
Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nonalcoholic Fatty Liver Disease (NAFLD)
2.1. Epidemiology and Burden of NAFLD
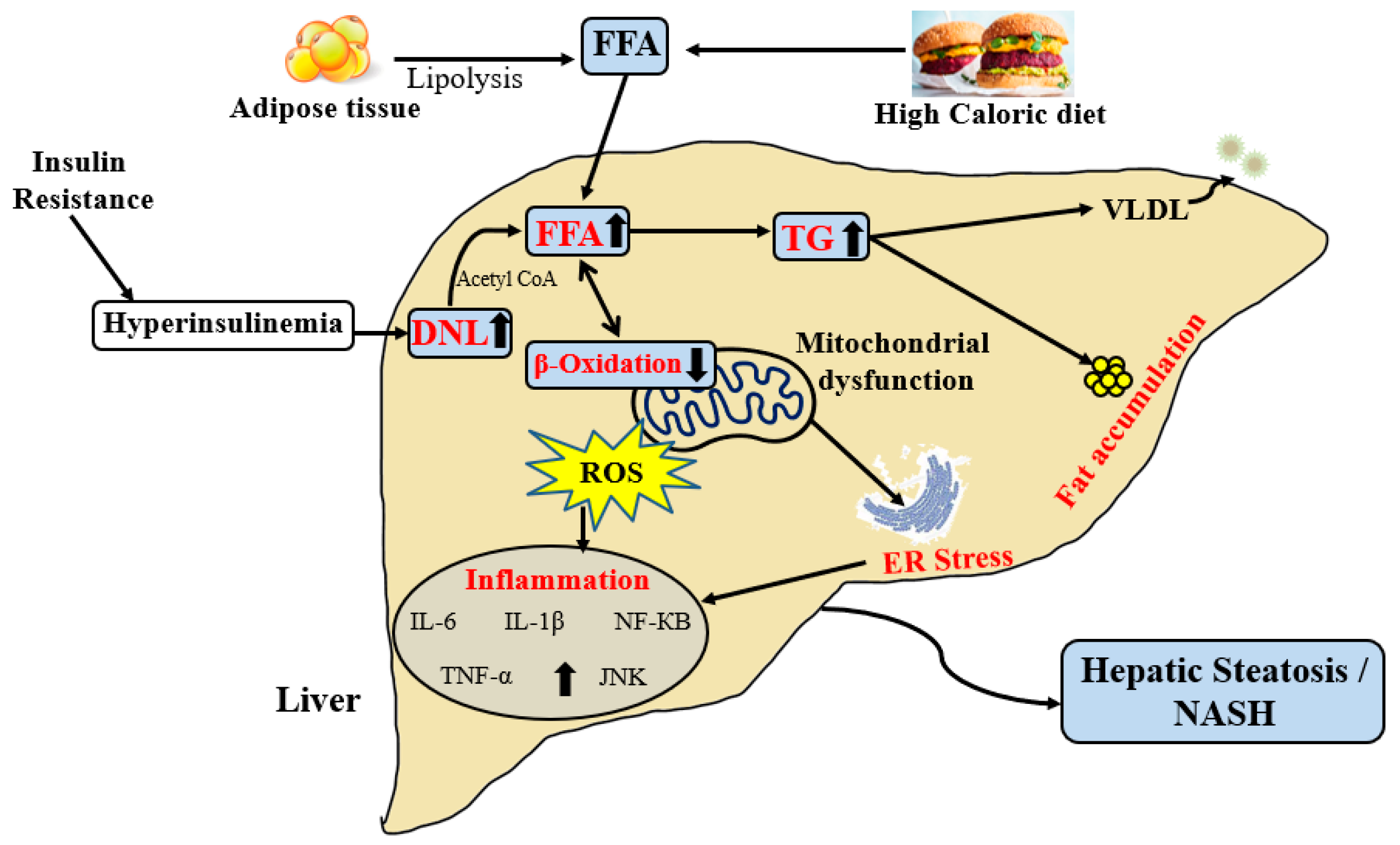
2.2. Pathogenesis of NAFLD
2.3. Diagnosis of NAFLD
2.4. Management of NAFLD
2.4.1. Diet Restriction
2.4.2. Weight Loss
2.4.3. Exercise
2.4.4. Pharmacological Interventions
3. Mitochondria and NAFLD
3.1. Mitochondrial Fatty Acid Oxidation
3.2. Mitochondrial Dynamics, Biogenesis and Mitophagy
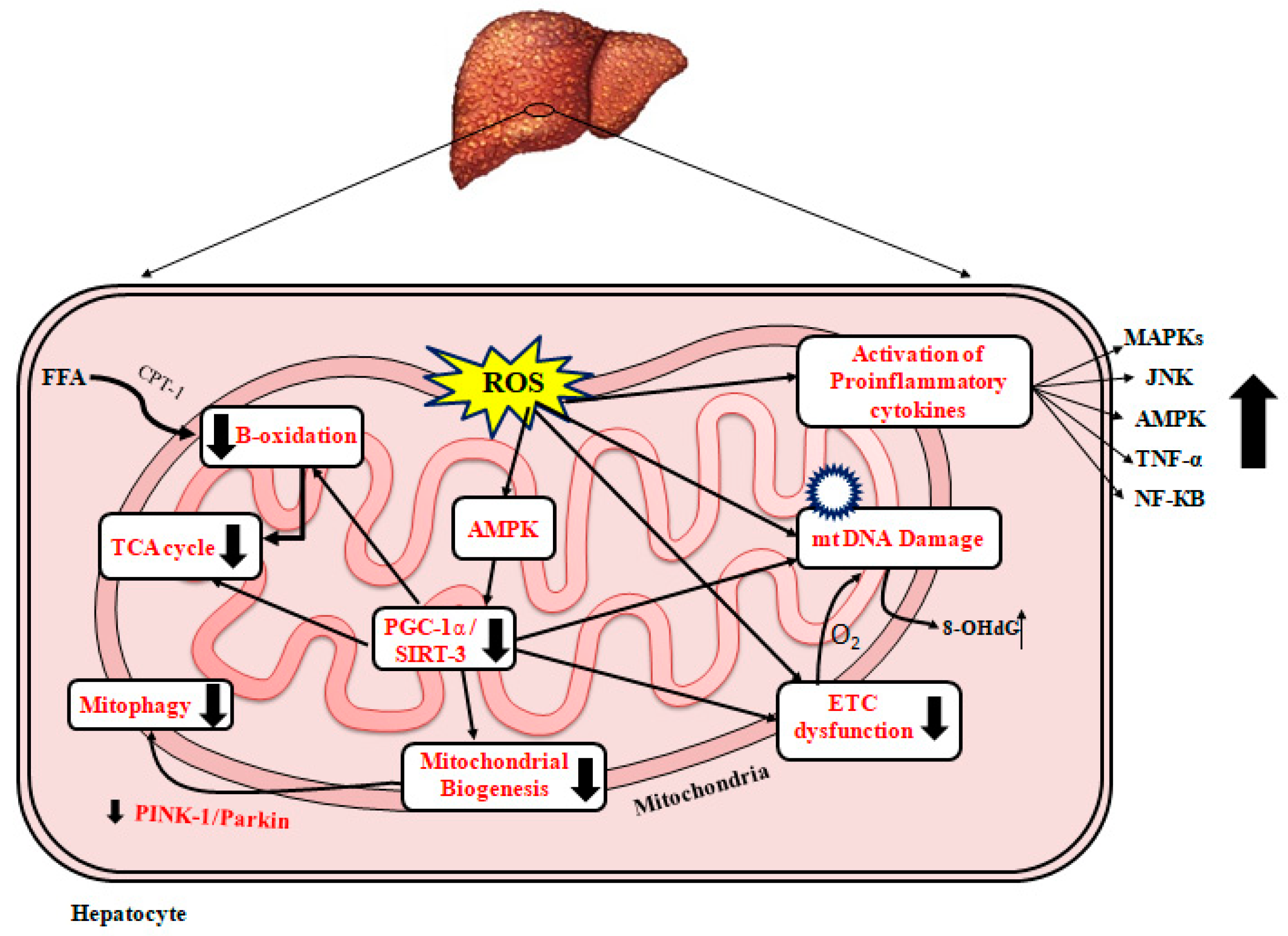
3.3. Role of Mitochondrial Dysfunction in NAFLD
3.3.1. FAO and NAFLD
3.3.2. Insulin Resistance and NAFLD
3.3.3. ROS and NAFLD
3.3.4. Mitochondrial Biogenesis and NAFLD
3.3.5. Mitophagy and NAFLD
3.3.6. Targeting Mitochondria for Treatment of NAFLD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Shirazi, F.; Wang, J.; Wong, R.J. Nonalcoholic Steatohepatitis Becomes the Leading Indication for Liver Transplant Registrants Among US Adults Born Between 1945 and 1965. J. Clin. Exp. Hepatol. 2020, 10, 30–36. [Google Scholar] [CrossRef]
- Noureddin, M.; Vipani, A.; Bresee, C.; Todo, T.; Kim, I.K.; Alkhouri, N.; Setiawan, V.W.; Tran, T.; Ayoub, W.S.; Lu, S.C.; et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications for Liver Transplant and Ethnic and Gender Variances. Am. J. Gastroenterol. 2018, 113, 1649–1659. [Google Scholar] [CrossRef]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients with Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099.e1091. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Ong, J.; Trimble, G.; AlQahtani, S.; Younossi, I.; Ahmed, A.; Racila, A.; Henry, L. Nonalcoholic Steatohepatitis Is the Most Rapidly Increasing Indication for Liver Transplantation in the United States. Clin. Gastroenterol. Hepatol. 2021, 19, 580–589.e585. [Google Scholar] [CrossRef]
- Mendes, F.D.; Suzuki, A.; Sanderson, S.O.; Lindor, K.D.; Angulo, P. Prevalence and Indicators of Portal Hypertension in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2012, 10, 1028–1033.e1022. [Google Scholar] [CrossRef] [Green Version]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Fotbolcu, H.; Zorlu, E. Nonalcoholic fatty liver disease as a multi-systemic disease. World J. Gastroenterol. 2016, 22, 4079–4090. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; on behalf of the International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.-G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Angulo, P.; Lindor, K.D. Non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2002, 17, S186–S190. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Hallsworth, K.; Adams, L.A. Lifestyle modification in NAFLD/NASH: Facts and figures. JHEP Rep. 2019, 1, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef]
- Estes, C.; Chan, H.L.Y.; Chien, R.N.; Chuang, W.-L.; Fung, J.; Goh, B.B.G.; Hu, T.H.; Huang, J.-F.; Jang, B.K.; Jun, D.W.; et al. Modelling NAFLD disease burden in four Asian regions-2019-2030. Aliment. Pharmacol. Ther. 2020, 51, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.M.; Fryar, C.D.; Carroll, M.D.; Freedman, D.S.; Ogden, C.L. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007-2008 to 2015-2016. JAMA J. Am. Med. Assoc. 2018, 319, 1723–1725. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: Revisited after a decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Eccleston, H.B.; Andringa, K.K.; Betancourt, A.M.; King, A.L.; Mantena, S.K.; Swain, T.M.; Tinsley, H.N.; Nolte, R.N.; Nagy, T.R.; Abrams, G.A.; et al. Chronic Exposure to a High-Fat Diet Induces Hepatic Steatosis, Impairs Nitric Oxide Bioavailability, and Modifies the Mitochondrial Proteome in Mice. Antioxid. Redox Signal. 2011, 15, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Conlon, M.A.; Bird, A.R. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef]
- Quesada-Vázquez, S.; Aragonès, G.; Del Bas, J.M.; Escoté, X. Diet, Gut Microbiota and Non-Alcoholic Fatty Liver Disease: Three Parts of the Same Axis. Cells 2020, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 8, 35–44. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K. Role of Insulin Resistance and Lipotoxicity in Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, M.W. Lipid Metabolism and Liver Inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium 2020, 94, 102336. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. S2), 83–92. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Y.; Zhang, H.; Guo, J.; Lam, C.W.K.; Wang, C.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef]
- Del Río, L.A.; Lopez-Huertas, E. ROS Generation in Peroxisomes and its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.-H.; Jang, S.; Gonzalez, F.J.; Song, B.-J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, D.; Nakao, Y.; Mauer, A.S.; Thompson, J.M.; Sehrawat, T.S.; Liao, C.-Y.; Krishnan, A.; Lucien, F.; Guo, Q.; Liu, M.; et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice with Steatohepatitis. Gastroenterology 2020, 159, 1487–1503.e1417. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of Nonalcoholic Fatty Liver Disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb1, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef]
- Dyson, J.K.; Anstee, Q.M.; McPherson, S. Non-alcoholic fatty liver disease: A practical approach to diagnosis and staging. Front. Gastroenterol. 2014, 5, 211–218. [Google Scholar] [CrossRef]
- Ferraioli, G.; Soares Monteiro, L.B. Ultrasound-based techniques for the diagnosis of liver steatosis. World J. Gastroenterol. 2019, 25, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Croce, A.C.; Ferrigno, A.; Di Pasqua, L.G.; Berardo, C.; Piccolini, V.M.; Bertone, V.; Bottiroli, G.; Vairetti, M. Autofluorescence discrimination of metabolic fingerprint in nutritional and genetic fatty liver models. J. Photochem. Photobiol. B Biol. 2016, 164, 13–20. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Heurtier, A.; Gombert, S.; Giral, P.; Bruckert, E.; Grimaldi, A.; Capron, F.; Poynard, T. Sampling Variability of Liver Biopsy in Nonalcoholic Fatty Liver Disease. Gastroenterology 2005, 128, 1898–1906. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Adams, L.A.; De Lédinghen, V.; Wong, G.L.-H.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef]
- Nobili, V.; Parkes, J.; Bottazzo, G.; Marcellini, M.; Cross, R.; Newman, D.; Vizzutti, F.; Pinzani, M.; Rosenberg, W.M. Performance of ELF Serum Markers in Predicting Fibrosis Stage in Pediatric Non-Alcoholic Fatty Liver Disease. Gastroenterology 2009, 136, 160–167. [Google Scholar] [CrossRef]
- Boyle, M.; Tiniakos, D.; Schattenberg, J.M.; Ratziu, V.; Bugianessi, E.; Petta, S.; Oliveira, C.P.; Govaere, O.; Younes, R.; McPherson, S.; et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. 2019, 1, 188–198. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Irles, M.; Wong, G.L.-H.; Shili, S.; Chan, A.W.-H.; Merrouche, W.; Shu, S.S.-T.; Foucher, J.; Le Bail, B.; Chan, W.K.; et al. Unified interpretation of liver stiffness measurement by M and XL probes in non-alcoholic fatty liver disease. Gut 2019, 68, 2057–2064. [Google Scholar] [CrossRef]
- Newsome, P.N.; Sasso, M.; Deeks, J.J.; Paredes, A.; Boursier, J.; Chan, W.-K.; Yilmaz, Y.; Czernichow, S.; Zheng, M.-H.; Wong, V.W.-S.; et al. FibroScan-AST (FAST) score for the non-invasive identification of patients with non-alcoholic steatohepatitis with significant activity and fibrosis: A prospective derivation and global validation study. Lancet Gastroenterol. Hepatol. 2020, 5, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Muir, A.J.; Dieterich, D.T.; Falck-Ytter, Y.T. American Gastroenterological Association Institute Technical Review on the Role of Elastography in Chronic Liver Diseases. Gastroenterology 2017, 152, 1544–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e365. [Google Scholar] [CrossRef] [PubMed]
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.-C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V.; et al. Bariatric Surgery Provides Long-term Resolution of Nonalcoholic Steatohepatitis and Regression of Fibrosis. Gastroenterology 2020, 159, 1290–1301.e1295. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Sullivan, S. Implications of diet on nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2010, 26, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Eslamparast, T.; Tandon, P.; Raman, M. Dietary Composition Independent of Weight Loss in the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 800. [Google Scholar] [CrossRef] [Green Version]
- Plaz Torres, M.C.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean Diet and NAFLD: What We Know and Questions That Still Need to Be Answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.-H.; Ferrell, L.D.; Tan, V.; Yeh, M.M.; Sarkar, M.; Gill, R.M. Aggressive non-alcoholic steatohepatitis following rapid weight loss and/or malnutrition. Mod. Pathol. 2017, 30, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Li, Z.; Xiang, Y.; Peng, H.; Yang, P.; Yuan, S.; Zhang, X.; Wu, Y.; Huang, M.; Li, J. Effect of Intermittent Fasting on Non-Alcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 709683. [Google Scholar] [CrossRef]
- Wei, H.; Qu, H.; Wang, H.; Deng, H. Associations between sitting time and non-alcoholic fatty liver diseases in Chinese male workers: A cross-sectional study. BMJ Open 2016, 6, e011939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, C.; Motta, V.; Torres, T.D.S.; Aguila, M.; Mandarim-De-Lacerda, C. Beneficial effects of exercise training (treadmill) on insulin resistance and nonalcoholic fatty liver disease in high-fat fed C57BL/6 mice. Braz. J. Med Biol. Res. 2010, 43, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreenivasa Baba, C.; Alexander, G.; Kalyani, B.; Pandey, R.; Rastogi, S.; Pandey, A.; Choudhuri, G. Effect of exercise and dietary modification on serum aminotransferase levels in patients with nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2006, 21, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Uptergrove, G.M.; Morris, E.M.; Borengasser, S.J.; Laughlin, M.H.; Booth, F.W.; Thyfault, J.P.; Ibdah, J.A. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G874–G883. [Google Scholar] [CrossRef] [Green Version]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G619–G626. [Google Scholar] [CrossRef] [Green Version]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 668. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, S. NASH Therapy: Omega 3 supplementation, vitamin E, insulin sensitizers and statin drugs. Clin. Mol. Hepatol. 2017, 23, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2020, 50, 101134. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef]
- Xu, J.; Shen, J.; Yuan, R.; Jia, B.; Zhang, Y.; Wang, S.; Liu, M.; Wang, T. Mitochondrial Targeting Therapeutics: Promising Role of Natural Products in Non-alcoholic Fatty Liver Disease. Front. Pharmacol. 2021, 12, 796207. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Sumneang, N.; Siri-Angkul, N.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 2020, 680, 108241. [Google Scholar] [CrossRef]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; Xu, R.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, e12447. [Google Scholar] [CrossRef]
- Ding, M.; Liu, C.; Shi, R.; Yu, M.; Zeng, K.; Kang, J.; Fu, F.; Mi, M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2020, 229, e13428. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Cerveny, K.L.; Tamura, Y.; Zhang, Z.; Jensen, R.E.; Sesaki, H. Regulation of mitochondrial fusion and division. Trends Cell Biol. 2007, 17, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. et Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.-B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [Green Version]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Islam, H.; Edgett, B.A.; Gurd, B.J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A re-evaluation. Metabolism 2018, 79, 42–51. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguère, V.; Kelly, D.P. Estrogen-Related Receptor α Directs Peroxisome Proliferator-Activated Receptor α Signaling in the Transcriptional Control of Energy Metabolism in Cardiac and Skeletal Muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [Green Version]
- Garesse, R.; Vallejo, C.G. Animal mitochondrial biogenesis and function: A regulatory cross-talk between two genomes. Gene 2001, 263, 1–16. [Google Scholar] [CrossRef]
- Larsson, N.-G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Birkenfeld, A.L.; Lee, H.-Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the Mammalian INDY Homolog Mimics Aspects of Dietary Restriction and Protects against Adiposity and Insulin Resistance in Mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Gusdon, A.M.; Song, K.-X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Ucar, F.; Sezer, S.; Erdogan, S.; Akyol, S.; Armutcu, F.; Akyol, O. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox Rep. 2013, 18, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice Heterozygous for a Defect in Mitochondrial Trifunctional Protein Develop Hepatic Steatosis and Insulin Resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef]
- Rector, R.S.; Morris, E.M.; Ridenhour, S.; Meers, G.M.; Hsu, F.-F.; Turk, J.; Ibdah, J.A. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 2013, 57, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Ibdah, J.A. Acute fatty liver of pregnancy: An update on pathogenesis and clinical implications. World J. Gastroenterol. 2006, 12, 7397–7404. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Paul, H.; Zhao, Y.; Binford, S.; Salleng, K.; Cline, M.; Matern, D.; Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig. 2001, 107, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Nassir, F.; Arndt, J.J.; Johnson, S.A.; Ibdah, J.A. Regulation of mitochondrial trifunctional protein modulates nonalcoholic fatty liver disease in mice. J. Lipid Res. 2018, 59, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N. Reduced Hepatic Mitochondrial Fatty Acid Oxidation in Veterans with Non-Alcoholic Fatty Liver Disease and Hepatic Fibrosis. In Hepatology; Wiley: Hoboken, NJ, USA, 2021; pp. 1121A–1122A. [Google Scholar]
- Reinecke, F.; Smeitink, J.A.; van der Westhuizen, F.H. OXPHOS gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta 2009, 1792, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; Del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver: Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of Subsarcolemmal Mitochondria in Obesity and Type 2 Diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of Acute Changes of Plasma Free Fatty Acids on Intramyocellular Fat Content and Insulin Resistance in Healthy Subjects. Diabetes 2001, 50, 1612–1617. [Google Scholar] [CrossRef] [Green Version]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone Induces Mitochondrial Biogenesis in Human Subcutaneous Adipose Tissue In Vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingerslev, B.; Hansen, J.S.; Hoffmann, C.; Clemmesen, J.O.; Secher, N.H.; Scheler, M.; de Angelis, M.H.; Häring, H.U.; Pedersen, B.K.; Weigert, C.; et al. Angiopoietin-like protein 4 is an exercise-induced hepatokine in humans, regulated by glucagon and cAMP. Mol. Metab. 2017, 6, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Catoire, M.; Alex, S.; Paraskevopulos, N.; Mattijssen, F.; Gogh, I.E.-V.; Schaart, G.; Jeppesen, J.; Kneppers, A.; Mensink, M.; Voshol, P.J.; et al. Fatty acid-inducible ANGPTL4 governs lipid metabolic response to exercise. Proc. Natl. Acad. Sci. USA 2014, 111, E1043–E1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandard, S.; Zandbergen, F.; van Straten, E.; Wahli, W.; Kuipers, F.; Müller, M.; Kersten, S. The Fasting-induced Adipose Factor/Angiopoietin-like Protein 4 Is Physically Associated with Lipoproteins and Governs Plasma Lipid Levels and Adiposity. J. Biol. Chem. 2006, 281, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Staiger, H.; Haas, C.; Machann, J.; Werner, R.; Weisser, M.; Schick, F.; Machicao, F.; Stefan, N.; Fritsche, A.; Häring, H.-U. Muscle-Derived Angiopoietin-Like Protein 4 Is Induced by Fatty Acids via Peroxisome Proliferator–Activated Receptor (PPAR)-δ and Is of Metabolic Relevance in Humans. Diabetes 2009, 58, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Dijk, W.; Beigneux, A.P.; Larsson, M.; Bensadoun, A.; Young, S.G.; Kersten, S. Angiopoietin-like 4 promotes intracellular degradation of lipoprotein lipase in adipocytes. J. Lipid Res. 2016, 57, 1670–1683. [Google Scholar] [CrossRef] [Green Version]
- Norheim, F.; Hjorth, M.; Langleite, T.M.; Lee, S.; Holen, T.; Bindesbøll, C.; Stadheim, H.K.; Gulseth, H.L.; Birkeland, K.I.; Kielland, A.; et al. Regulation of angiopoietin-like protein 4 production during and after exercise. Physiol. Rep. 2014, 2, e12109. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J. Invited Review: Effects of acute exercise and exercise training on insulin resistance. J. Appl. Physiol. 2002, 93, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Constable, S.H.; Favier, R.J.; McLane, J.A.; Fell, R.D.; Chen, M.; Holloszy, J.O. Energy metabolism in contracting rat skeletal muscle: Adaptation to exercise training. Am. J. Physiol. Physiol. 1987, 253, C316–C322. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.S.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef] [Green Version]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-Associated Reductions in AMP-Activated Protein Kinase Activity and Mitochondrial Biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Méndez-Sánchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965–6971. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal-Casellas, A.; Amengual-Cladera, E.; Proenza, A.M.; Lladó, I.; Gianotti, M. Long-term High-fat-diet Feeding Impairs Mitochondrial Biogenesis in Liver of Male and Female Rats. Cell. Physiol. Biochem. 2010, 26, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschöp, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [Green Version]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2015, 63, 1190–1204. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1α, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Pessayre, D.; Mansouri, A.; Fromenty, B.V. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G193–G199. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E. Novel Insights into the Pathophysiology of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2010, 30, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2014, 132, 443–451. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; He, C.; Carter, A. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020, 33, 101426. [Google Scholar] [CrossRef]
- Sheldon, R.; Meers, G.M.; Morris, E.M.; Linden, M.; Cunningham, R.P.; Ibdah, J.A.; Thyfault, J.P.; Laughlin, M.H.; Rector, R.S. eNOS deletion impairs mitochondrial quality control and exacerbates Western diet-induced NASH. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E605–E616. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.-H.; Kim, H.-S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.-X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Lv, J.; Ren, J.; Sun, D. Mechanism and Regulation of Selective Mitophagy in Cardiometabolic Disease. In Autophagy and Cardiometabolic Diseases; Academic Press: Cambridge, MA, USA, 2018; pp. 43–52. ISBN 9780128052532. [Google Scholar] [CrossRef]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Lira, V.A.; Brown, D.L.; Lira, A.K.; Kavazis, A.N.; Soltow, Q.A.; Zeanah, E.H.; Criswell, D.S. Nitric oxide and AMPK cooperatively regulate PGC-1α in skeletal muscle cells. J. Physiol. 2010, 588, 3551–3566. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Tengan, C.H.; Moraes, C.T. NO control of mitochondrial function in normal and transformed cells. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 573–581. [Google Scholar] [CrossRef]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [Green Version]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.L.; Leveille, G.A. Development of lipogenesis and insulin sensitivity in tissues of the ob/ob mouse. Am. J. Physiol. Metab. 1981, 240, E101–E107. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnasamy, Y.; Gooz, M.; Li, L.; Lemasters, J.J.; Zhong, Z. Role of mitochondrial depolarization and disrupted mitochondrial homeostasis in non-alcoholic steatohepatitis and fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 190–204. [Google Scholar] [PubMed]
- Liu, W.; Ye, C.; Cheng, Q.; Zhang, X.; Yao, L.; Li, Q.; Huang, J.; Liu, Y.; Zou, Z.; Wang, H.; et al. Macrophage Raptor Deficiency-Induced Lysosome Dysfunction Exacerbates Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 211–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.; Ju, L.; Qiu, M.; Xie, Q.; Cheng, Y.; Shen, W.; Sun, W.; Wang, W.; Tian, J. Liraglutide ameliorates non-alcoholic fatty liver disease by enhancing mitochondrial architecture and promoting autophagy through the SIRT1/SIRT3-FOXO3a pathway. Hepatol. Res. 2016, 46, 933–943. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef]
- Glick, D.; Zhang, W.; Beaton, M.; Marsboom, G.; Gruber, M.; Simon, M.C.; Hart, J.; Dorn, G.W.; Brady, M.J.; Macleod, K.F. BNip3 Regulates Mitochondrial Function and Lipid Metabolism in the Liver. Mol. Cell. Biol. 2012, 32, 2570–2584. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, I.O.; Passos, E.; Diogo, C.V.; Rocha-Rodrigues, S.; Alves, E.; Oliveira, P.; Ascensao, A.; Magalhães, J. Exercise mitigates mitochondrial permeability transition pore and quality control mechanisms alterations in nonalcoholic steatohepatitis. Appl. Physiol. Nutr. Metab. 2016, 41, 298–306. [Google Scholar] [CrossRef]
- Anekwe, C.; Chandrasekaran, P.; Stanford, F.C. Ketogenic Diet-induced Elevated Cholesterol, Elevated Liver Enzymes and Potential Non-alcoholic Fatty Liver Disease. Cureus 2020, 12, e6605. [Google Scholar] [CrossRef] [Green Version]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Zhu, L.; Shen, Y.; Mou, Q.; Lin, T.; Feng, H. Protection of hepatocyte mitochondrial function by blueberry juice and probiotics via SIRT1 regulation in non-alcoholic fatty liver disease. Food Funct. 2019, 10, 1540–1551. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Du, W.; Li, Y.; Shi, C.; Hu, N.; Ma, S.; Wang, W.; Ren, J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018, 64, e12450. [Google Scholar] [CrossRef]
- Sangineto, M.; Bukke, V.; Bellanti, F.; Tamborra, R.; Moola, A.; Duda, L.; Villani, R.; Romano, A.; Serviddio, G. A Novel Nutraceuticals Mixture Improves Liver Steatosis by Preventing Oxidative Stress and Mitochondrial Dysfunction in a NAFLD Model. Nutrients 2021, 13, 652. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lutchman, G.; Kleiner, D.; Ricks, M.; Feld, J.J.; Borg, B.B.; Modi, A.; Nagabhyru, P.; Sumner, A.E.; Liang, T.J.; et al. Clinical trial: Pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2009, 29, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Sun, Q.; Zhong, W.; Zhang, W.; Sun, X.; Zhou, Z. Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: A molecular mechanism of protection against alcoholic liver disease. Redox Biol. 2018, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Srivastava, A.; Johnson, M.S.; Benavides, G.A.; Chang, M.J.; Ye, Y.; Jhala, N.; Murphy, M.P.; Kalyanaraman, B.; Darley-Usmar, V.M. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology 2011, 54, 153–163. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. https://doi.org/10.3390/ijms23137280
Ramanathan R, Ali AH, Ibdah JA. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(13):7280. https://doi.org/10.3390/ijms23137280
Chicago/Turabian StyleRamanathan, Raghu, Ahmad Hassan Ali, and Jamal A. Ibdah. 2022. "Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 13: 7280. https://doi.org/10.3390/ijms23137280