, E.M.; Siddiqi, A.-E.-A.; Tsai, J. Hospitalizations of adults≥ 60 years of age with venous thromboembolism. Clin. Appl. Thromb. Hemost. 2014, 20, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Boutitie, F.; Pinede, L.; Schulman, S.; Agnelli, G.; Raskob, G.; Julian, J.; Hirsh, J.; Kearon, C. Influence of preceding length of anticoagulant treatment and initial presentation of venous thromboembolism on risk of recurrence after stopping treatment: Analysis of individual participants’ data from seven trials. BMJ 2011, 342, d3036. [Google Scholar] [CrossRef] [Green Version]
- Kearon, C. Natural history of venous thromboembolism. Circulation 2003, 107, I-22–I-30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heit, J.A. The epidemiology of venous thromboembolism in the community. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 370–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heit, J.A.; Cohen, A.T.; Anderson, F.A. Estimated Annual Number of Incident and Recurrent, Non-Fatal and Fatal Venous ThromboEmbolism (VTE) Events in the US. Blood 2005, 106, 910. [Google Scholar] [CrossRef]
- National Blood Clot Alliance. Blood Clots in the United States. Available online: https://www.stoptheclot.org/blood-clot-information/blood-clots-in-the-united-states/ (accessed on 8 July 2019).
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; Meade, T. Aspirin in the Primary and Secondary Prevention of Vascular Disease: Collaborative Meta-Analysis of Individual Participant Data from Randomised Trials. Lancet 2009, 373, 1849–1860. [Google Scholar]
- Hankey, G.; Sudlow, C.L.; Dunbabin, D.W. Thienopyridine derivatives (ticlopidine, clopidogrel) versus aspirin for preventing stroke and other serious vascular events in high vascular risk patients. Cochrane Database Syst. Rev. 2000, 2, CD001246. [Google Scholar] [CrossRef]
- Kim, H.-H.; Liao, J.K. Translational therapeutics of dipyridamole. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s39–s42. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Freedman, J.E. Dipyridamole, cerebrovascular disease, and the vasculature. Vasc. Pharmacol. 2008, 48, 143–149. [Google Scholar] [CrossRef]
- Vestergaard, P.; Steinberg, T.H.; Schwarz, P.; Jørgensen, N.R. Use of the oral platelet inhibitors dipyridamole and acetylsalicylic acid is associated with increased risk of fracture. Int. J. Cardiol. 2012, 160, 36–40. [Google Scholar] [CrossRef]
- Fujita, M.; Mizuno, K.; Ho, M.; Tsukahara, R.; Miyamoto, A.; Miki, O.; Ishii, K.; Miwa, K. Sarpogrelate treatment reduces restenosis after coronary stenting. Am. Heart J. 2003, 145, E16. [Google Scholar] [CrossRef]
- Almaula, N.; Ebersole, B.J.; Zhang, D.; Weinstein, H.; Sealfon, S.C. Mapping the binding site pocket of the serotonin 5-Hydroxytryptamine2A receptor. Ser3.36 provides a second interaction site for the protonated amine of serotonin but not of lysergic acid diethylamide or bufotenin. J. Biol. Chem. 1996, 271, 14672–14675. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Khelashvili, G.; Mondal, S.; Mehler, E.L.; Weinstein, H. Ligand-dependent conformations and dynamics of the serotonin 5-HT(2A) receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput. Biol. 2012, 8, e1002473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.D.; Gallaher, T.K.; Shih, J.C. Site-directed mutagenesis of the serotonin 5-hydroxytrypamine2 receptor: Identification of amino acids necessary for ligand binding and receptor activation. Mol. Pharm. 1993, 43, 931–940. [Google Scholar]
- Foster, C.J.; Prosser, D.M.; Agans, J.M.; Zhai, Y.; Smith, M.D.; Lachowicz, J.E.; Zhang, F.L.; Gustafson, E.; Monsma, F.J., Jr.; Wiekowski, M.T.; et al. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J. Clin. Investig. 2001, 107, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F. The P2Y12 receptor as a therapeutic target in cardiovascular disease. Platelets 2001, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wallen, N.H.; Ladjevardi, M.; Hjemdahl, P. Effects of serotonin on platelet activation in whole blood. Blood Coagul. Fibrinolysis 1997, 8, 517–523. [Google Scholar] [CrossRef]
- Gammie, J.S.; Zenati, M.; Kormos, R.L.; Hattler, B.G.; Wei, L.M.; Pellegrini, R.V.; Griffith, B.P.; Dyke, C.M. Abciximab and excessive bleeding in patients undergoing emergency cardiac operations. Ann. Thorac. Surg. 1998, 65, 465–469. [Google Scholar] [CrossRef]
- Tcheng, J.E.; Ellis, S.G.; George, B.S.; Kereiakes, D.J.; Kleiman, N.S.; Talley, J.D.; Wang, A.L.; Weisman, H.F.; Califf, R.M.; Topol, E.J. Pharmacodynamics of chimeric glycoprotein IIb/IIIa integrin antiplatelet antibody Fab 7E3 in high-risk coronary angioplasty. Circulation 1994, 90, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Simoons, M.L.; de Boer, M.J.; van den Brand, M.J.; van Miltenburg, A.J.; Hoorntje, J.C.; Heyndrickx, G.R.; van der Wieken, L.R.; de Bono, D.; Rutsch, W.; Schaible, T.F.; et al. Randomized trial of a GPIIb/IIIa platelet receptor blocker in refractory unstable angina. European Cooperative Study Group. Circulation 1994, 89, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Zimering, M.B.; Razzaki, T.; Tsang, T.; Shin, J.J. Inverse Association between Serotonin 2A Receptor Antagonist Medication Use and Mortality in Severe COVID-19 Infection. Endocrinol. Diabetes Metab. J. 2020, 4, 1–5. [Google Scholar]
- Ziu, E.; Mercado, C.P.; Li, Y.; Singh, P.; Ahmed, B.A.; Freyaldenhoven, S.; Lensing, S.; Ware, J.; Kilic, F. Down-regulation of the serotonin transporter in hyperreactive platelets counteracts the pro-thrombotic effect of serotonin. J. Mol. Cell Cardiol. 2012, 52, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekemeier, H.; Hirschelmann, R. Influence of serotonin, serotonin antagonists, some vasoactive substances and temperature on carrageenin-induced tail thrombosis in rats and mice. Agents Actions 1986, 18, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Kattelman, E.J.; Venton, D.L.; Le Breton, G.C. Characterization of U46619 binding in unactivated, intact human platelets and determination of binding site affinities of four TXA2/PGH2 receptor antagonists (13-APA, BM 13.177, ONO 3708 and SQ 29,548). Thromb. Res. 1986, 41, 471–481. [Google Scholar] [CrossRef]
- Hung, S.C.; Ghali, N.I.; Venton, D.L.; Le Breton, G.C. Specific binding of the thromboxane A2 antagonist 13-azaprostanoic acid to human platelet membranes. Biochim. Biophys. Acta 1983, 728, 171–178. [Google Scholar] [CrossRef]
- Karim, Z.A.; Alshbool, F.Z.; Vemana, H.P.; Conlon, C.; Druey, K.M.; Khasawneh, F.T. CXCL12 regulates platelet activation via the regulator of G-protein signaling 16. Biochim. Biophys. Acta 2016, 1863, 314–321. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, J.E.M.; Alarabi, A.B.; Khasawneh, F.T.; Alshbool, F.Z. A Novel Antibody Targeting the Second Extracellular Loop of the Serotonin 5-HT2A Receptor Inhibits Platelet Function. Int. J. Mol. Sci. 2022, 23, 8794. https://doi.org/10.3390/ijms23158794
Ramirez JEM, Alarabi AB, Khasawneh FT, Alshbool FZ. A Novel Antibody Targeting the Second Extracellular Loop of the Serotonin 5-HT2A Receptor Inhibits Platelet Function. International Journal of Molecular Sciences. 2022; 23(15):8794. https://doi.org/10.3390/ijms23158794
Chicago/Turabian StyleRamirez, Jean E. M., Ahmed B. Alarabi, Fadi T. Khasawneh, and Fatima Z. Alshbool. 2022. "A Novel Antibody Targeting the Second Extracellular Loop of the Serotonin 5-HT2A Receptor Inhibits Platelet Function" International Journal of Molecular Sciences 23, no. 15: 8794. https://doi.org/10.3390/ijms23158794