
Complete Reaction Phenotyping of Propranolol and 4-Hydroxypropranolol with the 19 Enzymes of the Human UGT1 and UGT2 Families


Abstract
:1. Introduction
2. Results
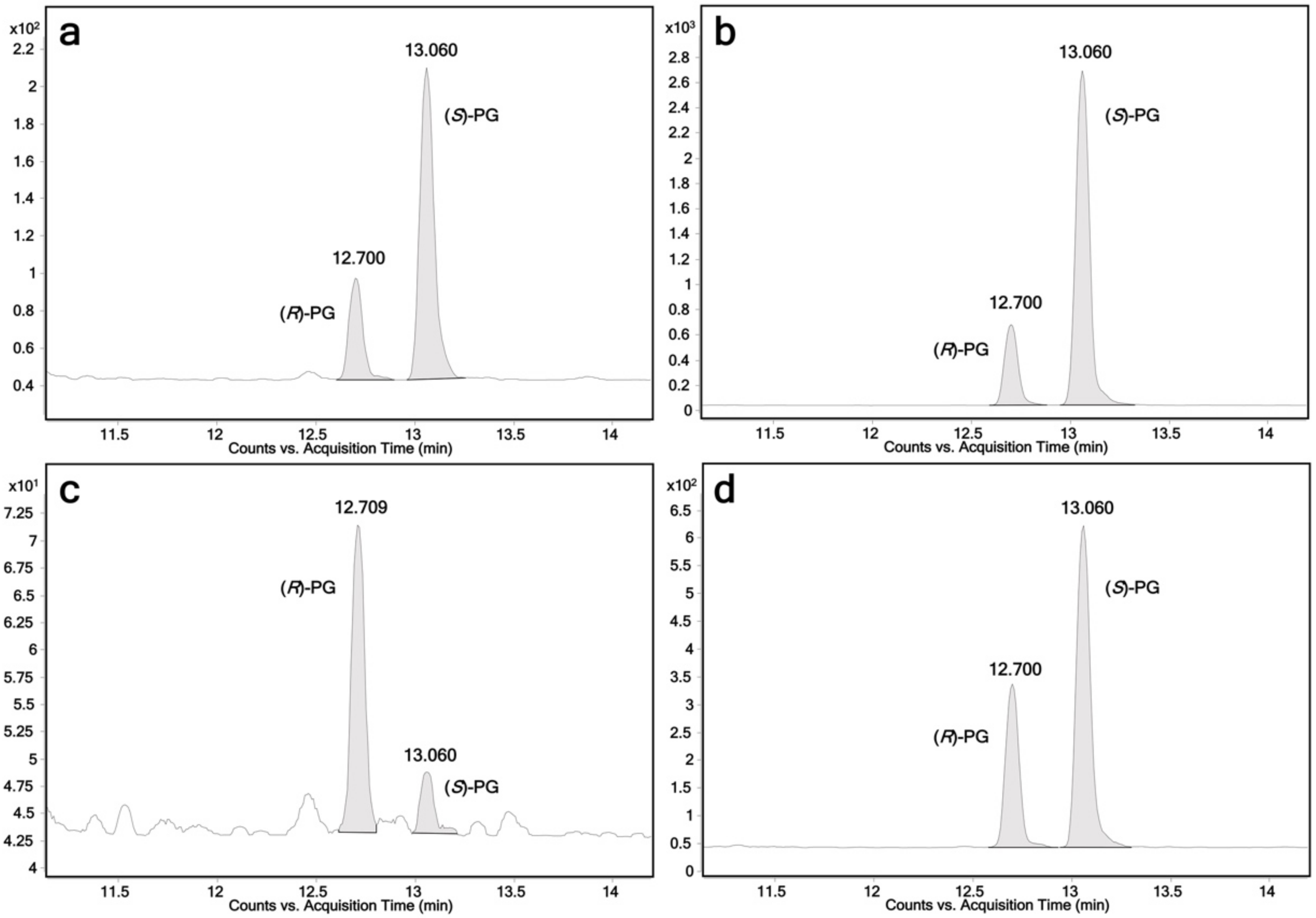
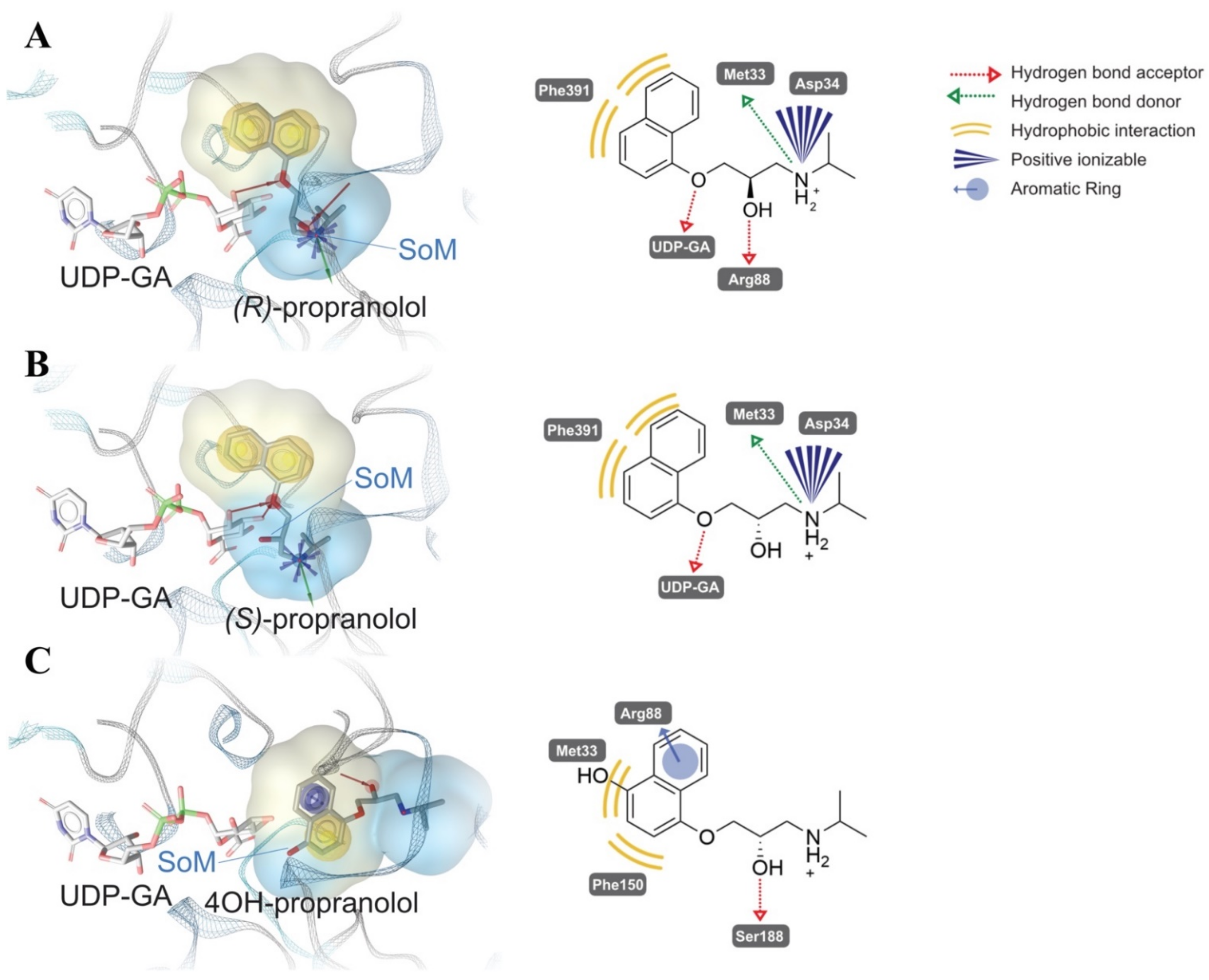
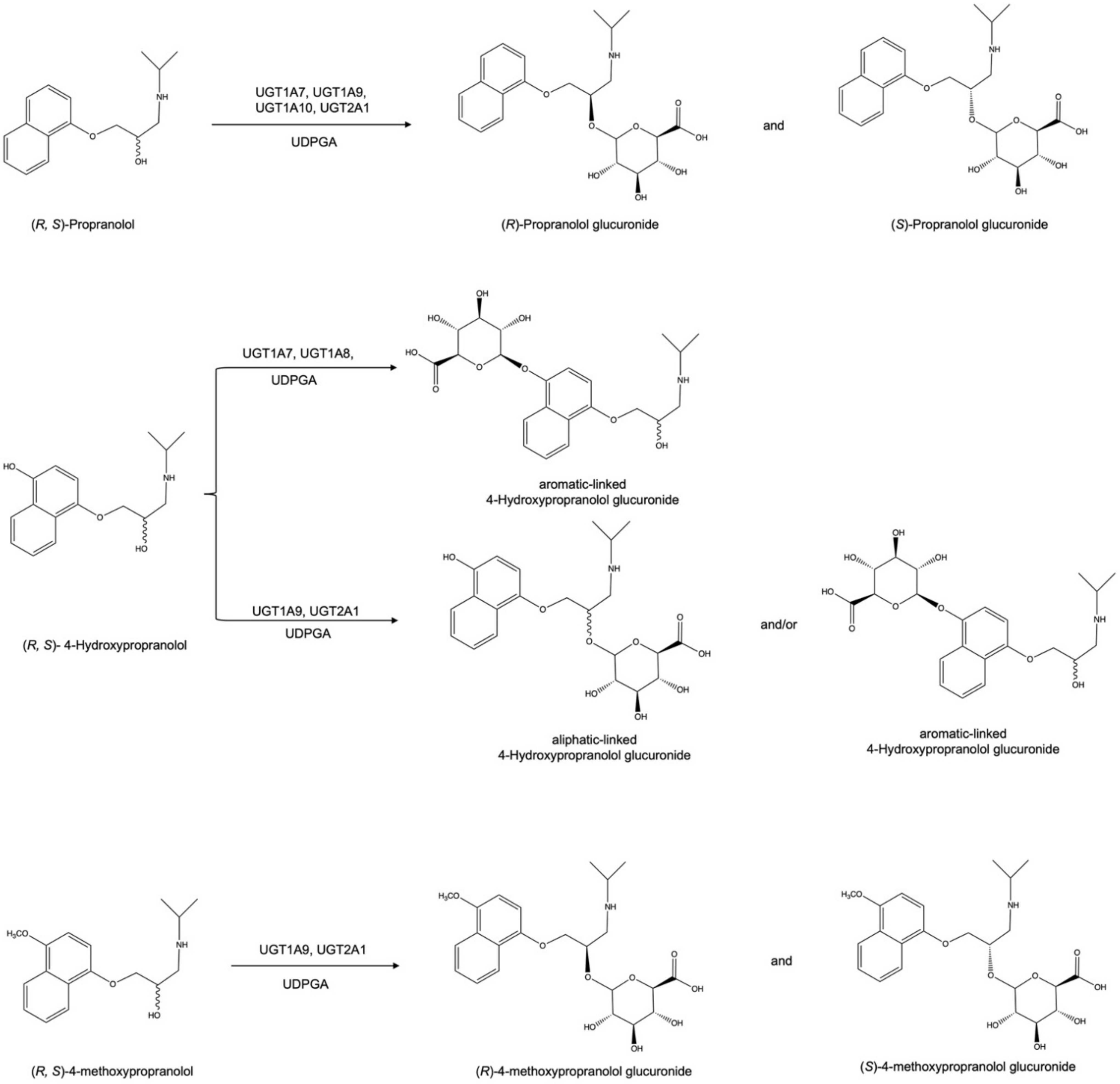
2.1. Glucuronidation of Racemic Propranolol and Enantiopure (R)-Propranolol
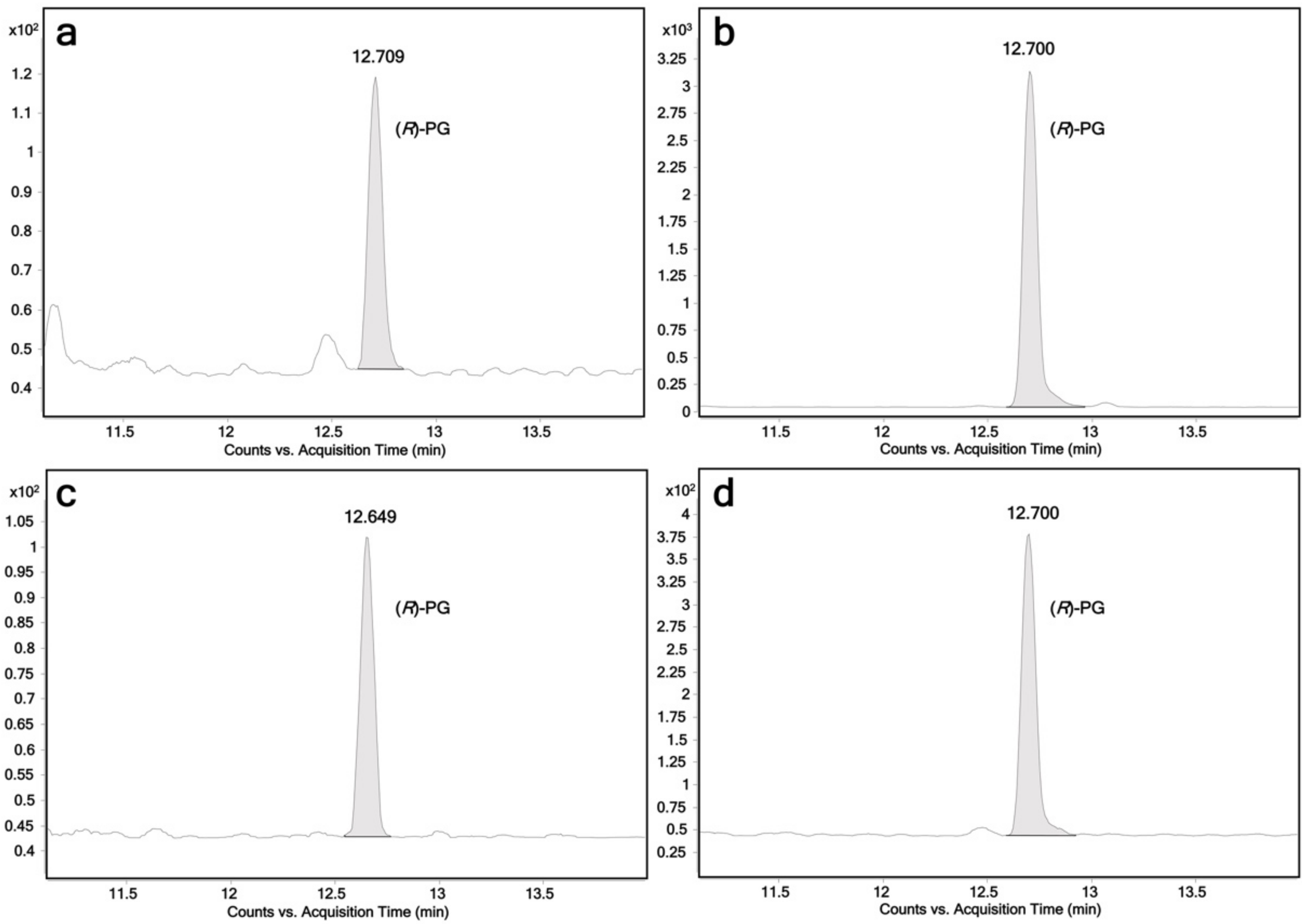
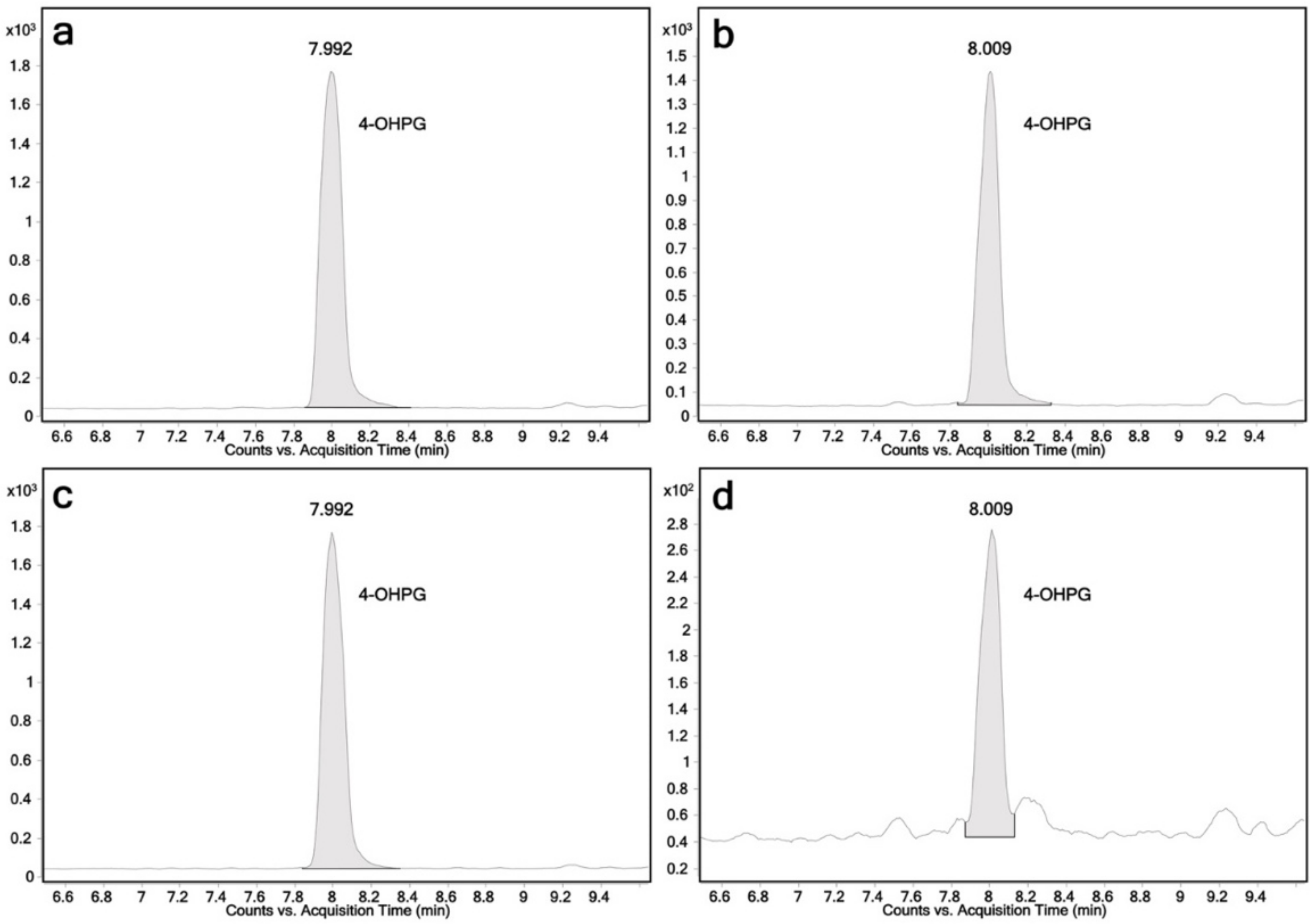
2.2. Glucuronidation Assay of Racemic 4-Hydroxypropranolol
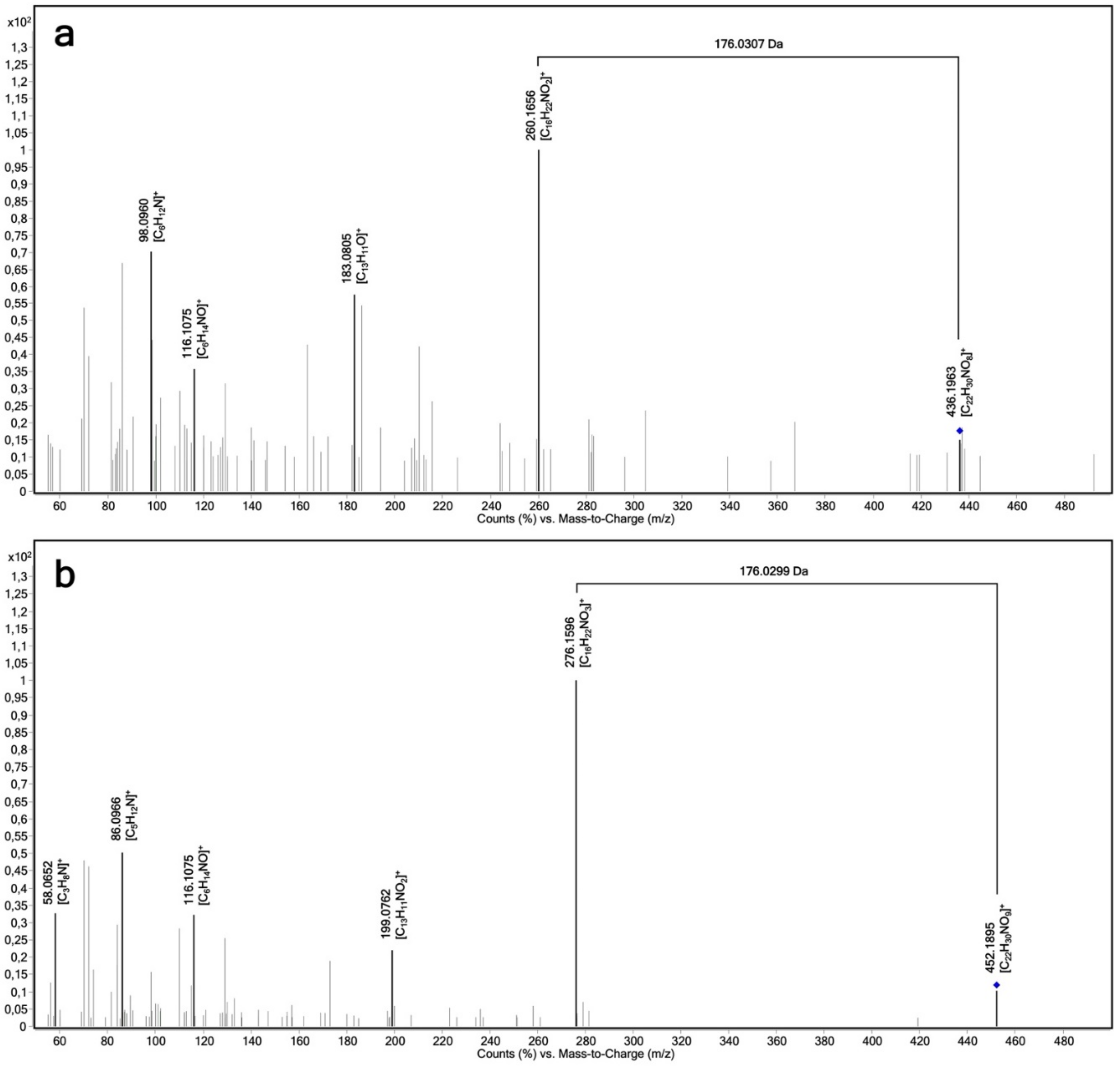
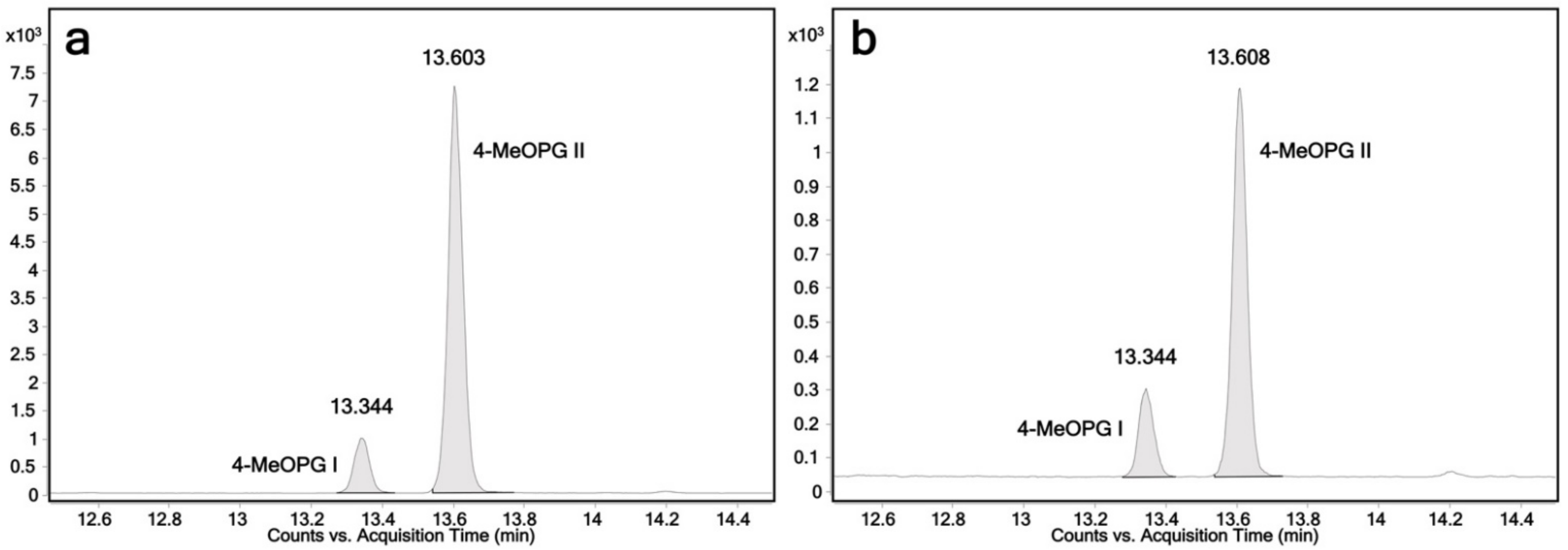
2.3. Determination of the Regioselectivity of 4-Hydroxypropranolol Glucuronidation
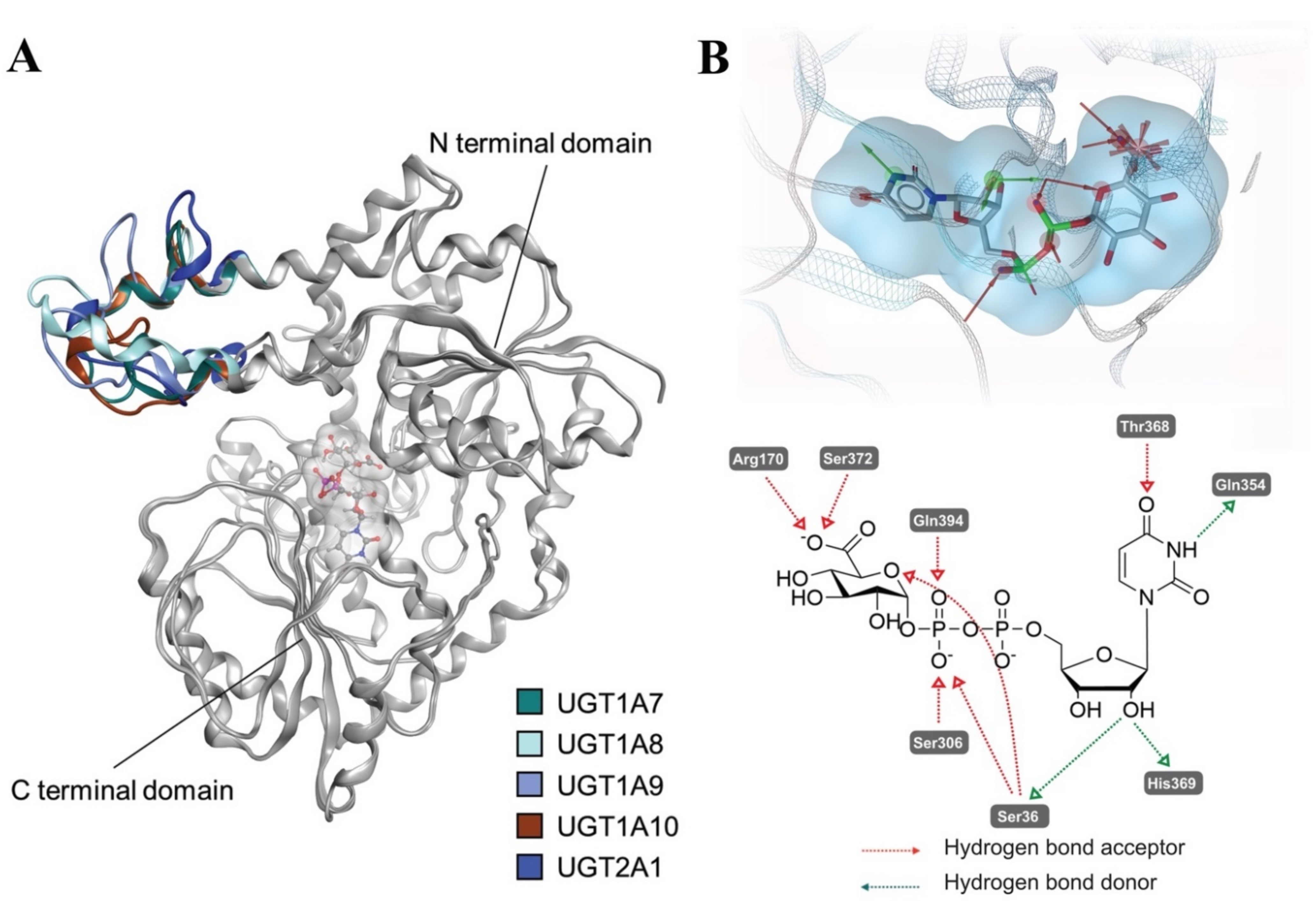
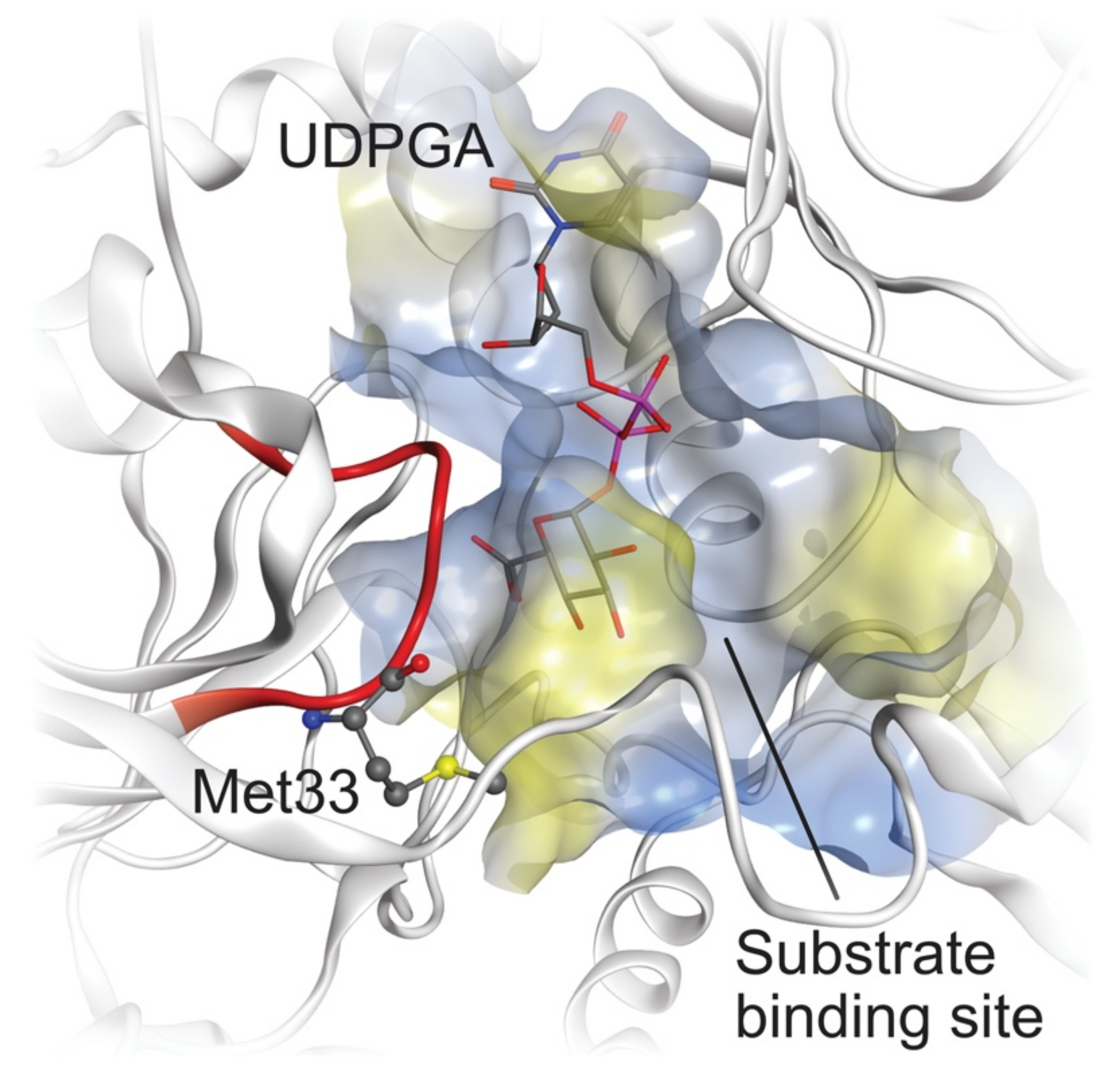
2.4. Comparative Mechanistic Modeling for Human UGTs
3. Discussion
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Synthesis of 4-Methoxypropranolol
4.3. GC-MS Analysis of 4-Methoxypropranolol
4.4. Biotransformation with UGT Enzyme Bags
4.5. LC-MS/MS Analysis of Propranolol Glucuronides, 4-Hydroxypropranolol Glucuronides and 4-Methoxypropranolol Glucuronides
4.6. Method Performance Characteristics
4.7. Homology Modeling of UGT1A7, UGT1A8, UGT1A9, UGT1A10 and UGT2A1
4.8. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dragan, C.A.; Buchheit, D.; Bischoff, D.; Ebner, T.; Bureik, M. Glucuronide production by whole-cell biotransformation using genetically engineered fission yeast Schizosaccharomyces pombe. Drug Metab. Dispos. 2010, 38, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouleau, M.; Tourancheau, A.; Girard-Bock, C.; Villeneuve, L.; Vaucher, J.; Duperre, A.M.; Audet-Delage, Y.; Gilbert, I.; Popa, I.; Droit, A.; et al. Divergent Expression and Metabolic Functions of Human Glucuronosyltransferases through Alternative Splicing. Cell Rep. 2016, 17, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Kallionpää, R.A.; Järvinen, E.; Finel, M. Glucuronidation of estrone and 16α-hydroxyestrone by human UGT enzymes: The key roles of UGT1A10 and UGT2B7. J. Steroid Biochem. Mol. Biol. 2015, 154, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, C.; Levesque, E.; Rouleau, M. Pharmacogenomics of human uridine diphospho-glucuronosyltransferases and clinical implications. Clin. Pharmacol. Ther. 2014, 96, 324–339. [Google Scholar] [CrossRef] [Green Version]
- Mazerska, Z.; Mroz, A.; Pawlowska, M.; Augustin, E. The role of glucuronidation in drug resistance. Pharmacol. Ther. 2016, 159, 35–55. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Owens, I.S.; Burchell, B.; Bock, K.W.; Bairoch, A.; Belanger, A.; Fournel-Gigleux, S.; Green, M.; Hum, D.W.; Iyanagi, T.; et al. The UDP glycosyltransferase gene superfamily: Recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 1997, 7, 255–269. [Google Scholar] [CrossRef]
- Miners, J.O.; Mackenzie, P.I.; Knights, K.M. The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro–in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug Metab. Rev. 2010, 42, 196–208. [Google Scholar] [CrossRef]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef]
- Radominska-Pandya, A.; Bratton, S.; Little, J. A Historical Overview of the Heterologous Expression of Mammalian UDP-Glucuronosyltransferase Isoforms Over the Past Twenty Years. Curr. Drug Metab. 2005, 6, 141–160. [Google Scholar] [CrossRef]
- Ikushiro, S.; Nishikawa, M.; Masuyama, Y.; Shouji, T.; Fujii, M.; Hamada, M.; Nakajima, N.; Finel, M.; Yasuda, K.; Kamakura, M.; et al. Biosynthesis of Drug Glucuronide Metabolites in the Budding Yeast Saccharomyces cerevisiae. Mol. Pharm. 2016, 13, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Machalz, D.; Wang, S.; Li, Z.; Wolber, G.; Bureik, M. A common polymorphic variant of UGT1A5 displays increased activity due to optimized cofactor binding. FEBS Lett. 2018, 592, 1837–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Durairaj, P.; Bureik, M. Rapid and convenient biotransformation procedure for human drug metabolizing enzymes using permeabilized fission yeast cells. Anal. Biochem. 2020, 607, 113704. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Machalz, D.; Wolber, G.; Parr, M.K.; Bureik, M. Functional Expression of All Human Sulfotransferases in Fission Yeast, Assay Development, and Structural Models for Isoforms SULT4A1 and SULT6B1. Biomolecules 2020, 10, 1517. [Google Scholar] [CrossRef]
- Partani, P.; Modhave, Y.; Gurule, S.; Khuroo, A.; Monif, T. Simultaneous determination of propranolol and 4-hydroxy propranolol in human plasma by solid phase extraction and liquid chromatography/electrospray tandem mass spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 966–976. [Google Scholar] [CrossRef]
- Routledge, P.A.; Shand, D.G. Clinical pharmacokinetics of propranolol. Clin. Pharmacokinet. 1979, 4, 73–90. [Google Scholar] [CrossRef]
- Steenen, S.A.; van Wijk, A.J.; van der Heijden, G.J.; van Westrhenen, R.; de Lange, J.; de Jongh, A. Propranolol for the treatment of anxiety disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2016, 30, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Luan, L.; Shao, Q.; Zeng, S. Direct determination of S-(-)- and R-(+)-propranolol glucuronide in rat hepatic microsomes by RP-HPLC. Biomed. Chromatogr. 2004, 18, 833–837. [Google Scholar] [CrossRef]
- Li, X.; Zeng, S. Stereoselective propranolol metabolism in two drug induced rat hepatic microsomes. World J. Gastroenterol. 2000, 6, 74–78. [Google Scholar] [CrossRef]
- Zhou, Q.; Yao, T.W.; Zeng, S. Chiral reversed phase high-performance liquid chromatography for determining propranolol enantiomers in transgenic Chinese hamster CHL cell lines expressing human cytochrome P450. J. Biochem. Biophys. Methods 2002, 54, 369–376. [Google Scholar] [CrossRef]
- Brosen, K. Drug interactions and the cytochrome P450 system. The role of cytochrome P450 1A2. Clin. Pharm. 1995, 29 (Suppl. S1), 20–25. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Echizen, H.; Chiba, K.; Tani, M.; Ishizaki, T. Identification of human CYP isoforms involved in the metabolism of propranolol enantiomers–N-desisopropylation is mediated mainly by CYP1A2. Br. J. Clin. Pharmacol. 1995, 39, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sten, T.; Qvisen, S.; Uutela, P.; Luukkanen, L.; Kostiainen, R.; Finel, M. Prominent but reverse stereoselectivity in propranolol glucuronidation by human UDP-glucuronosyltransferases 1A9 and 1A10. Drug Metab. Dispos. 2006, 34, 1488–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, B.; Riegelman, S. Stereospecific assay for (-)-and (+)-propranolol in human and dog plasma. J. Pharmacol. Exp. Ther. 1980, 215, 643–648. [Google Scholar]
- Walle, T.; Walle, U.K.; Olanoff, L.S. Quantitative account of propranolol metabolism in urine of normal man. Drug Metab. Dispos. Biol. Fate Chem. 1985, 13, 204–209. [Google Scholar]
- Luan, L.J.; Shao, Q.; Ma, J.Y.; Zeng, S. Stereoselective urinary excretion of S-(-)- and R-(+)-propranolol glucuronide following oral administration of RS-propranolol in Chinese Han subjects. World J. Gastroenterol. 2005, 11, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- Otton, S.V.; Gillam, E.M.; Lennard, M.S.; Tucker, G.T.; Woods, H.F. Propranolol oxidation by human liver microsomes-the use of cumene hydroperoxide to probe isoenzyme specificity and regio-and stereoselectivity. Br. J. Clin. Pharmacol. 1990, 30, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.A.; Hull, J.E.; Norris, K.J. Glucuronidation of propranolol and 4′-hydroxypropranolol. Substrate specificity and stereoselectivity of rat liver microsomal glucuronyltransferases. Drug Metab. Dispos. Biol. Fate Chem. 1981, 9, 466–471. [Google Scholar]
- Cleaveland, C.R.; Shand, D.G. Effect of route of administration on the relationship between β-adrenergic blockade and plasma propranolol level. Clin. Pharmacol. Ther. 1972, 13, 181–185. [Google Scholar] [CrossRef]
- Brown, A.K.; Wong, C.S. Simultaneous quantification of propranolol and sulfamethoxazole and major human metabolite conjugates 4-hydroxy-propranolol sulfate and sulfamethoxazole-β-glucuronide in municipal wastewater—A framework for multiple classes of drugs and conjugates. J. Chromatogr. A 2016, 1471, 34–44. [Google Scholar] [CrossRef]
- Buchheit, D.; Dragan, C.A.; Schmitt, E.I.; Bureik, M. Production of Ibuprofen Acyl Glucosides by Human UGT2B7. Drug Metab. Dispos. 2011, 39, 2174–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchheit, D.; Schmitt, E.I.; Bischoff, D.; Ebner, T.; Bureik, M. S-Glucuronidation of 7-mercapto-4-methylcoumarin by human UDP glycosyltransferases in genetically engineered fission yeast cells. Biol. Chem. 2011, 392, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Oatis, J.E.; Russell, M.P.; Knapp, D.R.; Walle, T. Ring-hydroxylated propranolol: Synthesis and .beta.-receptor antagonist and vasodilating activities of the seven isomers. J. Med. Chem. 1981, 24, 309–314. [Google Scholar] [CrossRef]
- Harps, L.C.; Schipperges, S.; Bredendiek, F.; Wuest, B.; Borowiak, A.; Parr, M.K. Two dimensional chromatography mass spectrometry: Quantitation of chiral shifts in metabolism of propranolol in bioanalysis. J. Chromatogr. A 2020, 1617, 460828. [Google Scholar] [CrossRef]
- Breton, C.; Snajdrová, L.; Jeanneau, C.; Koca, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Miley, M.J.; Zielinska, A.K.; Keenan, J.E.; Bratton, S.M.; Radominska-Pandya, A.; Redinbo, M.R. Crystal Structure of the Cofactor-Binding Domain of the Human Phase II Drug-Metabolism Enzyme UDP-Glucuronosyltransferase 2B7. J. Mol. Biol. 2007, 369, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhu, L.; Qu, W.; Wu, F.; Hu, M.; Xie, W.; Liu, Z.; Wang, C. Insight into tartrate inhibition patterns in vitro and in vivo based on cocrystal structure with UDP-glucuronosyltransferase 2B15. Biochem. Pharmacol. 2020, 172, 113753. [Google Scholar] [CrossRef]
- Walle, T.; Oatis, J.E.; Walle, U.K.; Knapp, D.R. New ring-hydroxylated metabolites of propranolol: Species differences and stereospecific 7-hydroxylation. Drug Metab. Dispos. Biol. Fate Chem. 1982, 10, 122–127. [Google Scholar]
- Walle, T.; Conradi, E.C.; Walle, U.K.; Fagan, T.C.; Gaffney, T.E. Propranolol glucuronide cumulation during long-term propranolol therapy: A proposed storage mechanism for propranolol. Clin. Pharmacol. Ther. 1979, 26, 686–695. [Google Scholar] [CrossRef]
- Fitzgerald, J.D.; O’Donnell, S.L.R. Pharmacology of 4-hydroxypropranolol, a metabolite of propranolol. Br. J. Pharmacol. 1971, 43, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.A.; Walle, T. Isolation, purification, and structure identification of glucuronic acid conjugates of propranolol and alprenolol and their ring-hydroxylated metabolites. Drug Metab. Dispos. 1984, 12, 749–754. [Google Scholar] [PubMed]
- Lampinen Salomonsson, M.; Bondesson, U.; Hedeland, M. In vitro formation of phase I and II metabolites of propranolol and determination of their structures using chemical derivatization and liquid chromatography-tandem mass spectrometry. J. Mass Spectrom. 2009, 44, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Machalz, D.; Zöllner, A.; Sorensen, E.J.; Wolber, G.; Bureik, M. Efficient substrate screening and inhibitor testing of human CYP4Z1 using permeabilized recombinant fission yeast. Biochem. Pharmacol. 2017, 146, 174–187. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, Y.; Feng, Y. Structural dissection of sterol glycosyltransferase UGT51 from Saccharomyces cerevisiae for substrate specificity. J. Struct. Biol. 2018, 204, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, S.; Onck, P.R.; van der Giessen, E. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: Piscataway, NJ, USA, 2006; p. 43. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UGTs | (R)-Propranolol | (S)-Propranolol | 4-Hydroxypropranolol | 4-Methoxypropranolol 1 |
|---|---|---|---|---|
| UGT1A3 | - | - | - | ** |
| UGT1A4 | - | - | - | ** |
| UGT1A5 | - | - | - | ** |
| UGT1A6 | - | - | - | ** |
| UGT1A7 | + + | + + + | + + + + | - |
| UGT1A8 | - | - | + + + + | - |
| UGT1A9 | + + + | + + + + | + + + + | + + + + |
| UGT1A10 | + + | + | - | - |
| UGT2A1 | + + + | + + + + | + + + | + + + + |
| UGT2A2 | - | - | - | ** |
| UGT2A3 | - | - | - | ** |
| UGT2B4 | - | - | - | ** |
| UGT2B7 | - | - | - | ** |
| UGT2B10 | - | - | - | ** |
| UGT2B11 | - | - | - | ** |
| UGT2B15 | - | - | - | ** |
| UGT2B17 | - | - | - | ** |
| UGT2B28 | - | - | - | ** |
| Analytes | Precursor Ions (m/z) | Product Ions (m/z) | CE (V) | ESI |
|---|---|---|---|---|
| Propranolol glucuronide | 436 | 258 | 12 | + |
| 436 | 183 | 16 | + | |
| 436 | 116 | 28 | + | |
| 4-Hydroxypropranolol glucuronide | 452 | 116 | 28 | + |
| 452 | 72 | 44 | + | |
| 4-Methoxypropranolol glucuronide | 466 | 288 | 12 | + |
| 466 | 116 | 28 | + | |
| 466 | 72 | 44 | + |
| Analytes | Level | Precision | |
|---|---|---|---|
| Mean Peak Area ± SD | CV (%) | ||
| (R)-Propranolol glucuronide | 0.02× | 31.19 ± 6.02 | 19.3 |
| 0.125× | 251.93 ± 20.08 | 7.9 | |
| 0.2× | 385.59 ± 20.61 | 5.3 | |
| n.d. | 1329.77 ± 91.07 | 6.8 | |
| (S)-Propranolol glucuronide | 0.02× | 120.67 ± 24.60 | 20.4 |
| 0.125× | 883.12 ± 68.86 | 7.8 | |
| 0.2× | 1362.62 ± 16.42 | 1.2 | |
| n.d. | 4880.75 ± 18.84 | 0.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Liu, S.; Wolber, G.; Bureik, M.; Parr, M.K. Complete Reaction Phenotyping of Propranolol and 4-Hydroxypropranolol with the 19 Enzymes of the Human UGT1 and UGT2 Families. Int. J. Mol. Sci. 2022, 23, 7476. https://doi.org/10.3390/ijms23137476
Yang F, Liu S, Wolber G, Bureik M, Parr MK. Complete Reaction Phenotyping of Propranolol and 4-Hydroxypropranolol with the 19 Enzymes of the Human UGT1 and UGT2 Families. International Journal of Molecular Sciences. 2022; 23(13):7476. https://doi.org/10.3390/ijms23137476
Chicago/Turabian StyleYang, Fan, Sijie Liu, Gerhard Wolber, Matthias Bureik, and Maria Kristina Parr. 2022. "Complete Reaction Phenotyping of Propranolol and 4-Hydroxypropranolol with the 19 Enzymes of the Human UGT1 and UGT2 Families" International Journal of Molecular Sciences 23, no. 13: 7476. https://doi.org/10.3390/ijms23137476