
The Inhibition of DNA Viruses by the Amphibian Antimicrobial Peptide Temporin G: A Virological Study Addressing HSV-1 and JPCyV

 , , , ,
, , , ,  , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
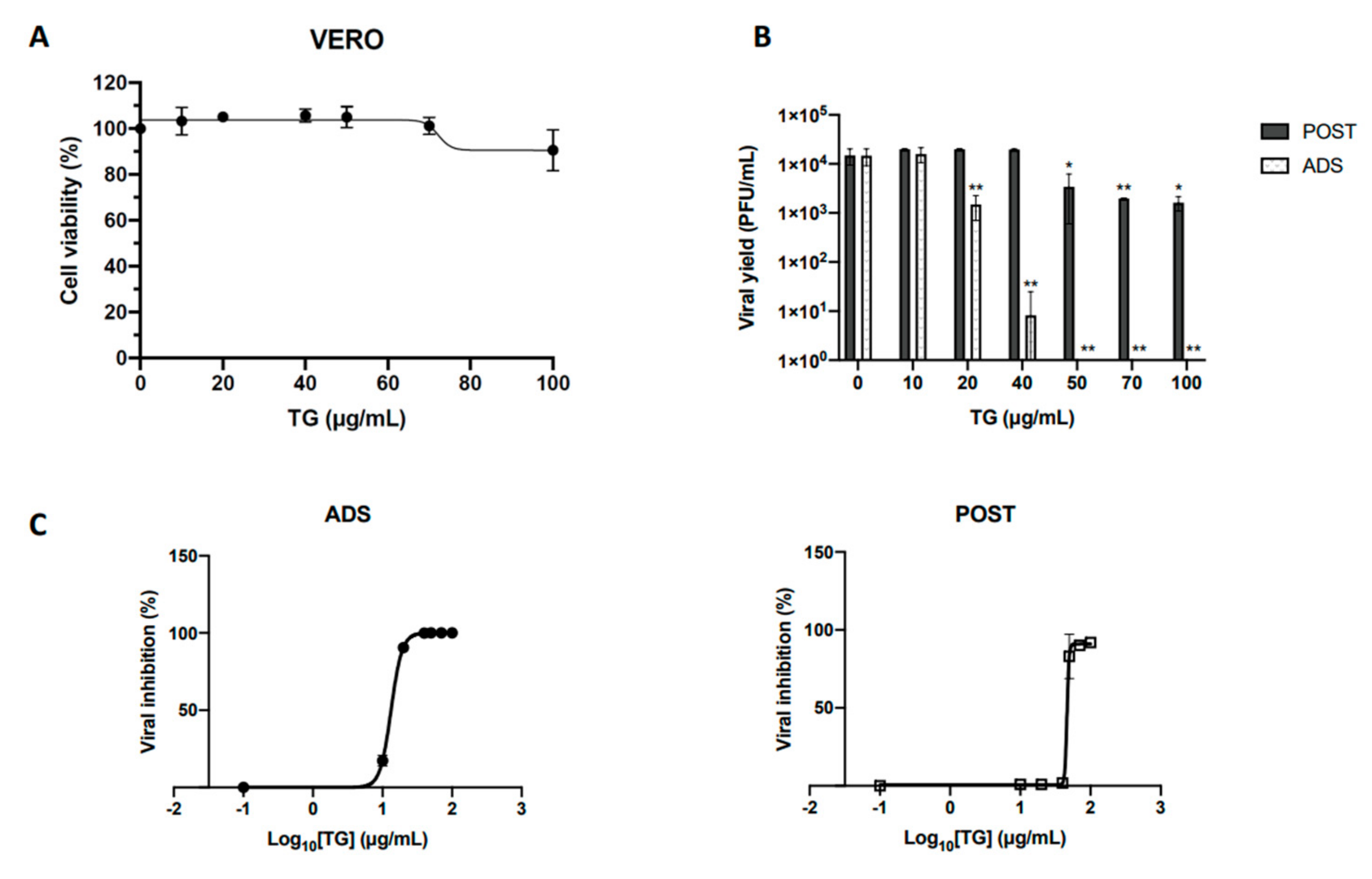
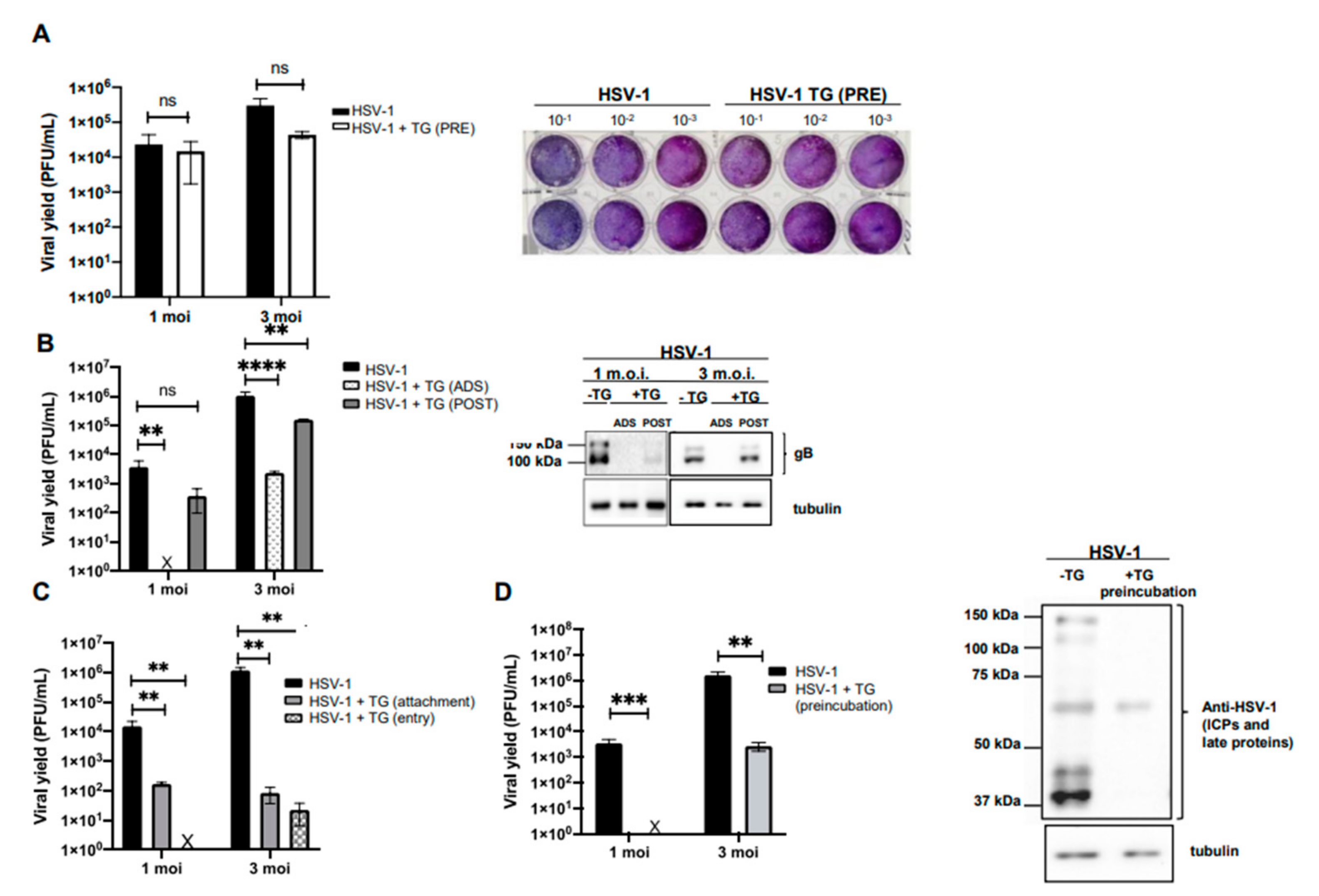
2.1. Temporin G Inhibits the Early Stages of the HSV-1 Replicative Cycle
2.2. TG Directly Affects the HSV-1 Virion
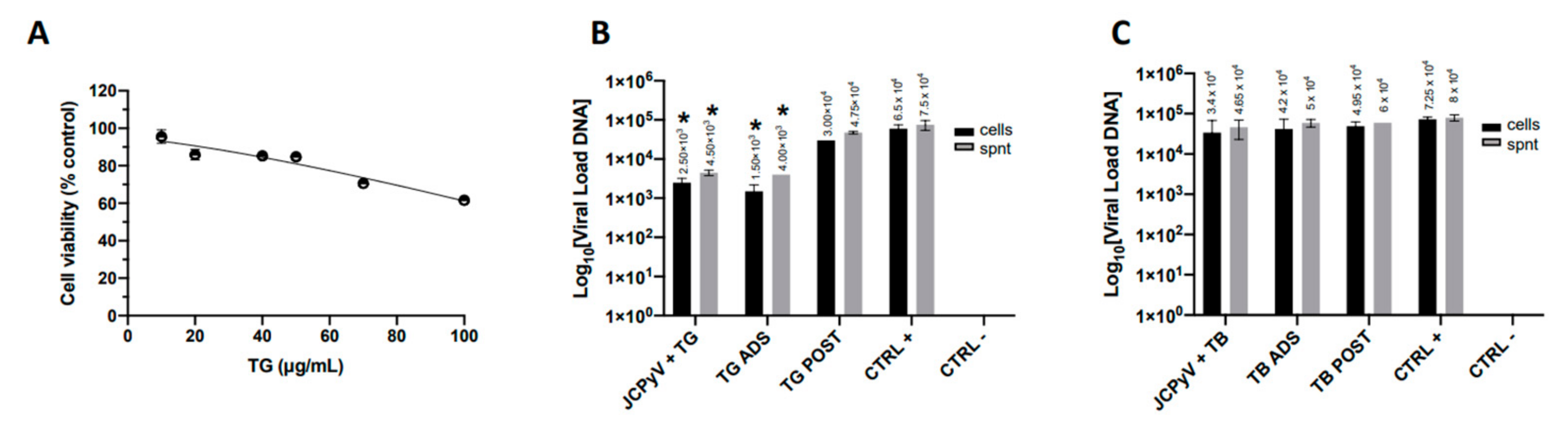
2.3. TG Exerts Slight Antiviral Activity against JCPyV Infection
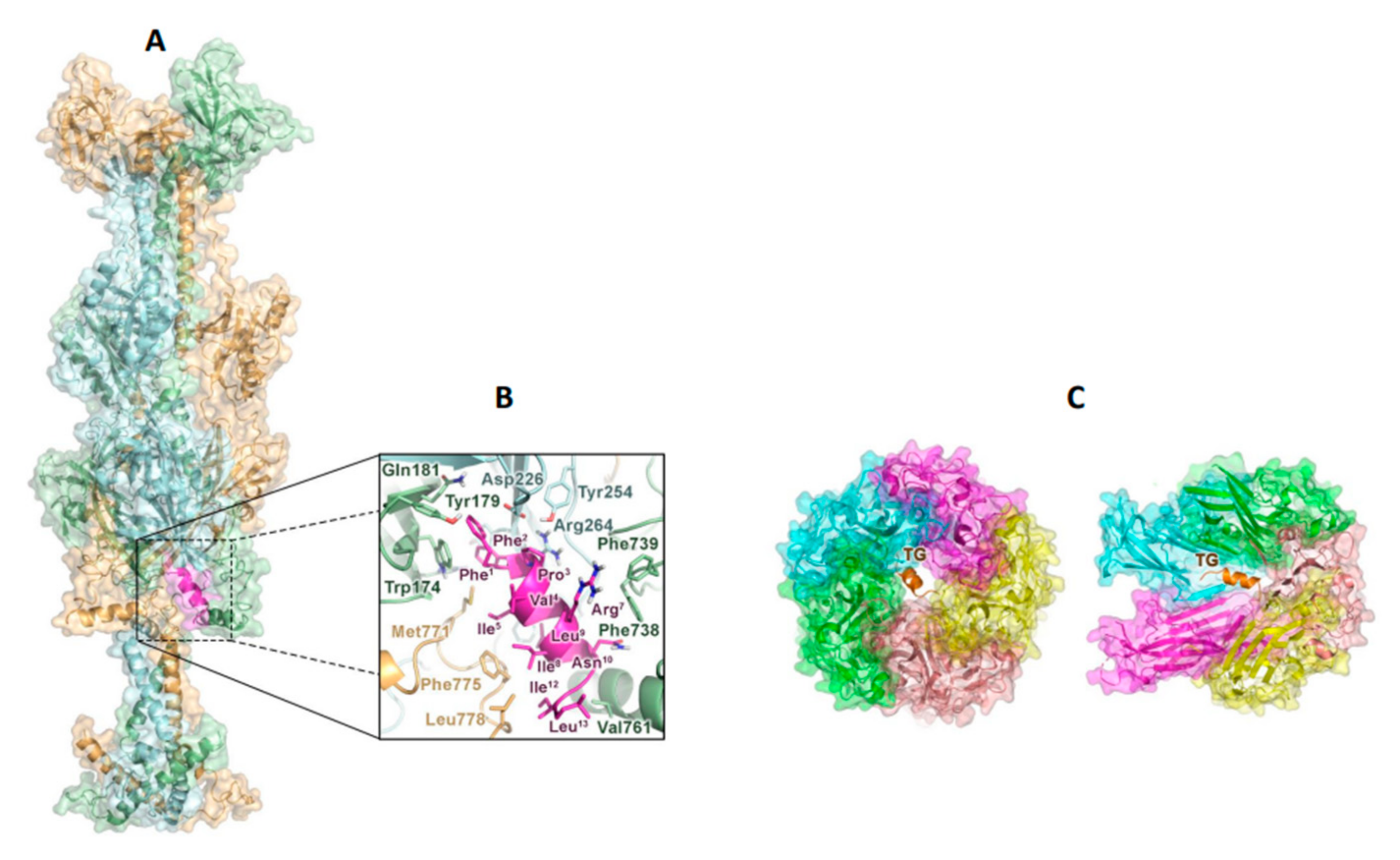
2.4. TG Interacts with the gB Glycoprotein of HSV-1 and with the Capsid Receptor VP1 of JCPyV
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Cell Cultures
4.3. HSV-1 and JCPyV Virion Production
4.4. Cellular Toxicity Assays
4.5. HSV-1 Infection and Determination of Viral Yields
4.6. JCPyV Infection and Quantification
4.7. Plaque Reduction Assay
4.8. Pre-Treatment Assay
4.9. Time-of-Addition Assay
4.10. Attachment Assay
4.11. Entry Assay
4.12. Virucidal Activity
4.13. Western Blot Analysis
4.14. Modeling of the Complexes of HSV1 and JCPyV Selected (Glyco)Proteins with TG
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Zuo, S.; Wang, B.; Zhang, K.; Wang, Y. Antimicrobial Mechanisms and Clinical Application Prospects of Antimicrobial Peptides. Molecules 2022, 27, 2675. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Marcellini, H.G.; Simmaco, M. Biological characterization and modes of action of temporins and bombinins H, multiple forms of short and mildly cationic anti-microbial peptides from amphibian skin. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2007, 13, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Islas-Rodriguez, A.E.; Marcellini, L.; Orioni, B.; Barra, D.; Stella, L.; Mangoni, M.L. Esculentin 1-21: A linear antimicrobial peptide from frog skin with inhibitory effect on bovine mastitis-causing bacteria. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2009, 15, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, B.; Cappiello, F.; Loffredo, M.R.; Ghirga, F.; Mangoni, M.L. The Potential of Frog Skin Peptides for Anti-Infective Therapies: The Case of Esculentin-1a(1-21)NH2. Curr. Med. Chem. 2020, 27, 1405–1419. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L. Temporins, anti-infective peptides with expanding properties. Cell. Mol. Life Sci. CMLS 2006, 63, 1060–1069. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Grazia, A.D.; Cappiello, F.; Casciaro, B.; Luca, V. Naturally Occurring Peptides from Rana temporaria: Antimicrobial Properties and More. Curr. Top. Med. Chem. 2016, 16, 54–64. [Google Scholar] [CrossRef]
- Varga, J.F.A.; Bui-Marinos, M.P.; Katzenback, B.A. Frog Skin Innate Immune Defences: Sensing and Surviving Pathogens. Front. Immunol. 2018, 9, 3128. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The Amphibian Antimicrobial Peptide Temporin B Inhibits In Vitro Herpes Simplex Virus 1 Infection. Antimicrob. Agents Chemother. 2018, 62, e02367-17. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.; Casciaro, B.; Genovese, A.; Brancaccio, D.; Marcocci, M.E.; Novellino, E.; Carotenuto, A.; Palamara, A.T.; Mangoni, M.L.; Nencioni, L. Temporin G, an amphibian antimicrobial peptide against influenza and parainfluenza respiratory viruses: Insights into biological activity and mechanism of action. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21358. [Google Scholar] [CrossRef]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef]
- Verzosa, A.L.; McGeever, L.A.; Bhark, S.J.; Delgado, T.; Salazar, N.; Sanchez, E.L. Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms. Front. Immunol. 2021, 12, 644664. [Google Scholar] [CrossRef] [PubMed]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2020, 10, 617578. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, G.; Vales, O.; Pineda, B.; Rodriguez, K.; Pane, C.; Sotelo, J. The presence of herpes simplex-1 and varicella zoster viruses is not related with clinical outcome of Bell’s Palsy. Virology 2020, 549, 85–88. [Google Scholar] [CrossRef]
- Protto, V.; Tramutola, A.; Fabiani, M.; Marcocci, M.E.; Napoletani, G.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Perluigi, M.; Di Domenico, F.; et al. Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms 2020, 8, 972. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Resistance of herpes simplex viruses to nucleoside analogues: Mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 2011, 55, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, V.; Prezioso, C.; Bagnato, F.; Antonelli, G. John Cunningham virus: An overview on biology and disease of the etiological agent of the progressive multifocal leukoencephalopathy. New Microbiol. 2018, 41, 179–186. [Google Scholar]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: Implications for viral latency. J. Virol. 1996, 70, 7004–7012. [Google Scholar] [CrossRef] [Green Version]
- Wollebo, H.S.; Melis, S.; Khalili, K.; Safak, M.; White, M.K. Cooperative roles of NF-kappaB and NFAT4 in polyomavirus JC regulation at the KB control element. Virology 2012, 432, 146–154. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Kaminski, R.; Khalili, K.; Wollebo, H.S. Rad51 activates polyomavirus JC early transcription. PLoS ONE 2014, 9, e110122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The human JC polyomavirus (JCPyV): Virological background and clinical implications. APMIS Acta Pathol. Microbiol. Et Immunol. Scand. 2013, 121, 685–727. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Stroh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Valle, L.; Pina-Oviedo, S. Human Polyomavirus JCPyV and Its Role in Progressive Multifocal Leukoencephalopathy and Oncogenesis. Front. Oncol. 2019, 9, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J. Clin. Microbiol. 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [Green Version]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Chapagain, M.L.; Nerurkar, V.R. Human polyomavirus JC (JCV) infection of human B lymphocytes: A possible mechanism for JCV transmigration across the blood-brain barrier. J. Infect. Dis. 2010, 202, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat. Rev. Neurol. 2021, 17, 37–51. [Google Scholar] [CrossRef]
- Tan, C.S.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: Clinical features and pathogenesis. Lancet. Neurol. 2010, 9, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Gheuens, S.; Wuthrich, C.; Koralnik, I.J. Progressive multifocal leukoencephalopathy: Why gray and white matter. Annu. Rev. Pathol. 2013, 8, 189–215. [Google Scholar] [CrossRef]
- Pavlovic, D.; Patera, A.C.; Nyberg, F.; Gerber, M.; Liu, M.; Progressive Multifocal Leukeoncephalopathy, C. Progressive multifocal leukoencephalopathy: Current treatment options and future perspectives. Ther. Adv. Neurol. Disord. 2015, 8, 255–273. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Sugimoto, C.; Kitamura, T.; Aoki, N.; Taguchi, F.; Yogo, Y. Archetype JC virus efficiently replicates in COS-7 cells, simian cells constitutively expressing simian virus 40 T antigen. J. Virol. 1998, 72, 5335–5342. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, B.; Grunewald, K. Herpesvirus membrane fusion—A team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannah, B.P.; Heldwein, E.E.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 2007, 81, 4858–4865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.D.; Stroh, L.J.; Gee, G.V.; O’Hara, B.A.; Stehle, T.; Atwood, W.J. Modulation of a pore in the capsid of JC polyomavirus reduces infectivity and prevents exposure of the minor capsid proteins. J. Virol. 2015, 89, 3910–3921. [Google Scholar] [CrossRef] [Green Version]
- Kane, J.R.; Fong, S.; Shaul, J.; Frommlet, A.; Frank, A.O.; Knapp, M.; Bussiere, D.E.; Kim, P.; Ornelas, E.; Cuellar, C.; et al. A polyomavirus peptide binds to the capsid VP1 pore and has potent antiviral activity against BK and JC polyomaviruses. eLife 2020, 9, e50722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, C.; Zhang, H.; Liu, G.D.; Xue, C.; Cao, Y. Targeting Hemagglutinin: Approaches for Broad Protection against the Influenza A Virus. Viruses 2019, 11, 405. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.S.; Georgieva, E.R.; Borbat, P.P.; Freed, J.H.; Heldwein, E.E. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat. Struct. Mol. Biol. 2018, 25, 416–424. [Google Scholar] [CrossRef]
- Zins, S.R.; Nelson, C.D.; Maginnis, M.S.; Banerjee, R.; O’Hara, B.A.; Atwood, W.J. The human alpha defensin HD5 neutralizes JC polyomavirus infection by reducing endoplasmic reticulum traffic and stabilizing the viral capsid. J. Virol. 2014, 88, 948–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gounder, A.P.; Wiens, M.E.; Wilson, S.S.; Lu, W.; Smith, J.G. Critical determinants of human alpha-defensin 5 activity against non-enveloped viruses. J. Biol. Chem. 2012, 287, 24554–24562. [Google Scholar] [CrossRef] [Green Version]
- Kollias, C.M.; Huneke, R.B.; Wigdahl, B.; Jennings, S.R. Animal models of herpes simplex virus immunity and pathogenesis. J. Neurovirol. 2015, 21, 8–23. [Google Scholar] [CrossRef]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, H.; Solis, M.; Kack-Kack, W.; Soulier, E.; Velay, A.; Fafi-Kremer, S. In Vitro and In Vivo Models for the Study of Human Polyomavirus Infection. Viruses 2016, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreras, P.; Pamies, D.; Monaco, M.C.; Munoz, L.S.; Zhong, X.; Major, E.O.; Hogberg, H.T.; Hartung, T.; Pardo, C.A. A human-derived 3D brain organoid model to study JC virus infection. J. Neurovirol. 2022, 28, 17–26. [Google Scholar] [CrossRef]
- Prezioso, C.; Scribano, D.; Bellizzi, A.; Anzivino, E.; Rodio, D.M.; Trancassini, M.; Palamara, A.T.; Pietropaolo, V. Efficient propagation of archetype JC polyomavirus in COS-7 cells: Evaluation of rearrangements within the NCCR structural organization after transfection. Arch. Virol. 2017, 162, 3745–3752. [Google Scholar] [CrossRef] [Green Version]
- Napoletani, G.; Protto, V.; Marcocci, M.E.; Nencioni, L.; Palamara, A.T.; De Chiara, G. Recurrent Herpes Simplex Virus Type 1 (HSV-1) Infection Modulates Neuronal Aging Marks in In Vitro and In Vivo Models. Int. J. Mol. Sci. 2021, 22, 6279. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001. Appendix 3, Appendix 3B. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Killlington, R.A.; Powell, K.L. Growth, assay and purification of herpes viruses. In Virology, a Practical Approach; Mahy, B.W.J., Ed.; IRL Press: Oxford, UK, 1985; pp. 207–236. [Google Scholar]
- Prezioso, C.; Scribano, D.; Rodio, D.M.; Ambrosi, C.; Trancassini, M.; Palamara, A.T.; Pietropaolo, V. COS-7-based model: Methodological approach to study John Cunningham virus replication cycle. Virol. J. 2018, 15, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbue, S.; Branchetti, E.; Boldorini, R.; Vago, L.; Zerbi, P.; Veggiani, C.; Tremolada, S.; Ferrante, P. Presence and expression of JCV early gene large T Antigen in the brains of immunocompromised and immunocompetent individuals. J. Med. Virol. 2008, 80, 2147–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, R.K.; Bugawan, T.L.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature 1986, 324, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.; Nana, H.M.; Ngane, R.A.; Gatsing, D.; Tchinda, A.T.; Rovito, R.; Cagno, V.; Cagliero, C.; Boyom, F.F.; Rubiolo, P.; et al. In vitro anti-Herpes simplex virus activity of crude extract of the roots of Nauclea latifolia Smith (Rubiaceae). BMC Complement. Altern. Med. 2013, 13, 266. [Google Scholar] [CrossRef]
- Rosenthal, K.S.; Perez, R.; Hodnichak, C. Inhibition of herpes simplex virus type 1 penetration by cytochalasins B and D. J. Gen. Virol. 1985, 66 Pt 7, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yan, J.; Lu, G.; Guo, Z.; Fan, Z.; Wang, J.; Shi, Y.; Qi, J.; Gao, G.F. Binding of herpes simplex virus glycoprotein D to nectin-1 exploits host cell adhesion. Nat. Commun. 2011, 2, 577. [Google Scholar] [CrossRef]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | H Score a | RMSD b | VdW c | Eletr d | Desolv e | Restr f | BSA g |
|---|---|---|---|---|---|---|---|
| gB/TG | −178.0 ± 9.7 | 0.3 ± 0.2 | −66.7 ± 2.1 | −27.9 ± 13.0 | −105.8 ± 10.4 | 0.9 ± 0.2 | 1795 ± 14 |
| gD/TG | −93.2 ± 1.6 | 0.3 ± 0.2 | −51.0 ± 2.3 | −48.9 ± 6.3 | −32.5 ± 1.0 | 0.0 ± 0.0 | 1200 ± 21 |
| gH-gL/TG | −93.9 ± 1.6 | 0.3 ± 0.2 | −72.6 ± 2.8 | −21.6 ± 5.4 | −17.0 ± 2.1 | 0.2 ± 0.2 | 1714 ± 48 |
| VP1/TG | −48.7 ± 6.6 | 0.2 ± 0.1 | −70.7 ± 1.3 | −62.0 ± 10.8 | 33.8 ± 5.3 | 5.4 ± 0.9 | 2134 ± 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcocci, M.E.; Jackowska, B.G.; Prezioso, C.; Protto, V.; De Angelis, M.; Di Leva, F.S.; Casciaro, B.; Carotenuto, A.; Mangoni, M.L.; Palamara, A.T.; et al. The Inhibition of DNA Viruses by the Amphibian Antimicrobial Peptide Temporin G: A Virological Study Addressing HSV-1 and JPCyV. Int. J. Mol. Sci. 2022, 23, 7194. https://doi.org/10.3390/ijms23137194
Marcocci ME, Jackowska BG, Prezioso C, Protto V, De Angelis M, Di Leva FS, Casciaro B, Carotenuto A, Mangoni ML, Palamara AT, et al. The Inhibition of DNA Viruses by the Amphibian Antimicrobial Peptide Temporin G: A Virological Study Addressing HSV-1 and JPCyV. International Journal of Molecular Sciences. 2022; 23(13):7194. https://doi.org/10.3390/ijms23137194
Chicago/Turabian StyleMarcocci, Maria Elena, Bianka Gabriela Jackowska, Carla Prezioso, Virginia Protto, Marta De Angelis, Francesco Saverio Di Leva, Bruno Casciaro, Alfonso Carotenuto, Maria Luisa Mangoni, Anna Teresa Palamara, and et al. 2022. "The Inhibition of DNA Viruses by the Amphibian Antimicrobial Peptide Temporin G: A Virological Study Addressing HSV-1 and JPCyV" International Journal of Molecular Sciences 23, no. 13: 7194. https://doi.org/10.3390/ijms23137194