
Canagliflozin Inhibits Human Endothelial Cell Inflammation through the Induction of Heme Oxygenase-1


Abstract
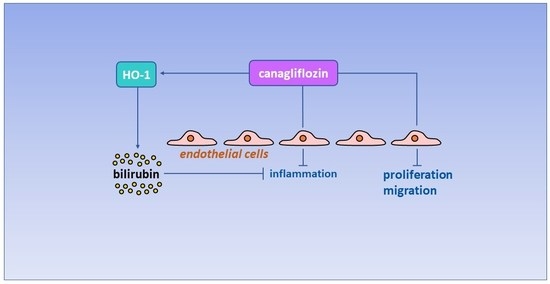
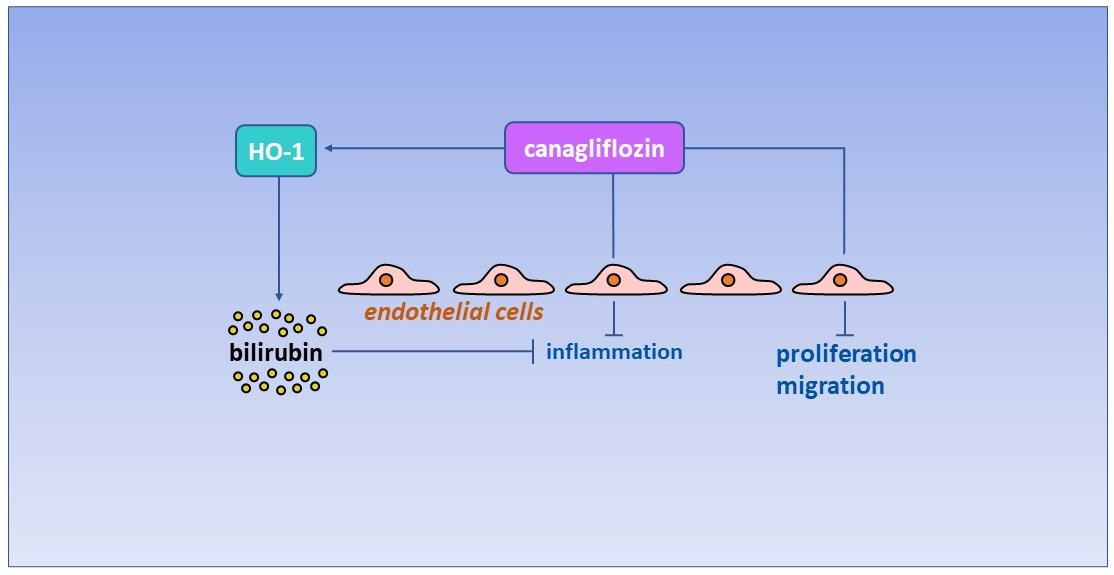
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
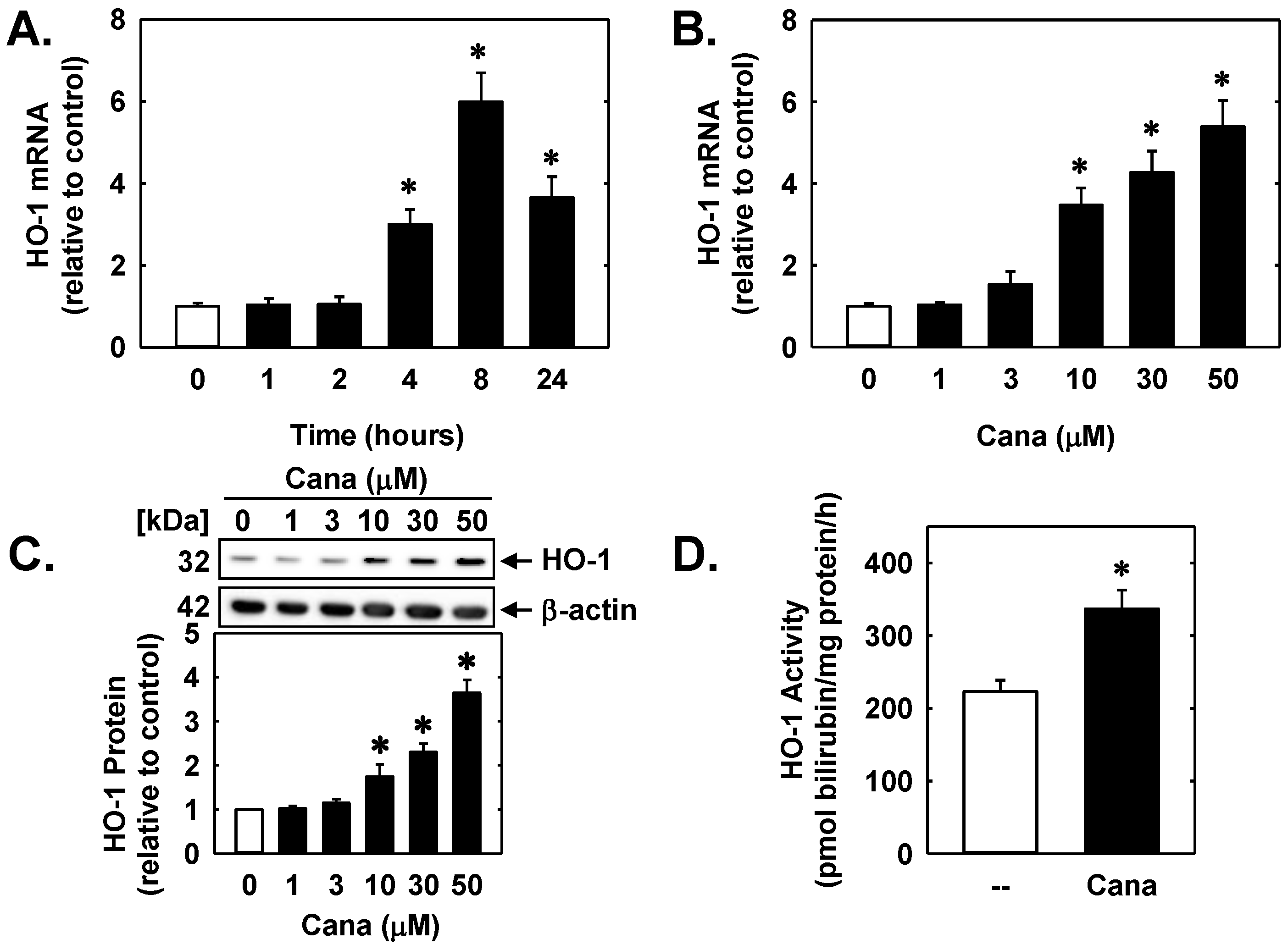
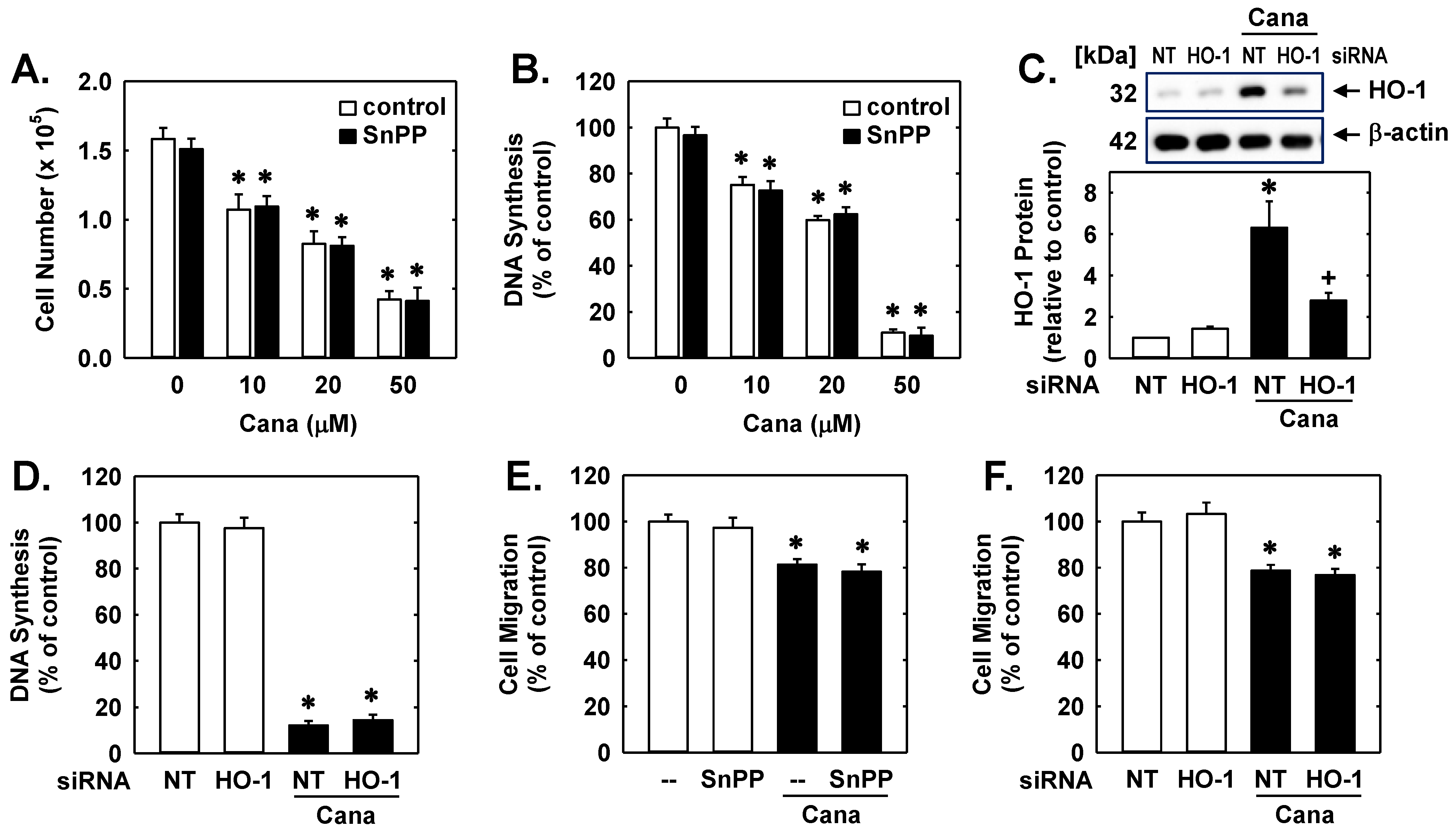
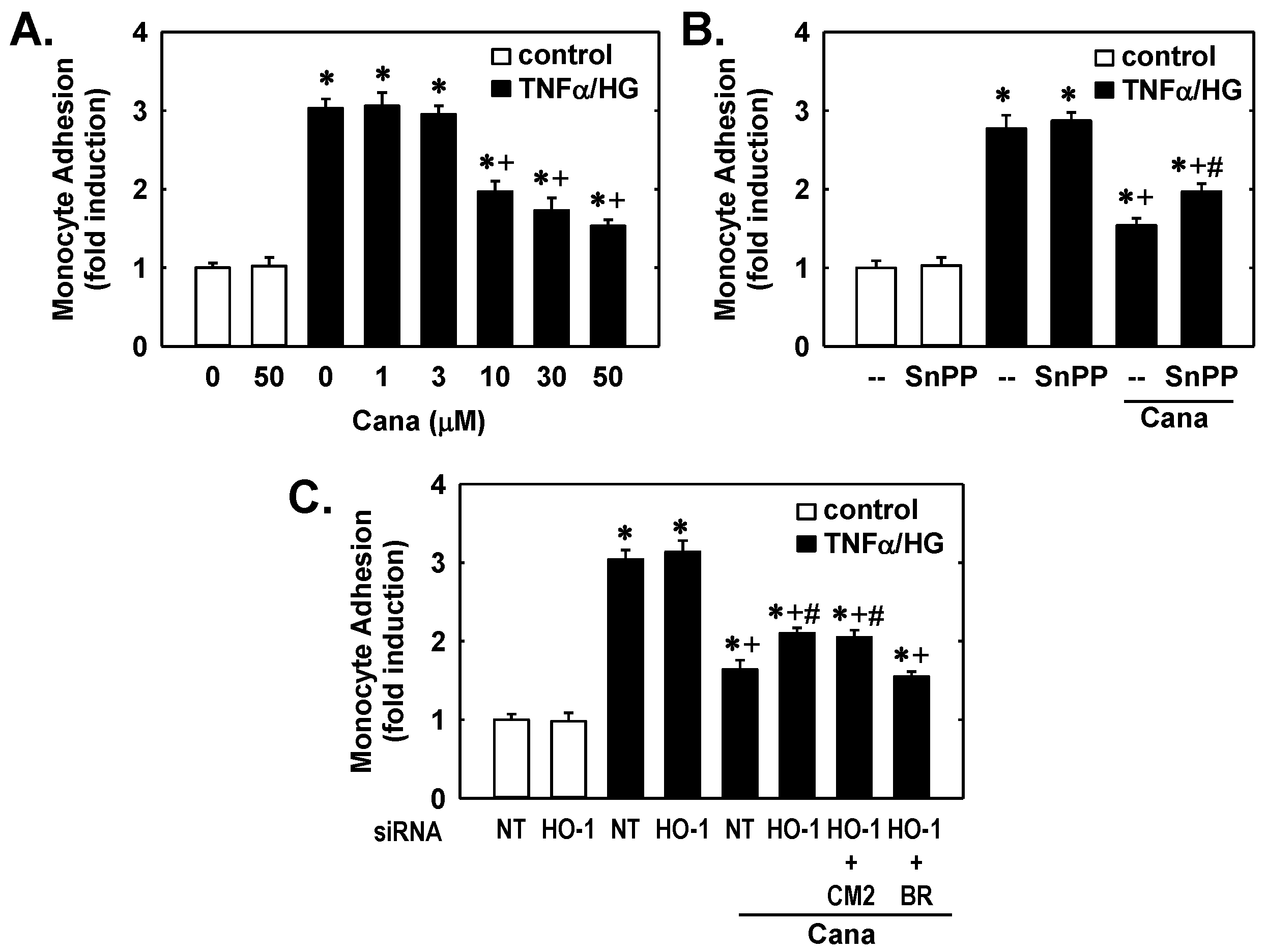
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation and DNA Synthesis
4.4. Cell Migration
4.5. Monocyte Adhesion
4.6. Western Blotting
4.7. Quantitative Real-Time PCR
4.8. HO Activity
4.9. Gene Silencing
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Foundation. IDF Diabetes Atlas. 2021. Available online: http://diabetesatlas.org/atlas/tenth-edition (accessed on 1 July 2022).
- Matheus, A.S.; Tannus, L.R.; Cobas, A.R.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, K.; Crowe, C.C.; Harris, M.I. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care 1998, 21, 1138–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnick, H.E.; Howard, B.V. Diabetes and cardiovascular disease. Annu. Rev. Med. 2002, 53, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.S.; Heneghan, C.J.; Farmer, A.J.; Fuller, A.M.; Adler, A.I.; Aronson, J.K.; Stevens, R.J. All-cause and cardiovascular mortality in middle-aged people with type 2 diabetes compared with people without diabetes in a large U.K. primary care database. Diabetes Care 2013, 36, 2366–2371. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef]
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Creager, M.A.; Luscher, T.E.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zou, M. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 2009, 120, 1266–1286. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, S. Sodium glucose co-transporter 2 (SGLT2) inhibitors: A review of their basic and clinical pharmacology. Diabetes Ther. 2014, 5, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelnicker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Zelnicker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systemic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Birkeland, K.I.; Jørgensen, M.E.; Carstensen, B.; Persson, F.; Gulseth, H.L.; Thuresson, M.; Fenici, P.; Nathanson, D.; Nyström, T.; Eriksson, J.W.; et al. Cardiovascular mortality and morbidity in patients with type 2 diabetes following initiation of sodium-glucose co-transporter-2 inhibitors versus other glucose-lowering drugs (CV-REAL-NORDIC): A multinational observational study. Lancet Diabetes Endocrinol. 2017, 5, 709–717. [Google Scholar] [CrossRef]
- Kosiborod, M.; Cavender, M.A.; Fu, A.Z.; Wilding, J.P.; Khunti, K.; Holl, R.W.; Norhammar, A.; Birkeland, K.I.; Jørgensen, M.E.; Thuresson, M.; et al. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: The CVD-REAL Study (comparative effectiveness of cardiovascular outcomes in new users of sodium-glucose cotransporter-2 inhibitors). Circulation 2017, 136, 249–259. [Google Scholar]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Heme oxygenase and the cardiovascular-renal system. Free Radic. Biol. Med. 2005, 39, 1–25. [Google Scholar] [CrossRef]
- Durante, W. Targeting heme oxygenase-1 in the arterial response to injury and disease. Antioxidants 2020, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; Ishikawa, K.; Sacerdoti, D.; Abraham, N.G. The emerging role of heme oxygenase-1 and its metabolites in the regulation of vascular function. Int. J. Hypertens. 2012, 2012, 593530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stoker, R. Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Perrella, M.A.; Lee, M.A.; Kourembanas, S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular smooth muscle. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [Green Version]
- Li Volti, G.; Wang, J.; Traganos, F.; Kappas, A.; Abraham, N.G. Differential effect of heme oxygenase-1 endothelial and smooth muscle cell cycle progression. Biochem. Biophys. Res. Commun. 2002, 296, 1077–1082. [Google Scholar] [CrossRef]
- Peyton, K.J.; Reyna, S.V.; Chapman, G.B.; Ensenat, D.; Liu, X.; Wang, H.; Schafer, A.I.; Durante, W. Heme oxygenase-1-derived carbon monoxide inhibitor of vascular smooth muscle cell growth. Blood 2002, 51, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Behnammanesh, G.; Durante, G.L.; Khanna, Y.P.; Peyton, K.J.; Durante, W. Canagliflozin inhibits vascular smooth muscle cell proliferation and migration: Role of heme oxygenase-1. Redox Biol. 2020, 32, 101527. [Google Scholar] [CrossRef]
- Cavaiola, T.S.; Pettus, J. Cardiovascular effects of sodium glucose cotransporter 2 inhibitors. Diabetes Metab. Syndr. Obes. 2018, 11, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Verma, S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Ann. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Durante, W. Effects of sodium-glucose co-transporter2 inhibitors on vascular cell function and arterial remodeling. Int. J. Mol. Sci. 2021, 22, 8786. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: A state-of-the-art review. J. Am. Coll. Cardiol. Basic Trans. Sci. 2020, 5, 632–644. [Google Scholar]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Alshnbari, A.S.; Millar, S.A.; O’Sullivan, S.E.; Idris, I. Effect of sodium-glucose cotransporter 2 inhibitors on endothelial function: A systematic review of preclinical studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef] [PubMed]
- Ugusman, A.; Kumar, J.; Aminuddin, A. Endothelial function and dysfunction: Impact of sodium-glucose cotransporter 2 inhibitors. Pharmacol. Ther. 2021, 224, 107832. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Caturano, A.; Galiero, R.; Martino, A.D.; Albanese, G.; Vetrano, E.; Sardu, C.; Marfella, R.; Rinaldi, L.; Sasso, F.C. Cardiovascular benefits from gliflozins: Effects on endothelial function. Biomedicines 2021, 9, 1356. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.J.; Boyd, D.; Katwan, O.J.; Strembitska, A.; Almabrouk, T.A.; Kennedy, S.; Palmer, T.M.; Salt, I.P. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMPK-activated protein kinase-dependent and independent mechanisms. Sci. Rep. 2018, 8, 5276. [Google Scholar] [CrossRef] [Green Version]
- Behnammanesh, G.; Durante, Z.E.; Peyton, K.J.; Martinez-Lemus, L.A.; Brown, S.M.; Bender, S.B.; Durante, W. Canagliflozin inhibits human endothelial cell proliferation and tube formation. Front. Pharmacol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Uthman, L.; Kuschma, M.; Romer, G.; Boomsma, M.; Kessler, J.; Hermanides, J.; Hollmann, M.W.; Preckel, B.; Zuurbier, C.J.; Weber, N.C. Novel anti-inflammatory effects of canagliflozin involving hexokinase II in lipopolysaccharide-stimulated human coronary endothelial cells. Cardiovasc. Drugs Ther. 2020, 35, 1083–1094. [Google Scholar] [CrossRef]
- Devineni, D.; Polidori, D. Clinical pharmacokinetic, pharmacodynamic, and drug-drug interaction profile of canagliflozin, a sodium-glucose co-transporter 2 inhibitor. Clin. Pharm. 2015, 54, 1027–1041. [Google Scholar] [CrossRef]
- Pattanawongsa, A.; Chau, N.; Rowland, A.; Miner, J.O. Inhibition of human UDP-glucuronosyltransferase enzymes by canagliflozin and dapagliflozin: Implications for drug-drug interactions. Drug Metab. Dispos. 2015, 43, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, W.T.; Leiter, L.A.; Yoon, K.-H.; Arias, P.; Niskanen, L.; Xie, J.; Balis, D.A.; Canovatchel, W.; Meininger, G. Efficacy and safety of cangagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomized, double-blind, phase 3 non-inferiority trial. Lancet 2013, 382, 941–950. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Chowdhury, F.I.; Nayan, S.I.; Rhaman, M.M.; Khan, F.; Subhan, N.; et al. Canagliflozin attenuates isoprenaline-induced cardiac oxidative stress by stimulating multiple antioxidant and anti-inflammatory signaling pathways. Sci. Rep. 2020, 10, 14459. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Furfaro, A.L.; Mann, G.E. Heme oxygenase dependent bilirubin generation in vascular cells: A role in preventing endothelial dysfunction and inflammation. Front. Physiol. 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ishikawa, K.; Wada, Y.; Kimura, S.; Matsumoto, H.; Kohro, T.; Itabe, H.; Kodama, T.; Maruyama, Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevkaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [Green Version]
- Durante, W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 2011, 16, 2372–2388. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, N.; Sardu, C.; Trotta, M.C.; Scisciola, L.; Turriziani, F.; Ferraraccio, F.; Panarese, I.; Petrella, L.; Fanelli, M.; Modugno, P.; et al. Sodium-glucose co-transporter2 expression and inflammatory activity in diabetic atherosclerotic plaques: Effects of sodium-glucose co-transporter2 inhibitor treatment. Metabolism 2022, 127, 154936. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Parvez, F.; Rahman, M.M.; Khan, F.; Subhan, N.; Alam, M.A. Canagliflozin ameliorates renal oxidative stress and inflammation by stimulating AMPK-Akt-eNOS pathway in the isoprenaline-induced oxidative stress model. Sci. Rep. 2020, 10, 14659. [Google Scholar] [CrossRef]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar] [CrossRef]
- Lin, F.; Song, C.; Zeng, Y.; Li, Y.; Li, H.; Liu, B.; Dai, M.; Pan, P. Canagliflozin alleviates LPS-induced acute lung injury by modulating alveolar macrophage polarization. Int. Immunopharmacol. 2020, 88, 106969. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhang, Y.; Zhang, W.; Lu, J.; Chen, Y.; Hao, W.; Zhou, J.; Wang, L.; Xie, W. Canagliflozin ameliorates NLRP3 inflammasome-mediated inflammation through inhibiting NF-κB signaling and upregulating Bif-1. Front. Pharmacol. 2022, 13, 820541. [Google Scholar] [CrossRef] [PubMed]
- True, A.L.; Olive, M.; Boehm, M.; San, H.; Westrick, R.J.; Raghavachari, N.; Xu, X.; Lynn, E.G.; Sack, M.N.; Munson, P.J.; et al. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ. Res. 2007, 101, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollinger, R.; Bilban, M.; Erat, A.; Froio, A.; McDavid, J.; Tyagi, S.; Csizmadia, E.; Gracaa-Souza, A.V.; Liloia, A.; Soares, M.P.; et al. Bilirubin: A natural inhibitor of vascular smooth muscle cell proliferation. Circulation 2005, 112, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Peyton, K.J.; Shebib, A.R.; Azam, M.A.; Liu, X.M.; Tulis, D.A.; Durante, W. Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front. Pharmacol. 2012, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Vogel, M.E.; Idelman, G.; Konaniah, E.S.; Zucker, S.D. Bilirubin prevents atherosclerosis lesion formation in low-density lipoprotein receptor-deficient mice by inhibiting endothelial VCAM-1 and ICAM-1. J. Am. Heart Assoc. 2017, 6, e004820. [Google Scholar] [CrossRef] [Green Version]
- Wen, G.; Yao, L.; Hao, Y.; Wang, J.; Liu, J. Bilirubin ameliorates murine atherosclerosis through inhibiting cholesterol synthesis and reshaping the immune system. J. Transl. Med. 2022, 20, 1. [Google Scholar] [CrossRef]
- McClung, J.A.; Levy, L.; Garcia, V.; Stec, D.E.; Peterson, S.J.; Abraham, N.G. Heme oxygenase and lipid mediators in obesity and associated cardiometabolic diseases: Therapeutic implications. Pharmacol. Ther. 2022, 231, 107975. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Stec, D.E. Bilirubin, a cardiometabolic signaling molecule. Hypertension 2018, 72, 788–795. [Google Scholar] [CrossRef]
- Stec, D.E.; Hosick, P.A.; Granger, J.P. Bilirubin, renal hemodynamics, and blood pressure. Front. Pharmacol. 2012, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin binding to PPARα inhibits lipid accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.M.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4, e123130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeShane, J.; Chen, S.; Caballero, S.; Grot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase-1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Ahmed, A.; Pemberton, H.; Landis, R.C.; Di Carlo, F.; Haskard, D.O.; Mason, J.C. Bifunctional role for vegf-induced heme oxygenase-1 in vivo: Induction of angiogenesis and inhibition of leukocyte infiltration. Blood 2004, 103, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Hamano, H.; Satoh, A.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Aihara, K.-I.; Tsuchiya, K.; Tamaki, T. Bilirubin exerts pro-angiogenic property through Akt-eNOS-dependent pathway. Hypertens. Res. 2015, 38, 733–740. [Google Scholar] [CrossRef]
- Batzlsperger, C.A.; Achatz, S.; Spreng, J.; Riegger, G.A.J.; Griese, D.P. Evidence for a possible inhibitory interaction between the HO-1/CO- and Akt/NO pathways in human endothelial cells. Cardiovasc. Drugs Ther. 2007, 21, 347–355. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Yang, B.; Yang, J.; Ruan, X.; Yang, Y.; Wakeland, E.K.; Li, Q.; Fang, X. Influence of carbon monoxide on growth and apoptosis of human umbilical artery smooth muscle cells and vein endothelial cells. Int. J. Biol. Sci. 2012, 8, 1431–1446. [Google Scholar] [CrossRef]
- Ahmad, S.; Hewitt, P.W.; Fujisawa, T.; Sissaoui, S.; Cai, M.; Gueron, G.; Al-Ani, B.; Cudmore, M.; Ahmed, S.F.; Wong, M.K.; et al. Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation. Thromb. Haemost. 2015, 113, 329–337. [Google Scholar]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Durante, W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J. Pharmacol. Exp. Ther. 2012, 342, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Behnammanesh, G.; Durante, W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018, 156, 204–214. [Google Scholar] [CrossRef]
- Secker, P.F.; Beneke, S.; Schlichenmaier, N.; Delp, J.; Gutbier, S.; Leist, M.; Dietrich, D.R. Canagliflozin mediated dual inhibition of mitochondrial glutamate dehydrogenase and complex I: An off-target adverse effect. Cell Death Dis. 2018, 9, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.M.; Peyton, K.J.; Durante, W. Physiologic cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.F.; Liu, X.M.; Peyton, K.J.; Durante, W. Heme oxygenase-1 counteracts contrast media-induced endothelial cell dysfunction. Biochem. Pharmacol. 2014, 87, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.M.; Peyton, K.J.; Durante, W. Ammonia promotes endothelial cell survival via the heme oxygenase-1-mediated release of carbon monoxide. Free Radic. Biol. Med. 2017, 102, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyton, K.J.; Behnammanesh, G.; Durante, G.L.; Durante, W. Canagliflozin Inhibits Human Endothelial Cell Inflammation through the Induction of Heme Oxygenase-1. Int. J. Mol. Sci. 2022, 23, 8777. https://doi.org/10.3390/ijms23158777
Peyton KJ, Behnammanesh G, Durante GL, Durante W. Canagliflozin Inhibits Human Endothelial Cell Inflammation through the Induction of Heme Oxygenase-1. International Journal of Molecular Sciences. 2022; 23(15):8777. https://doi.org/10.3390/ijms23158777
Chicago/Turabian StylePeyton, Kelly J., Ghazaleh Behnammanesh, Giovanna L. Durante, and William Durante. 2022. "Canagliflozin Inhibits Human Endothelial Cell Inflammation through the Induction of Heme Oxygenase-1" International Journal of Molecular Sciences 23, no. 15: 8777. https://doi.org/10.3390/ijms23158777