
Transcriptomic and Functional Evidence for Differential Effects of MCF-7 Breast Cancer Cell-Secretome on Vascular and Lymphatic Endothelial Cell Growth


Abstract
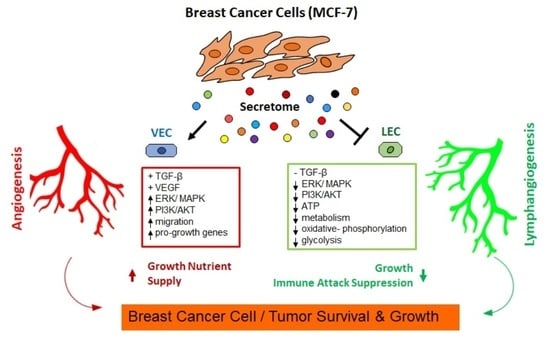
:
1. Introduction
2. Results
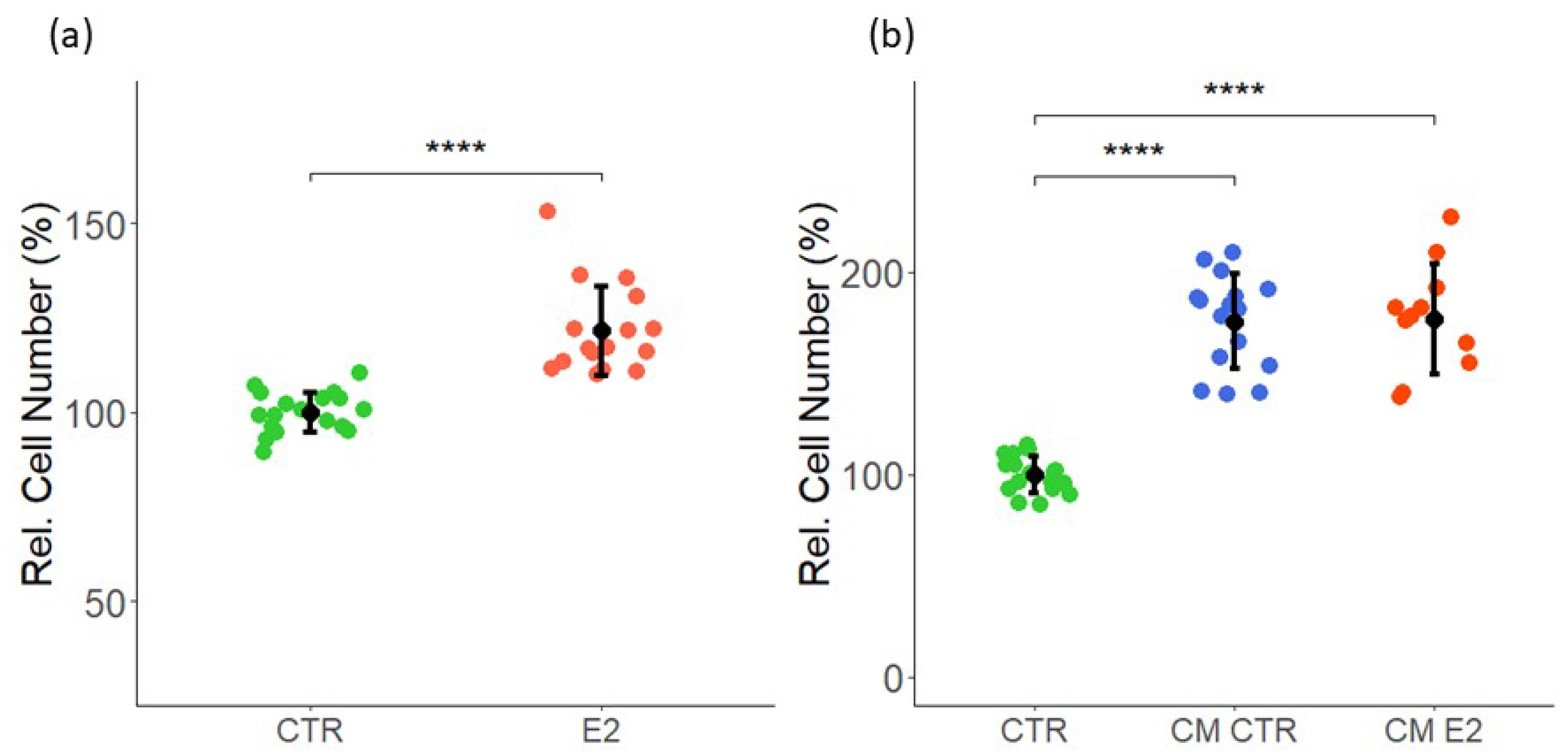
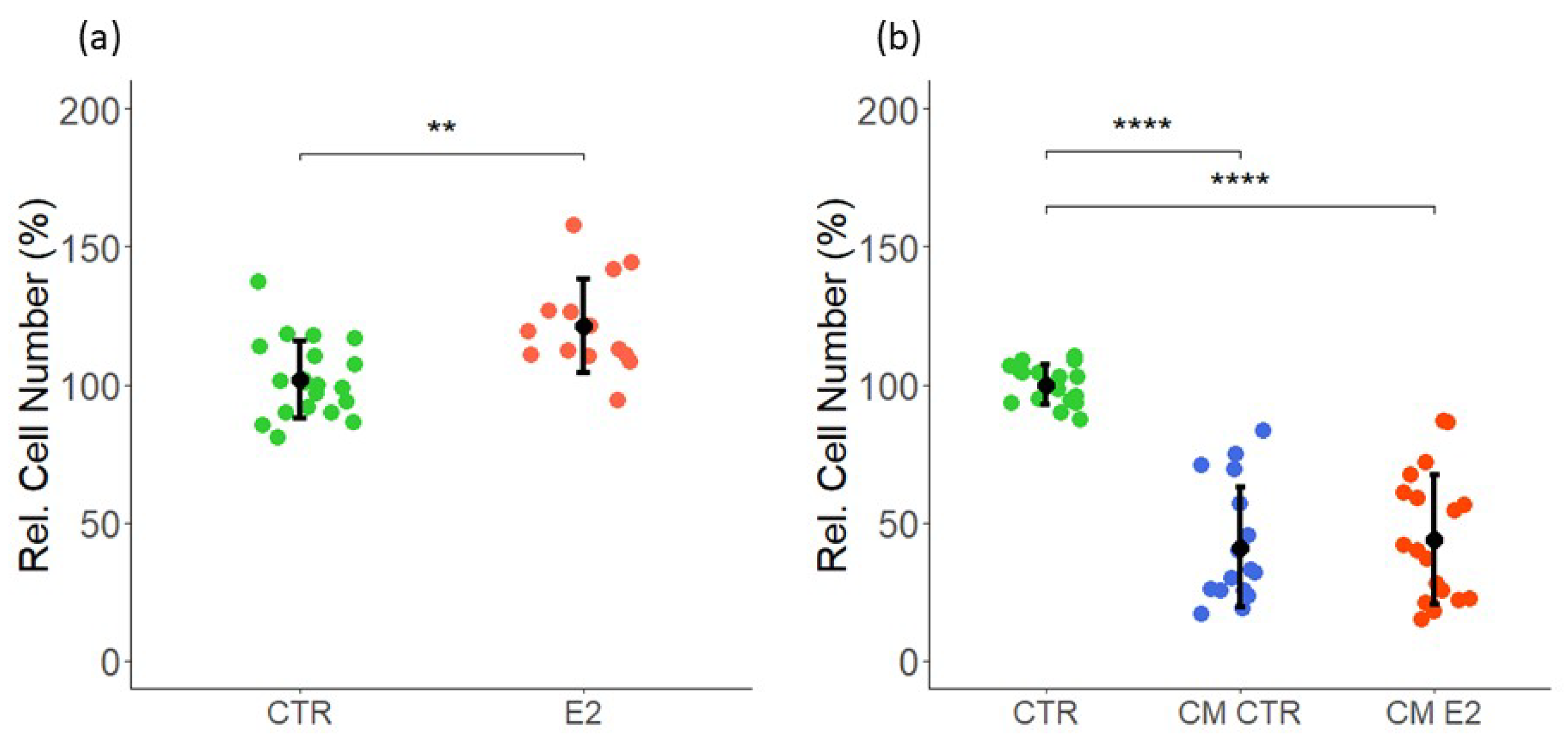
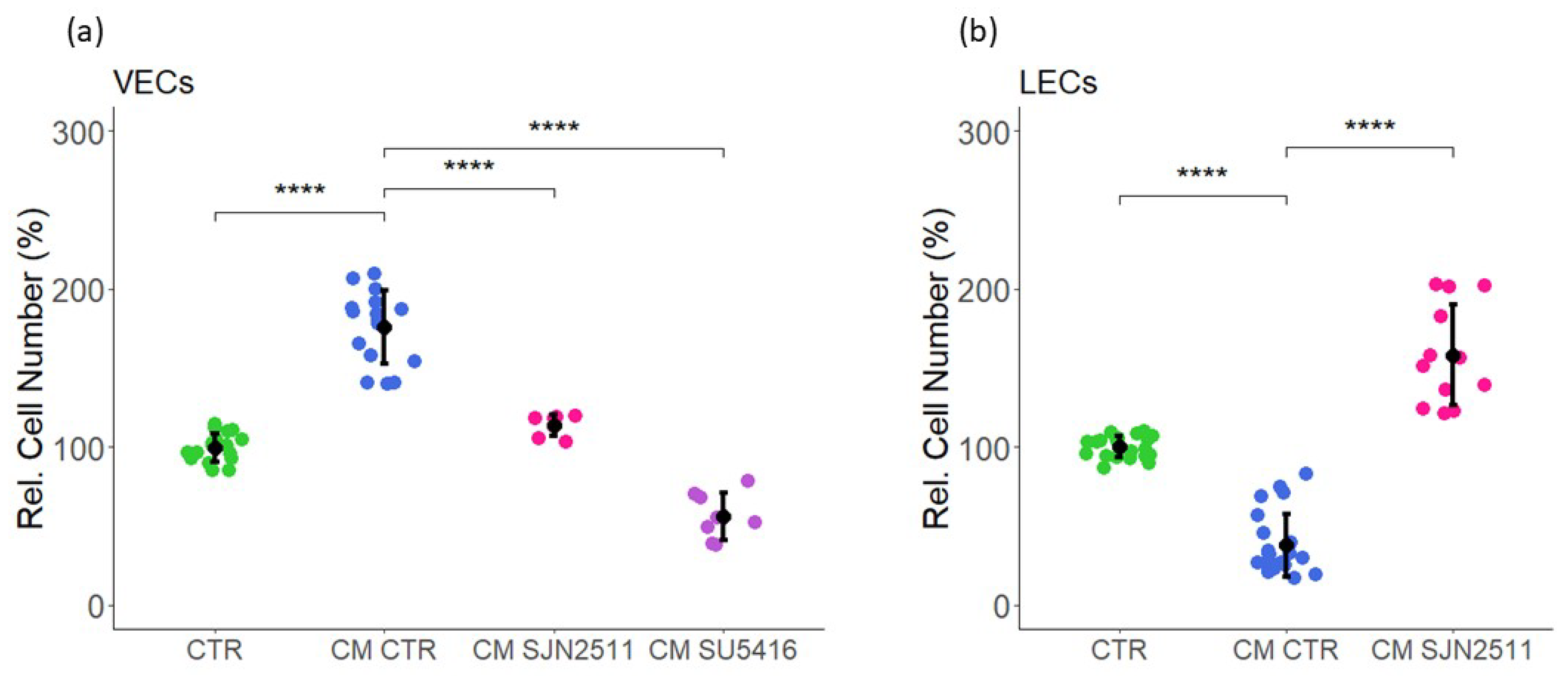
2.1. Effect of Estrogen and CM on VEC Proliferation
2.2. Effect of Estrogen and CM on LEC Proliferation
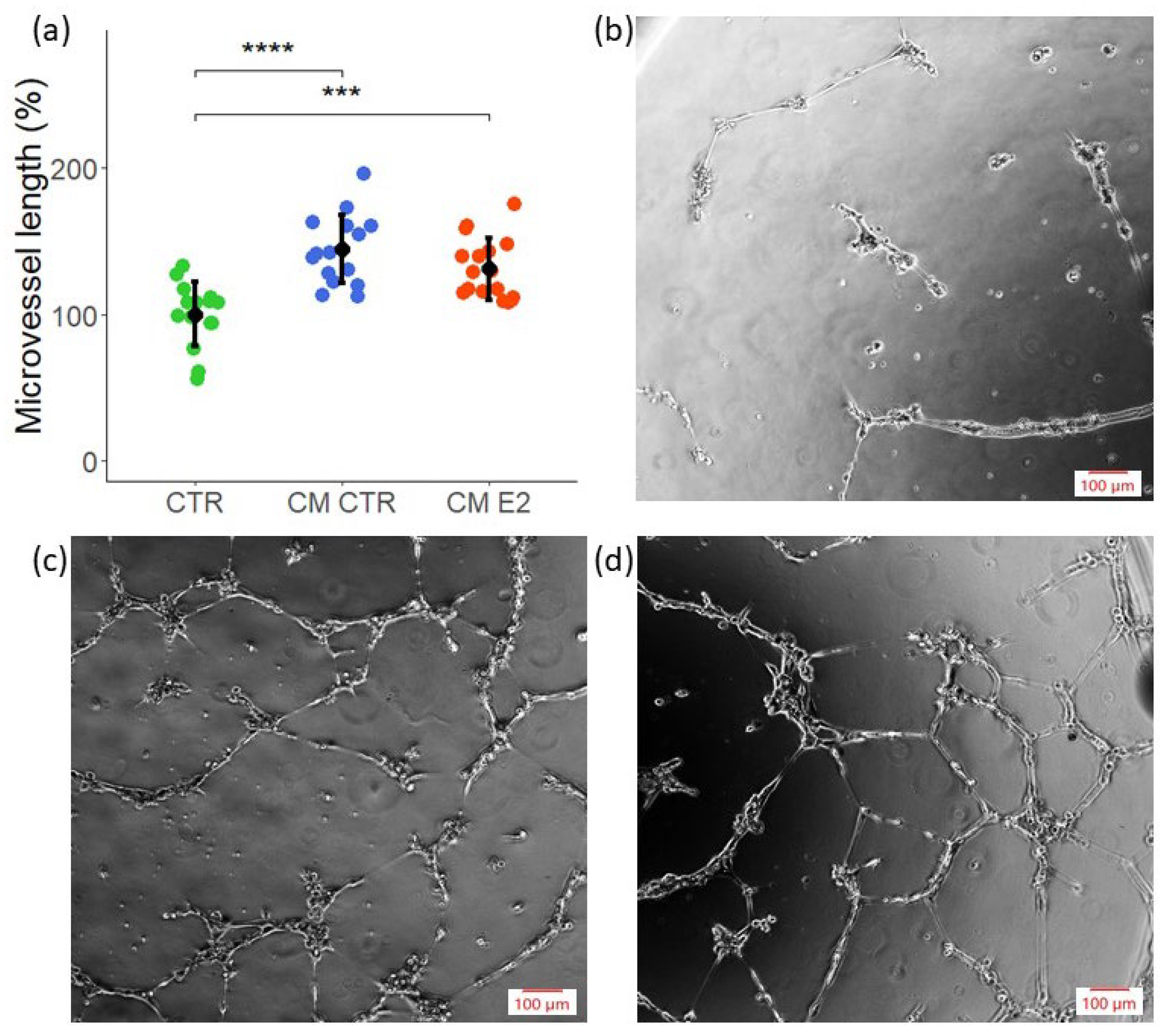
2.3. CM Exhibits Angiogenic Potential in Tube Formation Assay
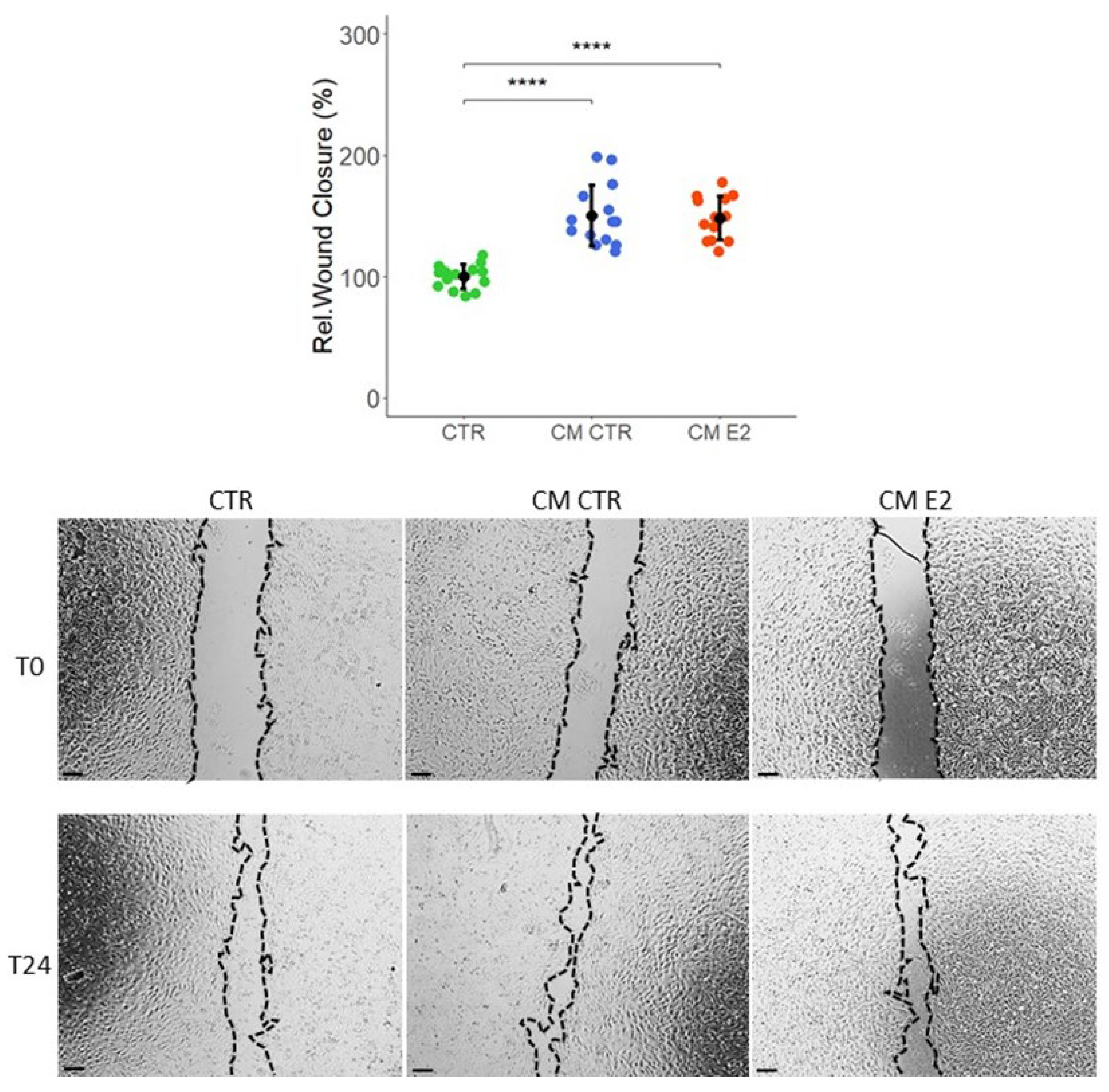
2.4. Effect of Conditioned Media on Cell Migration
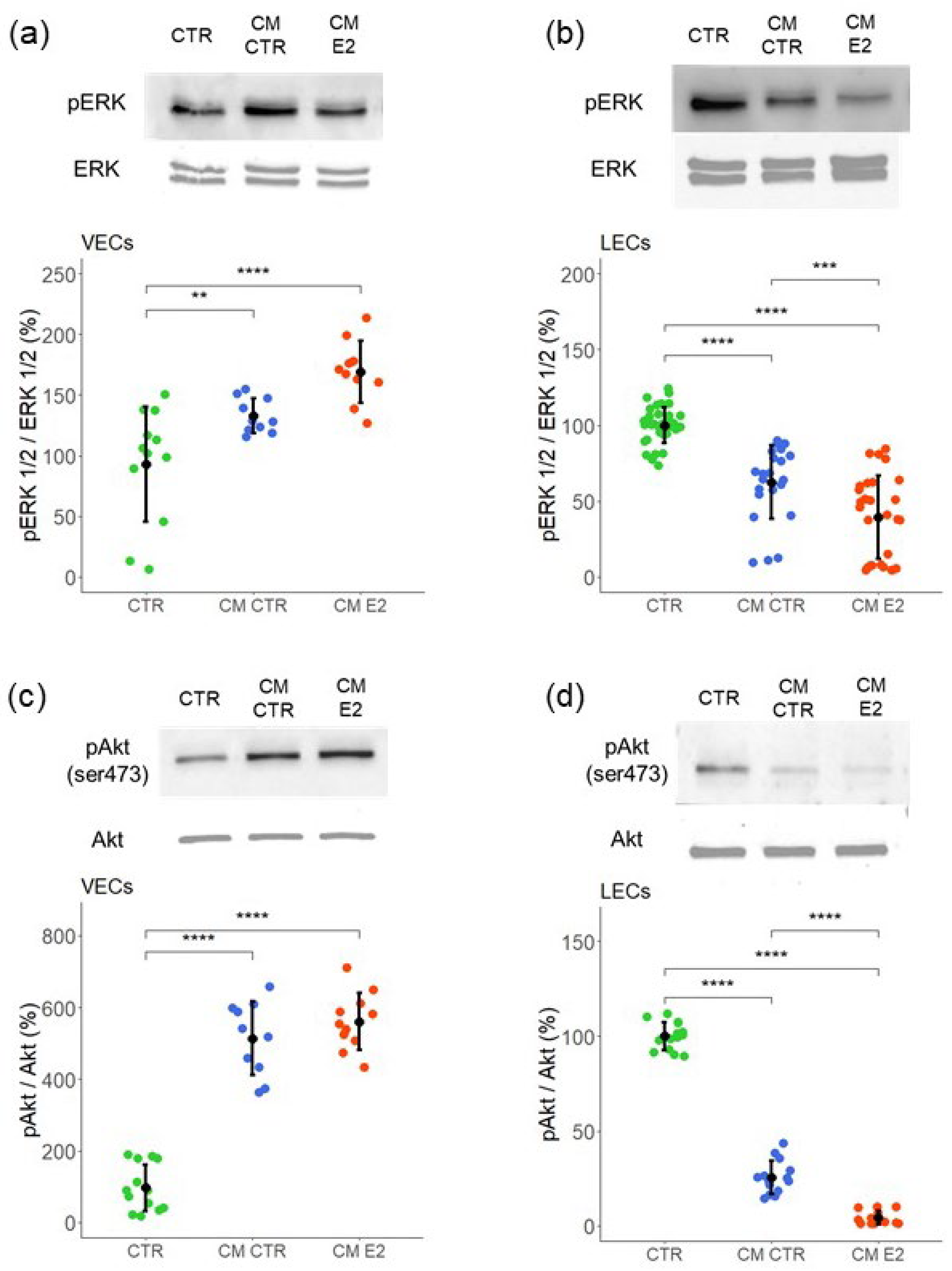
2.5. Role of MAPK (ERK1/2) and PI3K-Akt Pathways in Mediating the Effects of CM on VEC and LEC Proliferation
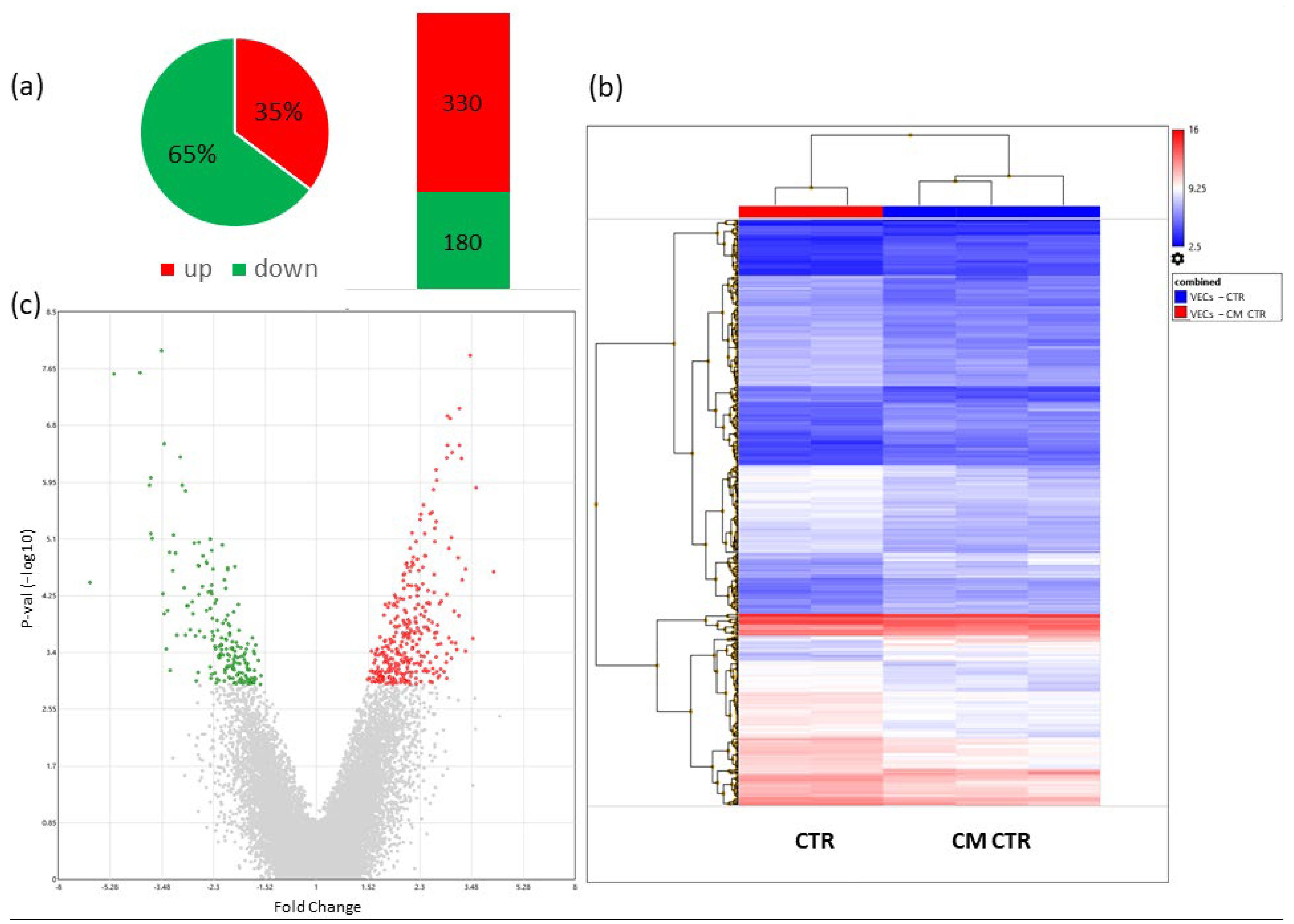
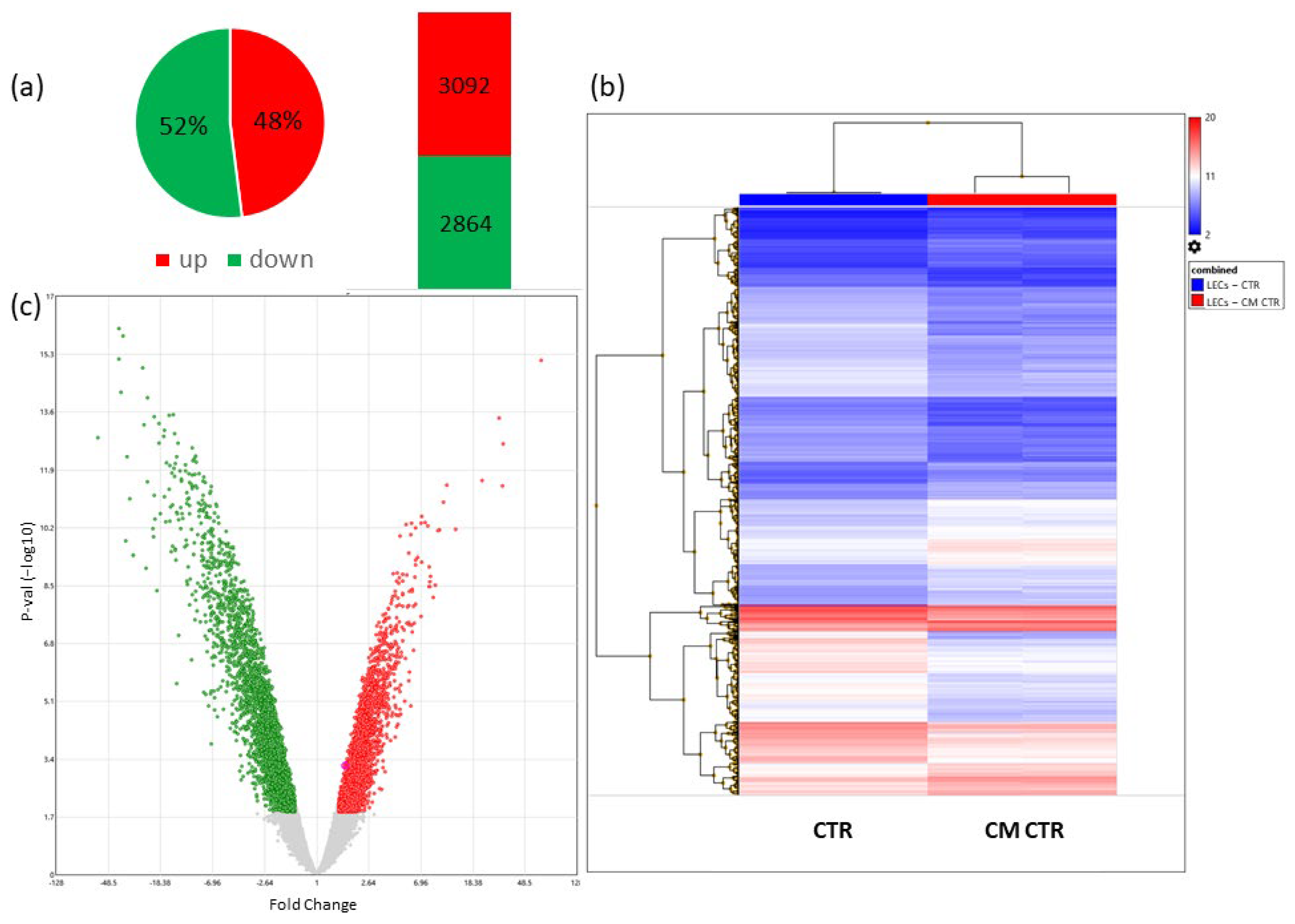
2.6. Microarray Analysis
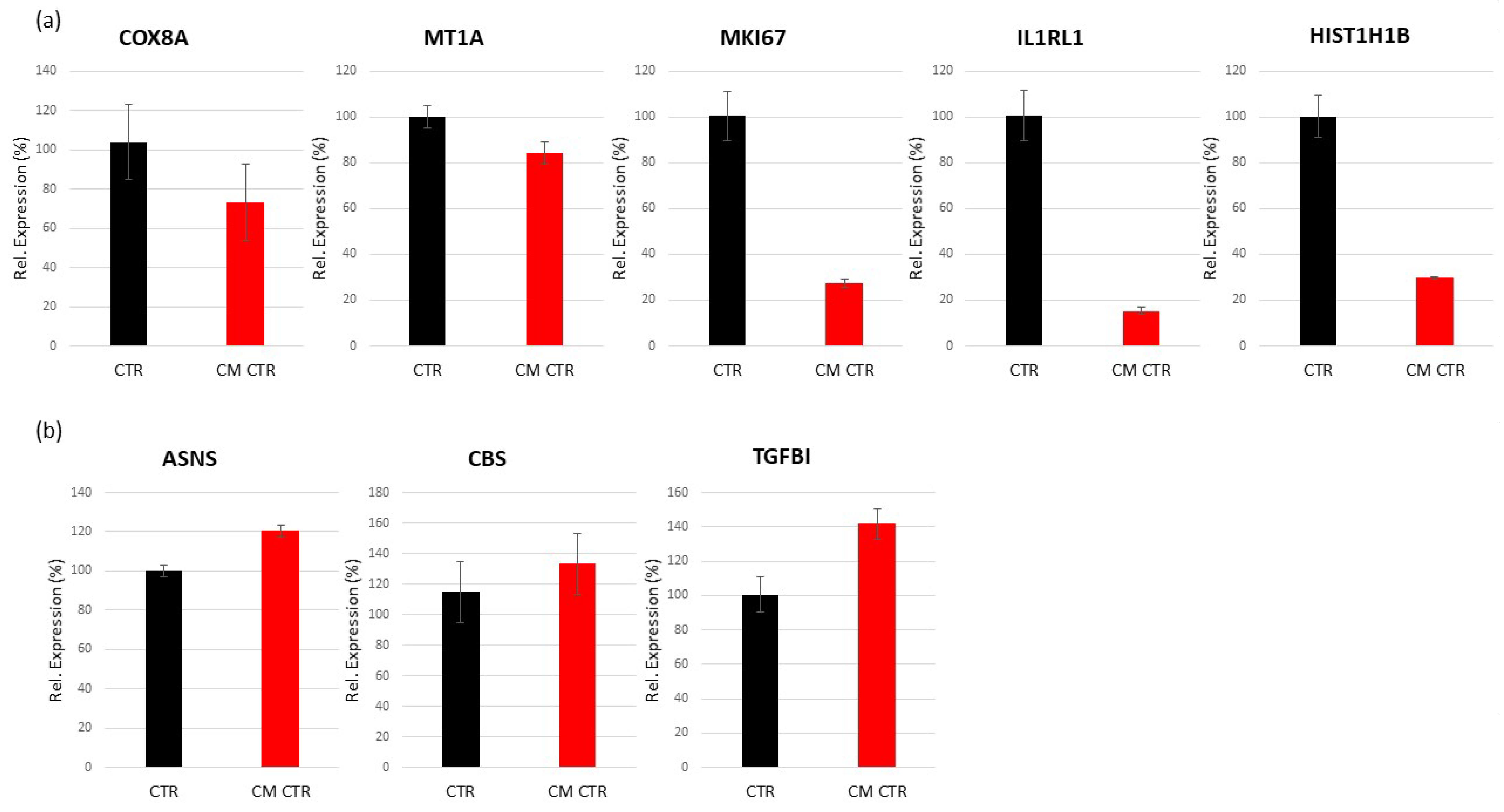
2.6.1. Differentially Regulated Genes in VECs Cultured in CM
Validation of Regulated Genes by Real-Time PCR
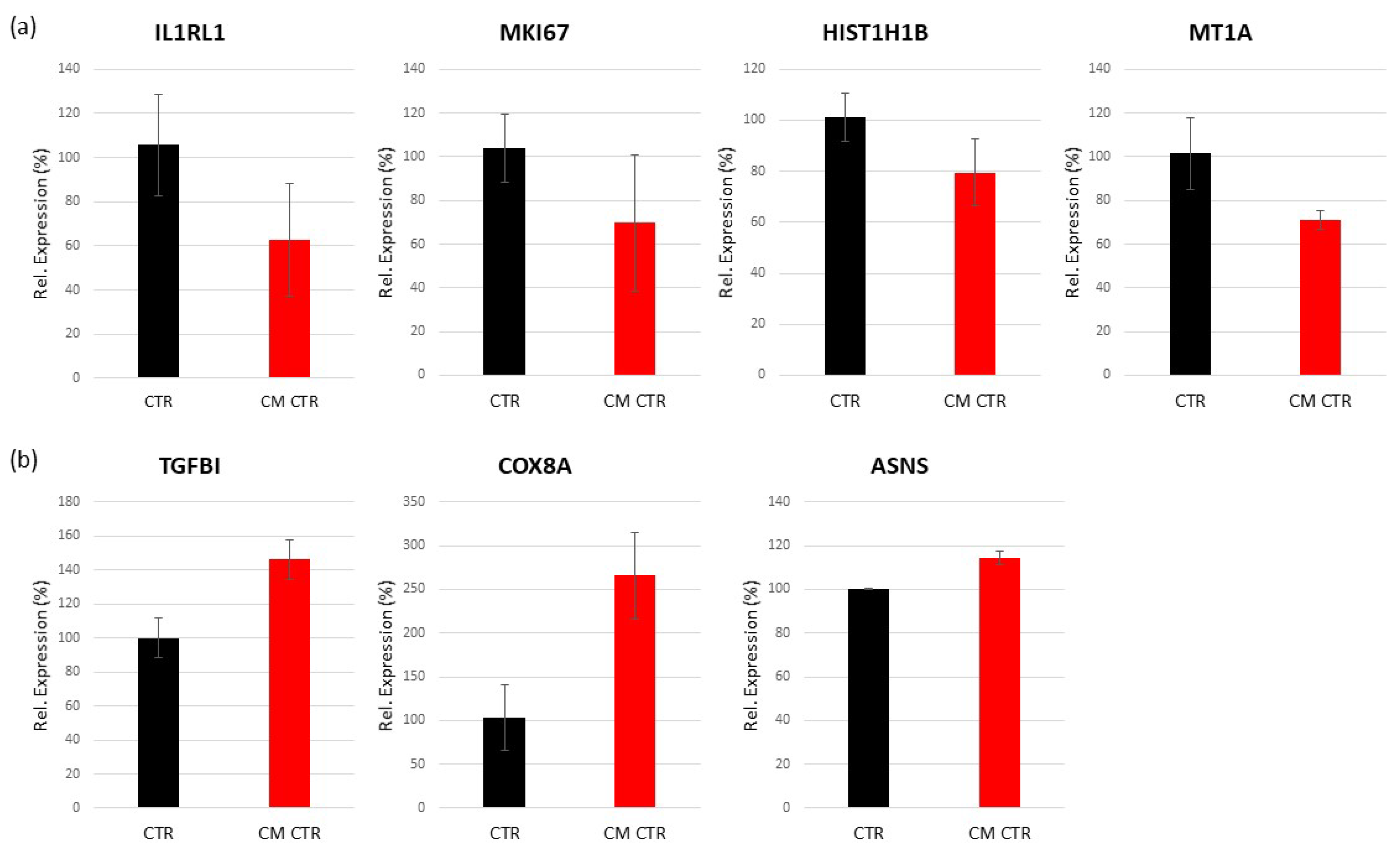
2.6.2. Differentially Regulated Genes in LECs Cultured in CM
Validation of Regulated Genes by Real-Time PCR
2.6.3. Pathway Enrichment Analysis of DRGs in VECs
Genes’ Identification in Biological Pathways
2.6.4. Pathway Enrichment Analysis of DRGs in LECs
Genes’ Identification in Biological Pathways
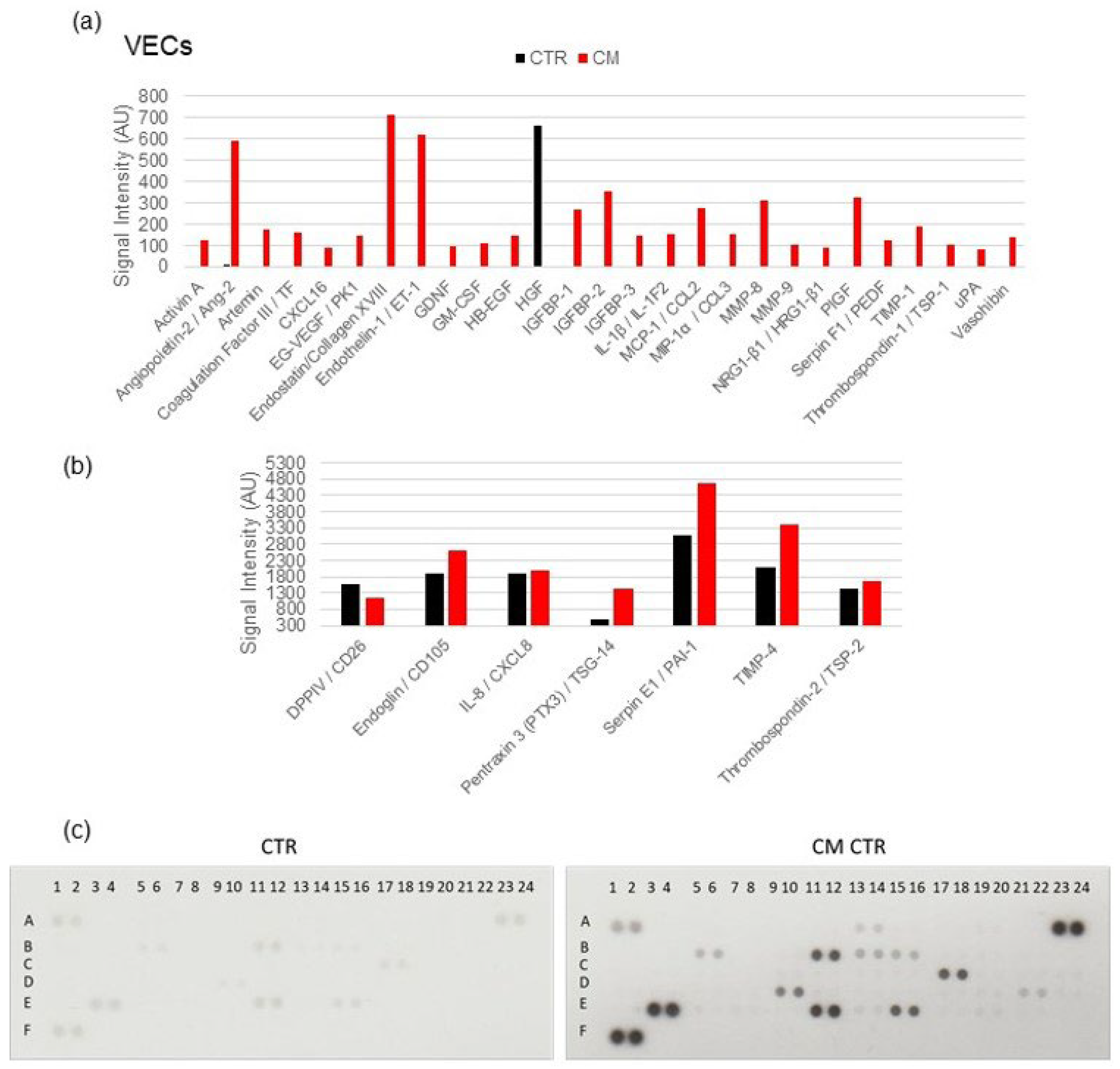
2.7. Angiogenesis Proteome Profiler in VECs
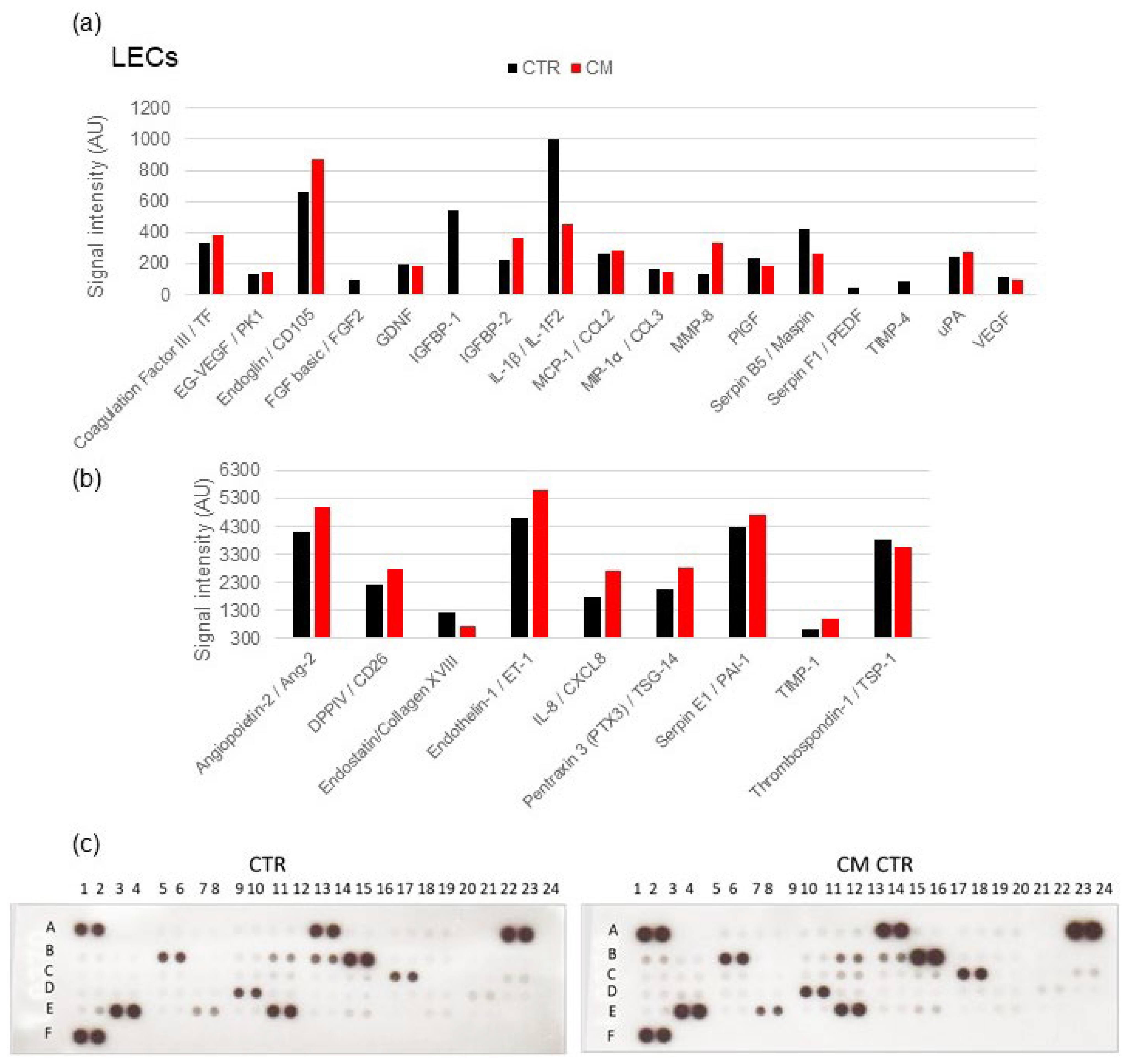
2.8. Angiogenesis Proteome Profiler in LECs

2.9. Role of TGF-β and VEGF on MCF-7-CM-Mediated Growth in VECs and LECs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
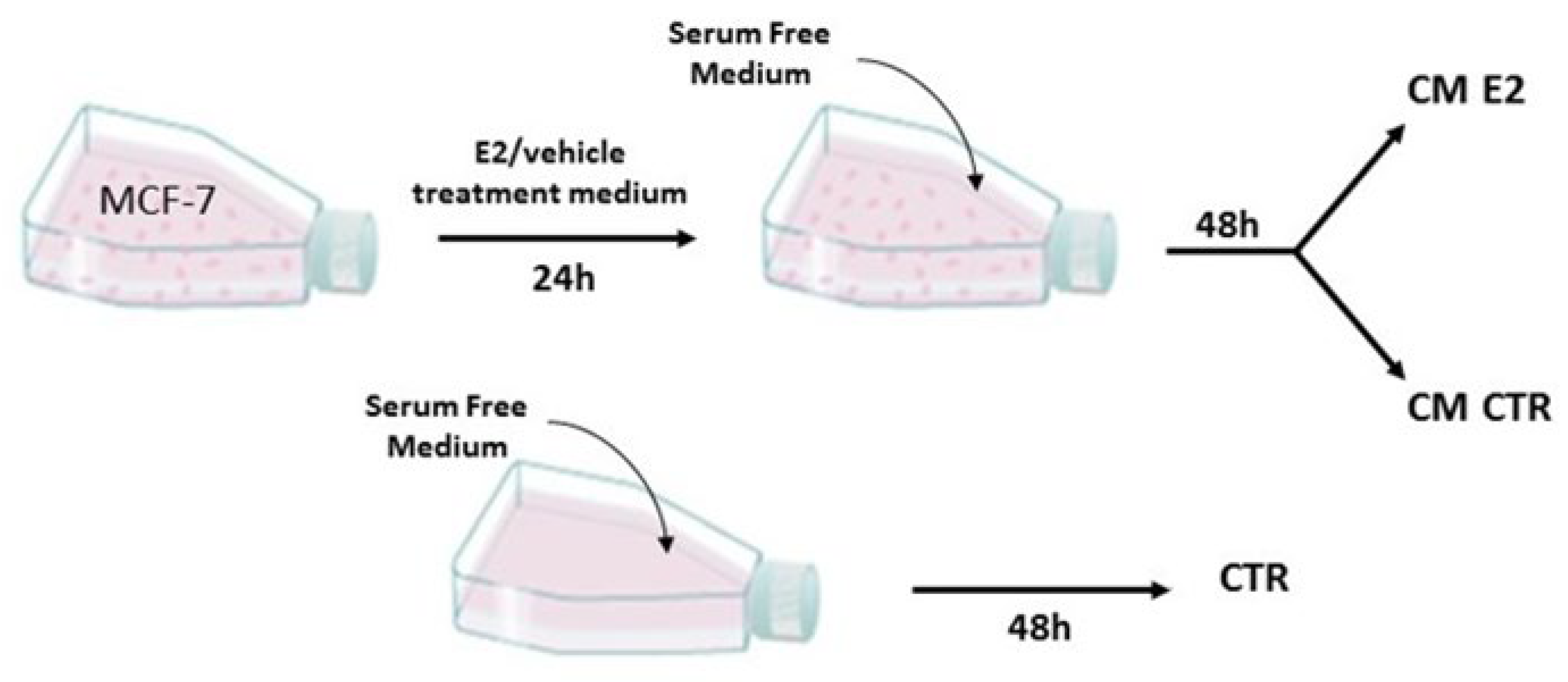
4.2. Conditioned Medium (CM)
4.3. Cell Proliferation Assay
4.4. Microvessel Formation Assay
4.5. Migration Studies
4.6. Western Blotting
4.7. Microarray
4.8. Quantitative RT-PCR
4.9. Angiogenesis Proteome Array
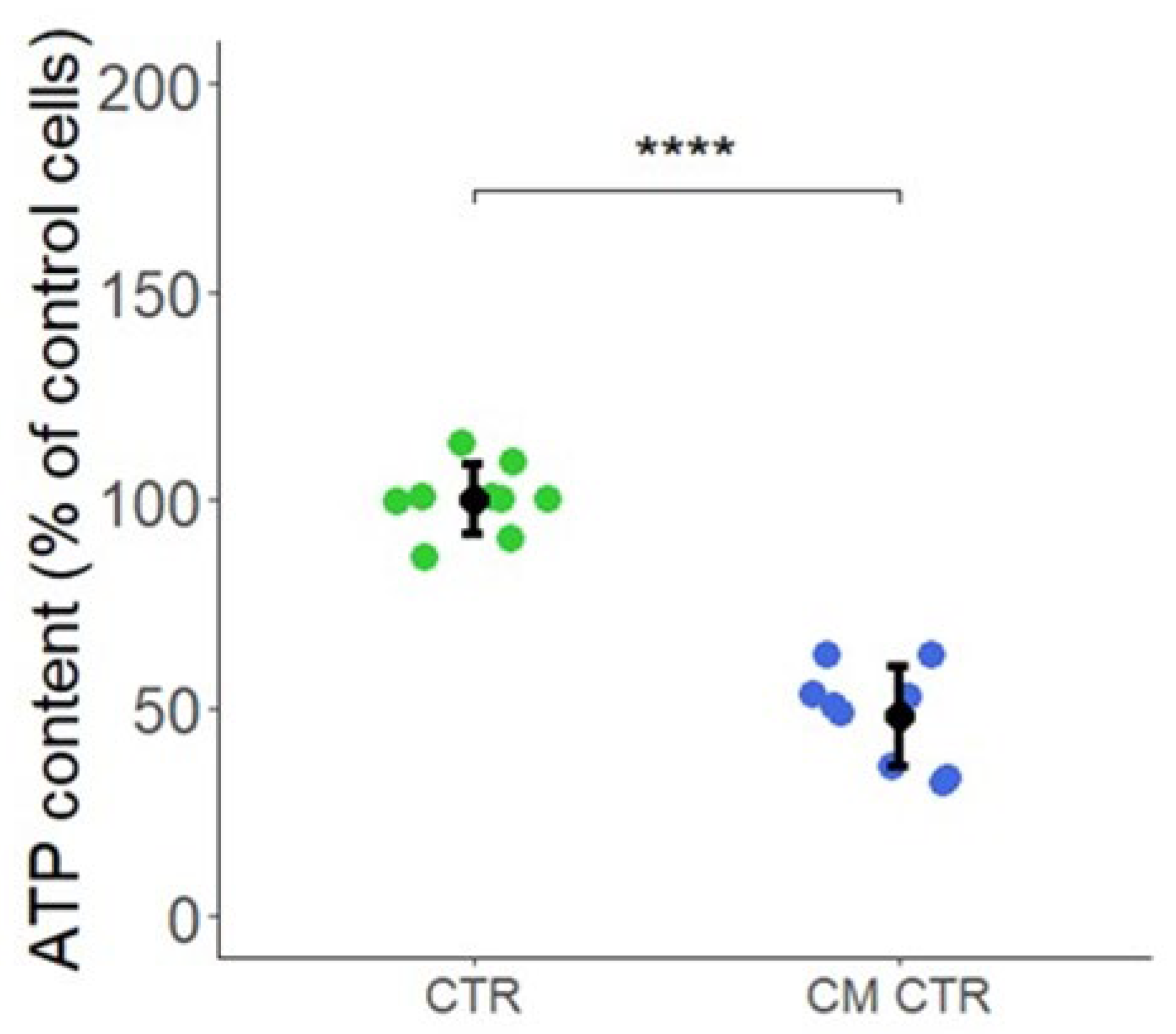
4.10. Intracellular ATP Content
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Enayatrad, M.; Amoori, N.; Salehiniya, H. Epidemiology and trends in breast cancer mortality in iran. Iran. J. Public Health 2015, 44, 430–431. [Google Scholar] [PubMed]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Physician 2010, 81, 1339–1346. [Google Scholar] [PubMed]
- Fahad Ullah, M. Breast Cancer: Current Perspectives on the Disease Status. Adv. Exp. Med. Biol. 2019, 1152, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Nishiyama, M.; Kasuya, Y.; Kimura, S.; Ishikura, H. Pathophysiology of tumor neovascularization. Vasc. Health Risk Manag. 2005, 1, 277–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, Y.; Uchino, J.; Chihara, Y.; Tamiya, N.; Kaneko, Y.; Yamada, T.; Takayama, K. Tumor Neovascularization and Developments in Therapeutics. Cancers 2019, 11, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Miranda, L.; Yaniv, K. Cellular Origins of the Lymphatic Endothelium: Implications for Cancer Lymphangiogenesis. Front. Physiol. 2020, 11, 577584. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncol. 2015, 20, 660–673. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Weidner, N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res. Treat. 1995, 36, 169–180. [Google Scholar] [CrossRef]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Ran, S.; Volk, L.; Hall, K.; Flister, M.J. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology 2010, 17, 229–251. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Lee, S.J.; Koskimaki, J.E.; Han, Z.; Pandey, N.B.; Popel, A.S. Inhibition of breast cancer growth and metastasis by a biomimetic peptide. Sci. Rep. 2014, 4, 7139. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.M.; Luo, B.; Ma, R.; Luo, X.X.; Chen, Y.S.; Li, Y. Lymphatic Endothelial Markers and Tumor Lymphangiogenesis Assessment in Human Breast Cancer. Diagnostics 2021, 12, 4. [Google Scholar] [CrossRef]
- Rezzola, S.; Sigmund, E.C.; Halin, C.; Ronca, R. The lymphatic vasculature: An active and dynamic player in cancer progression. Med. Res. Rev. 2022, 42, 576–614. [Google Scholar] [CrossRef]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—Partnering up with the immune system? Nat. Rev. Immunol. 2022, 1–13. [Google Scholar] [CrossRef]
- Jalkanen, S.; Salmi, M. Lymphatic endothelial cells of the lymph node. Nat. Rev. Immunol. 2020, 20, 566–578. [Google Scholar] [CrossRef]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Stachura, J.; Wachowska, M.; Kilarski, W.W.; Güç, E.; Golab, J.; Muchowicz, A. The dual role of tumor lymphatic vessels in dissemination of metastases and immune response development. Oncoimmunology 2016, 5, e1182278. [Google Scholar] [CrossRef] [Green Version]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [Green Version]
- Cunnick, G.H.; Jiang, W.G.; Douglas-Jones, T.; Watkins, G.; Gomez, K.F.; Morgan, M.J.; Subramanian, A.; Mokbel, K.; Mansel, R.E. Lymphangiogenesis and lymph node metastasis in breast cancer. Mol. Cancer 2008, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Zarzynska, J.M. Two faces of TGF-beta1 in breast cancer. Mediat. Inflamm. 2014, 2014, 141747. [Google Scholar] [CrossRef]
- Biselli-Chicote, P.M.; Oliveira, A.R.; Pavarino, E.C.; Goloni-Bertollo, E.M. VEGF gene alternative splicing: Pro- and anti-angiogenic isoforms in cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 363–370. [Google Scholar] [CrossRef]
- Gasparini, G. Prognostic value of vascular endothelial growth factor in breast cancer. Oncologist 2000, 5 (Suppl. 1), 37–44. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, K.D.; Rohlenova, K.; Luo, Y.; Carmeliet, P. The metabolic engine of endothelial cells. Nat. Metab. 2019, 1, 937–946. [Google Scholar] [CrossRef]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gil, H.J.; Liu, X.; Diebold, L.P.; Morgan, M.A.; Oxendine-Burns, M.J.; Gao, P.; Chandel, N.S.; Oliver, G. Mitochondrial respiration controls the Prox1-Vegfr3 feedback loop during lymphatic endothelial cell fate specification and maintenance. Sci. Adv. 2021, 7, 7359. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Lee, E.; Pandey, N.B.; Popel, A.S. Pre-treatment of mice with tumor-conditioned media accelerates metastasis to lymph nodes and lungs: A new spontaneous breast cancer metastasis model. Clin. Exp. Metastasis 2014, 31, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Sovijit, W.; Sovijit, W.; Ishii, Y.; Kambe, J.; Fujita, T.; Watanabe, G.; Yamaguchi, H.; Nagaoka, K. Estrogen promotes increased breast cancer cell proliferation and migration through downregulation of CPEB1 expression. Biochem. Biophys. Res. Commun. 2021, 534, 871–876. [Google Scholar] [CrossRef]
- Tian, J.M.; Ran, B.; Zhang, C.L.; Yan, D.M.; Li, X.H. Estrogen and progesterone promote breast cancer cell proliferation by inducing cyclin G1 expression. Braz. J. Med. Biol. Res. 2018, 51, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Cheung, M.; Testa, J.R. Diverse mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets 2013, 13, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Futakuchi, M.; Nannuru, K.C.; Varney, M.L.; Sadanandam, A.; Nakao, K.; Asai, K.; Shirai, T.; Sato, S.Y.; Singh, R.K. Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009, 100, 71–81. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, X.; Guan, S.; Yan, Y.; Lin, H.; Hua, Z.C. Curcumin inhibits angiogenesis and improves defective hematopoiesis induced by tumor-derived VEGF in tumor model through modulating VEGF-VEGFR2 signaling pathway. Oncotarget 2015, 6, 19469–19482. [Google Scholar] [CrossRef] [Green Version]
- Oka, M.; Iwata, C.; Suzuki, H.I.; Kiyono, K.; Morishita, Y.; Watabe, T.; Komuro, A.; Kano, M.R.; Miyazono, K. Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood 2008, 111, 4571–4579. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Yip, C.H.; Rhodes, A. Estrogen and progesterone receptors in breast cancer. Future Oncol. 2014, 10, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Boneberg, E.M.; Legler, D.F.; Hoefer, M.M.; Ohlschlegel, C.; Steininger, H.; Füzesi, L.; Beer, G.M.; Dupont-Lampert, V.; Otto, F.; Senn, H.J.; et al. Angiogenesis and lymphangiogenesis are downregulated in primary breast cancer. Br. J. Cancer 2009, 101, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Pandey, N.B.; Popel, A.S. Lymphatic endothelial cells support tumor growth in breast cancer. Sci. Rep. 2014, 4, 5853. [Google Scholar] [CrossRef] [Green Version]
- Lucas, E.D.; Tamburini, B.A.J. Lymph Node Lymphatic Endothelial Cell Expansion and Contraction and the Programming of the Immune Response. Front. Immunol. 2019, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Brandi, J.; Manfredi, M.; Speziali, G.; Gosetti, F.; Marengo, E.; Cecconi, D. Proteomic approaches to decipher cancer cell secretome. Semin. Cell Dev. Biol. 2018, 78, 93–101. [Google Scholar] [CrossRef]
- El-Badawy, A.; Ghoneim, M.A.; Gabr, M.M.; Salah, R.A.; Mohamed, I.K.; Amer, M.; El-Badri, N. Cancer cell-soluble factors reprogram mesenchymal stromal cells to slow cycling, chemoresistant cells with a more stem-like state. Stem Cell Res. Ther. 2017, 8, 254. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Uchiba, M.; Okajima, K.; Oike, Y.; Ito, Y.; Fukudome, K.; Isobe, H.; Suda, T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ. Res. 2004, 95, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006, 16, 461–466. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Finley, S.D. Mechanistic insight into activation of MAPK signaling by pro-angiogenic factors. BMC Syst. Biol. 2018, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, F.A.; Silljé, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Raab, C.A.; Raab, M.; Becker, S.; Strebhardt, K. Non-mitotic functions of polo-like kinases in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188467. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, G.; Kong, C. FOXM1 participates in PLK1-regulated cell cycle progression in renal cell cancer cells. Oncol. Lett. 2016, 11, 2685–2691. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Sanhaji, M.; Matthess, Y.; Hörlin, A.; Lorenz, I.; Dötsch, C.; Habbe, N.; Waidmann, O.; Kurunci-Csacsko, E.; Firestein, R.; et al. PLK1 has tumor-suppressive potential in APC-truncated colon cancer cells. Nat. Commun. 2018, 9, 1106. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef]
- Zou, Y.; Ruan, S.; Jin, L.; Chen, Z.; Han, H.; Zhang, Y.; Jian, Z.; Lin, Y.; Shi, N.; Jin, H. CDK1, CCNB1, and CCNB2 are Prognostic Biomarkers and Correlated with Immune Infiltration in Hepatocellular Carcinoma. Int. Med. J. Exp. Clin. Res. 2020, 26, e925289. [Google Scholar] [CrossRef]
- Haneke, K.; Schott, J.; Lindner, D.; Hollensen, A.K.; Damgaard, C.K.; Mongis, C.; Knop, M.; Palm, W.; Ruggieri, A.; Stoecklin, G. CDK1 couples proliferation with protein synthesis. J. Cell Biol. 2020, 219, e201906147. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Sui, D.; You, D.; Yao, S.; Zhang, L.; Wang, Y.; Zhao, J.; Zhang, Y. MiR-29a-5p inhibits proliferation and invasion and induces apoptosis in endometrial carcinoma via targeting TPX2. Cell Cycle 2018, 17, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Yang, L.; Zhang, Z.; Yu, J.; Dai, L.; Gao, M.; Shang, Z.; Niu, Y. KIF20A Affects the Prognosis of Bladder Cancer by Promoting the Proliferation and Metastasis of Bladder Cancer Cells. Dis. Markers 2019, 2019, 4863182. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, H.; Li, J.; Wei, S.; Liu, X.; Wan, H.; Zheng, P.; Zheng, H. Effect of Ect2 Expression on the Growth of Triple-Negative Breast Cancer Cells with Paclitaxel Intervention. OncoTargets Ther. 2020, 13, 12905–12918. [Google Scholar] [CrossRef]
- Zhu, L.J.; Pan, Y.; Chen, X.Y.; Hou, P.F. BUB1 promotes proliferation of liver cancer cells by activating SMAD2 phosphorylation. Oncol. Lett. 2020, 19, 3506–3512. [Google Scholar] [CrossRef]
- Chen, J.; Liao, A.; Powers, E.N.; Liao, H.; Kohlstaedt, L.A.; Evans, R.; Holly, R.M.; Kim, J.K.; Jovanovic, M.; Ünal, E. Aurora B-dependent Ndc80 degradation regulates kinetochore composition in meiosis. Genes Dev. 2020, 34, 209–225. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Li, Y.; Petersen, R.B.; Huang, K. Targeting mitosis exit: A brake for cancer cell proliferation. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 179–191. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Reviews. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Vadivel Gnanasundram, S.; Fåhraeus, R. Translation Stress Regulates Ribosome Synthesis and Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3757. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B. DNA damage-triggered apoptosis: Critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem. Pharmacol. 2003, 66, 1547–1554. [Google Scholar] [CrossRef]
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef]
- Kitao, H.; Iimori, M.; Kataoka, Y.; Wakasa, T.; Tokunaga, E.; Saeki, H.; Oki, E.; Maehara, Y. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018, 109, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Cui, K.; Liu, C.; Li, X.; Zhang, Q.; Li, Y. Comprehensive characterization of the rRNA metabolism-related genes in human cancer. Oncogene 2020, 39, 786–800. [Google Scholar] [CrossRef]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Hua, Y.; Sun, C.; Zhang, Y.; Yang, F.; Yang, M.; Yang, Y.; Li, J.; Huang, X.; Wu, H.; et al. Relationship between tRNA-derived fragments and human cancers. Int. J. Cancer 2020, 147, 3007–3018. [Google Scholar] [CrossRef]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2020, 19, 112–129. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell. Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimeloe, S.; Gubser, P.; Loeliger, J.; Frick, C.; Develioglu, L.; Fischer, M.; Marquardsen, F.; Bantug, G.R.; Thommen, D.; Lecoultre, Y.; et al. Tumor-derived TGF-β inhibits mitochondrial respiration to suppress IFN-γ production by human CD4(+) T cells. Sci. Signal. 2019, 12, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Kim, J.; Ahn, J.H.; Hong, Y.K.; Mäkinen, T.; Lim, D.S.; Koh, G.Y. YAP and TAZ Negatively Regulate Prox1 During Developmental and Pathologic Lymphangiogenesis. Circ. Res. 2019, 124, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, Y.; Zhang, Y.; Zhang, L.; Wang, Z.; Zhang, X.; Gui, L.; Huang, J. IFI6 Inhibits Apoptosis via Mitochondrial-Dependent Pathway in Dengue Virus 2 Infected Vascular Endothelial Cells. PLoS ONE 2015, 10, e0132743. [Google Scholar] [CrossRef] [Green Version]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wei, F.; Zhou, C.; Sumida, T.; Hamakawa, H.; Hu, Y.; Liu, S. Silencing Id-1 inhibits lymphangiogenesis through down-regulation of VEGF-C in oral squamous cell carcinoma. Oral Oncol. 2011, 47, 27–32. [Google Scholar] [CrossRef]
- Shang, M.; Yang, H.; Yang, R.; Chen, T.; Fu, Y.; Li, Y.; Fang, X.; Zhang, K.; Zhang, J.; Li, H.; et al. The folate cycle enzyme MTHFD2 induces cancer immune evasion through PD-L1 up-regulation. Nat. Commun. 2021, 12, 1940. [Google Scholar] [CrossRef]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat. Commun. 2014, 5, 4715. [Google Scholar] [CrossRef] [Green Version]
- Scavelli, C.; Vacca, A.; Di Pietro, G.; Dammacco, F.; Ribatti, D. Crosstalk between angiogenesis and lymphangiogenesis in tumor progression. Leukemia 2004, 18, 1054–1058. [Google Scholar] [CrossRef]
- Song, S.H.; Kim, K.L.; Lee, K.A.; Suh, W. Tie1 regulates the Tie2 agonistic role of angiopoietin-2 in human lymphatic endothelial cells. Biochem. Biophys. Res. Commun. 2012, 419, 281–286. [Google Scholar] [CrossRef]
- Presta, M.; Foglio, E.; Churruca Schuind, A.; Ronca, R. Long Pentraxin-3 Modulates the Angiogenic Activity of Fibroblast Growth Factor-2. Front. Immunol. 2018, 9, 2327. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.C.; Lee, T.H.; Tang, C.H.; Chen, L.P.; Chen, W.C.; Lee, M.S.; Chen, P.C.; Lin, C.Y.; Chi, C.W.; Chen, Y.J.; et al. Phomaketide A Inhibits Lymphangiogenesis in Human Lymphatic Endothelial Cells. Mar. Drugs 2019, 17, 215. [Google Scholar] [CrossRef] [Green Version]
- Kajiyama, H.; Kikkawa, F.; Suzuki, T.; Shibata, K.; Ino, K.; Mizutani, S. Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res. 2002, 62, 2753–2757. [Google Scholar]
- Pethiyagoda, C.L.; Welch, D.R.; Fleming, T.P. Dipeptidyl peptidase IV (DPPIV) inhibits cellular invasion of melanoma cells. Clin. Exp. Metastasis 2000, 18, 391–400. [Google Scholar] [CrossRef]
- Kyzas, P.A.; Agnantis, N.J.; Stefanou, D. Endoglin (CD105) as a prognostic factor in head and neck squamous cell carcinoma. Virchows Arch. 2006, 448, 768–775. [Google Scholar] [CrossRef]
- Duff, S.E.; Li, C.; Garland, J.M.; Kumar, S. CD105 is important for angiogenesis: Evidence and potential applications. FASEB J. 2003, 17, 984–992. [Google Scholar] [CrossRef]
- Juurikka, K.; Butler, G.S.; Salo, T.; Nyberg, P.; Åström, P. The Role of MMP8 in Cancer: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 4506. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Pufe, T.; Petersen, W.J.; Miosge, N.; Goldring, M.B.; Mentlein, R.; Varoga, D.J.; Tillmann, B.N. Endostatin/collagen XVIII--an inhibitor of angiogenesis--is expressed in cartilage and fibrocartilage. Matrix Biol. 2004, 23, 267–276. [Google Scholar] [CrossRef]
- Brideau, G.; Mäkinen, M.J.; Elamaa, H.; Tu, H.; Nilsson, G.; Alitalo, K.; Pihlajaniemi, T.; Heljasvaara, R. Endostatin overexpression inhibits lymphangiogenesis and lymph node metastasis in mice. Cancer Res. 2007, 67, 11528–11535. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Cho, C.S.; Kim, W.U. Role of placenta growth factor in cancer and inflammation. Exp. Mol. Med. 2012, 44, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Cheng, H.; Cai, J.; Cai, L.; Zhang, J.; Wang, Z. PlGF expression in pre-invasive and invasive lesions of uterine cervix is associated with angiogenesis and lymphangiogenesis. Apmis 2009, 117, 831–838. [Google Scholar] [CrossRef]
- Yang, S.F.; Yeh, C.B.; Chou, Y.E.; Lee, H.L.; Liu, Y.F. Serpin peptidase inhibitor (SERPINB5) haplotypes are associated with susceptibility to hepatocellular carcinoma. Sci. Rep. 2016, 6, 26605. [Google Scholar] [CrossRef]
- Shi, H.Y.; Liang, R.; Templeton, N.S.; Zhang, M. Inhibition of breast tumor progression by systemic delivery of the maspin gene in a syngeneic tumor model. Mol. Ther. 2002, 5, 755–761. [Google Scholar] [CrossRef]
- McColl, B.K.; Stacker, S.A.; Achen, M.G. Molecular regulation of the VEGF family -- inducers of angiogenesis and lymphangiogenesis. Apmis 2004, 112, 463–480. [Google Scholar] [CrossRef]
- Shibuya, M. VEGF-VEGFR System as a Target for Suppressing Inflammation and other Diseases. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 135–144. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Description | Log2 FC | FDR p-Value |
|---|---|---|---|
| TGFBI | transforming growth factor, beta-induced, 68 kDa | 2.05 | 0.0073 |
| ABCA1 | ATP binding cassette, subfamily A, member 1 | 1.85 | 0.0015 |
| EFNA1 | ephrin-A1 | 1.82 | 0.0251 |
| TMEM63B | transmembrane protein 63B | 1.78 | 0.0001 |
| CCL20 | chemokine (C-C motif) ligand 20 | 1.73 | 0.0072 |
| WDTC1 | WD and tetratricopeptide repeats 1 | 1.73 | 0.0308 |
| EDEM1 | ER degradation enhancer, mannosidase alpha-like 1 | 1.69 | 0.009 |
| NISCH | nischarin | 1.68 | 0.0008 |
| SORT1 | sortilin 1 | 1.66 | 0.0007 |
| MGP | matrix Gla protein | 1.65 | 0.0004 |
| Gene Symbol | Description | Log2 FC | FDR p-Value |
|---|---|---|---|
| IL1RL1 | interleukin 1 receptor-like 1 | −2.64 | 0.0096 |
| MKI67 | marker of proliferation Ki-67 | −2.36 | 0.0001 |
| HIST1H1B | histone cluster 1, H1b | −2.05 | 0.0001 |
| RRM2 | ribonucleotide reductase M2 | −1.95 | 0.0014 |
| ARHGAP11B; ARHGAP11A | Rho GTPase activating protein 11B Rho GTPase activating protein 11A | −1.93 | 0.0013 |
| KIF20B | kinesin family member 20B | −1.93 | 0.0045 |
| ANLN | anillin actin binding protein | −1.91 | 0.0048 |
| TOP2A | topoisomerase (DNA) II alpha | −1.8 | 0.0001 |
| SHCBP1 | SHC SH2-domain binding protein 1 | −1.79 | 0.0117 |
| NCAPG | non-SMC condensin I complex subunit G | −1.77 | 0.0007 |
| Gene Symbol | Description | Log2 FC | FDR p-Value |
|---|---|---|---|
| ASNS | asparagine synthetase (glutamine-hydrolyzing) | 6.03 | 4.11 × 10−12 |
| CBS | cystathionine-beta-synthase | 5.02 | 2.28 × 10−10 |
| GYPC | glycophorin C (Gerbich blood group) | 5 | 1.53 × 10−9 |
| PSAT1 | phosphoserine aminotransferase 1 | 4.9 | 7.12 × 10−11 |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, methenyltetrahydrofolate cyclohydrolase | 4.45 | 1.29 × 10−9 |
| STC2 | stanniocalcin 2 | 3.73 | 1.27 × 10−8 |
| ANKRD1 | ankyrin repeat domain 1 (cardiac muscle) | 3.51 | 1.53 × 10−9 |
| HOOK3 | hook microtubule-tethering protein 3 | 3.4 | 3.45 × 10−9 |
| ANKRD36B | ankyrin repeat domain 36B | 3.29 | 1.30 × 10−8 |
| CHD1 | chromodomain helicase DNA binding protein 1 | 3.26 | 1.35 × 10−8 |
| Gene Symbol | Description | Log2 FC | FDR p-Value |
|---|---|---|---|
| COX8A | cytochrome c oxidase subunit VIIIA (ubiquitous) | −5.88 | 1.82 × 10−10 |
| MT1B | metallothionein 1B | −5.32 | 1.53 × 10−12 |
| MT1A | metallothionein 1A | −5.31 | 4.11 × 10−12 |
| MT1X | metallothionein 1X | −5.27 | 2.29 × 10−11 |
| SNRPD3 | small nuclear ribonucleoprotein D3 polypeptide | −5.21 | 1.53 × 10−12 |
| ID1 | inhibitor of DNA binding 1, HLH protein | −5.13 | 2.33 × 10−8 |
| MT1L | metallothionein 1L (gene/pseudogene) | −5.1 | 4.66 × 10−10 |
| COX7B | cytochrome c oxidase subunit VII B | −5.03 | 2.98 × 10−9 |
| IFI6 | interferon, alpha-inducible protein 6 | −4.93 | 4.91 × 10−8 |
| TMEM88 | transmembrane protein 88 | −4.68 | 5.32 × 10−12 |
| Pathway | Overlap | Adj. p-Value |
|---|---|---|
| BioPlanet | ||
| Polo-like kinase 1 (PLK1) pathway | 10/46 | 1.99 × 10−4 |
| Cell cycle | 30/453 | 0.001296906 |
| p73 transcription factor network | 10/79 | 0.010895079 |
| Aurora B signaling | 7/39 | 0.012713335 |
| Lysosome | 12/121 | 0.013134648 |
| SRF and microRNAs in smooth muscle differentiation and proliferation | 4/12 | 0.024709241 |
| M phase pathway | 10/96 | 0.024709241 |
| p53 activity regulation | 11/118 | 0.027319292 |
| Systemic lupus erythematosus | 12/139 | 0.027319292 |
| Phosphorylation of Emi1 | 3/6 | 0.029461214 |
| MicroRNA regulation of DNA damage response | 8/70 | 0.036366012 |
| Cyclin A/B1-associated events during G2/M transition | 4/15 | 0.036438016 |
| Kinesins | 5/27 | 0.036987861 |
| FOXM1 transcription factor network | 6/41 | 0.036987861 |
| Integrated breast cancer pathway | 12/152 | 0.036987861 |
| Mitotic prometaphase | 6/43 | 0.044760715 |
| GO Biological Process | ||
| mitotic spindle organization (GO:0007052) | 17/157 | 0.001553571 |
| microtubule cytoskeleton organization involved in mitosis (GO:1902850) | 15/128 | 0.001553571 |
| mitotic cytokinesis (GO:0000281) | 8/49 | 0.030235267 |
| KEGG | ||
| Lysosome | 12/128 | 1.25 × 10−4 |
| Gene Symbol | Description | Log2 FC | FDR p-Value |
|---|---|---|---|
| ANLN | anillin actin binding protein | −1.91 | 0.0048 |
| AP3B2 | adaptor-related protein complex 3, beta 2 subunit | −0.92 | 0.0372 |
| ATP6AP1 | ATPase, H+ transporting, lysosomal accessory protein 1 | 0.67 | 0.0482 |
| AURKA | aurora kinase A | −0.72 | 0.0321 |
| BUB1 | BUB1 mitotic checkpoint serine/threonine kinase | −1.53 | 0.0242 |
| CD68 | CD68 molecule | 1.29 | 0.0315 |
| CDC20 | cell division cycle 20 | −0.99 | 0.0298 |
| CEP55 | centrosomal protein 55kDa | −1.24 | 0.0149 |
| CLASP1 | cytoplasmic linker associated protein 1 | 1.01 | 0.0092 |
| CTSA | cathepsin A | 0.84 | 0.03 |
| DLGAP5 | discs, large (Drosophila) homolog-associated protein 5 | −1.38 | 0.007 |
| ECT2 | epithelial cell transforming 2 | −0.98 | 0.03 |
| ENTPD4 | ectonucleoside triphosphate diphosphohydrolase 4 | 1.02 | 0.0197 |
| GBA | glucosidase, beta, acid | 0.79 | 0.0264 |
| IGF2R | insulin-like growth factor 2 receptor | 1.11 | 0.0425 |
| KIF11 | kinesin family member 11 | −0.82 | 0.0455 |
| KIF20A | kinesin family member 20A | −1.12 | 0.0322 |
| KIF23 | kinesin family member 23 | −1.22 | 0.0057 |
| KIF4A | kinesin family member 4A | −1.74 | 0.0157 |
| LIPA | lipase A, lysosomal acid, cholesterol esterase | −0.97 | 0.041 |
| MCOLN1 | mucolipin 1 | 0.88 | 0.0412 |
| NDC80 | NDC80 kinetochore complex component | −1.71 | 0.006 |
| NUSAP1 | nucleolar and spindle associated protein 1 | −1.16 | 0.0229 |
| PLK1 | polo-like kinase 1 | −1.42 | 0.0049 |
| PPT2 | palmitoyl-protein thioesterase 2 | 1.08 | 0.0072 |
| PRC1 | protein regulator of cytokinesis 1 | −1.37 | 0.0049 |
| RCC2 | regulator of chromosome condensation 2 | 0.99 | 0.0234 |
| SKA2 | spindle and kinetochore associated complex subunit 2 | −1.36 | 0.0231 |
| SLC11A2 | solute carrier family 11 member 2 | 1.44 | 0.0415 |
| SORT1 | sortilin 1 | 1.66 | 0.0007 |
| STIL | SCL/TAL1 interrupting locus | −1.28 | 0.0156 |
| TPX2 | TPX2, microtubule-associated | −1.28 | 0.007 |
| TTK | TTK protein kinase | −1.59 | 0.0008 |
| ZWILCH | zwilch kinetochore protein | −0.77 | 0.0497 |
| Pathway | Overlap | Adj. p-Value |
|---|---|---|
| BioPlanet | ||
| Translation | 117/151 | 9.03 × 10−30 |
| Cytoplasmic ribosomal proteins | 87/108 | 1.74 × 10−24 |
| Gene expression | 424/968 | 1.79 × 10−17 |
| Parkinson’s disease | 87/131 | 2.14 × 10−15 |
| Cell cycle | 222/453 | 4.52 × 10−15 |
| Oxidative phosphorylation | 86/136 | 2.42 × 10−13 |
| Proteasome degradation | 49/63 | 1.06 × 10−12 |
| DNA replication | 116/207 | 1.19 × 10−12 |
| Alzheimer’s disease | 99/169 | 2.03 × 10−12 |
| Huntington’s disease | 105/184 | 3.54 × 10−12 |
| Antigen processing: cross presentation | 52/79 | 4.99 × 10−9 |
| T-cell receptor regulation of apoptosis | 255/603 | 1.25 × 10−8 |
| S phase | 65/112 | 5.41 × 10−8 |
| Apoptosis regulation | 49/78 | 1.41 × 10−7 |
| Antigen presentation: folding, assembly, and peptide loading of class I MHC proteins | 120/255 | 5.27 × 10−7 |
| DNA replication pre-initiation | 52/88 | 7.94 × 10−7 |
| Mitotic G1-G1/S phases | 72/135 | 7.94 × 10−7 |
| Cell cycle checkpoints | 64/117 | 1.26 × 10−6 |
| Messenger RNA processing | 97/203 | 4.43 × 10−6 |
| Lysosome | 63/121 | 1.55 × 10−5 |
| Translation factors | 32/50 | 2.82 × 10−5 |
| M-phase pathway | 52/96 | 2.99 × 10−5 |
| Protein processing in the endoplasmic reticulum | 80/166 | 3.02 × 10−5 |
| Proteasome complex | 19/24 | 3.22 × 10−5 |
| Immune system signaling by interferons, interleukins, prolactin, and growth hormones | 122/280 | 5.09 × 10−5 |
| Adherens junction cell adhesion | 42/74 | 5.82 × 10−5 |
| Spliceosome | 63/127 | 1.07 × 10−4 |
| Apoptosis | 104/242 | 4.55 × 10−4 |
| Mitotic G2-G2/M phases | 45/87 | 5.31 × 10−4 |
| Cholesterol biosynthesis | 17/24 | 9.85 × 10−4 |
| Renal cell carcinoma | 37/70 | 0.001303568 |
| Transcription | 79/181 | 0.001762513 |
| Ubiquitin-mediated proteolysis | 62/136 | 0.002129516 |
| Pathogenic Escherichia coli infection | 30/57 | 0.005652786 |
| N-glycan biosynthesis | 26/48 | 0.007204858 |
| Messenger RNA splicing: major pathway | 34/68 | 0.007574914 |
| Glutathione metabolism | 27/51 | 0.008633558 |
| Colorectal cancer | 31/62 | 0.011858167 |
| Interleukin-2 signaling pathway | 299/847 | 0.01193764 |
| Focal adhesion | 93/233 | 0.014128026 |
| Mitotic prometaphase | 23/43 | 0.01576878 |
| Valine, leucine, and isoleucine degradation | 23/44 | 0.021845628 |
| Cellular response to hypoxia | 15/25 | 0.022274613 |
| Pancreatic cancer | 33/70 | 0.024629677 |
| Neurophilin interactions with VEGF and VEGF receptor | 05/05 | 0.02670186 |
| Endocytosis | 80/201 | 0.02766493 |
| Cell cycle progression regulation by PLK3 | 12/19 | 0.03136828 |
| Phagosome | 63/154 | 0.033723598 |
| Meiosis | 37/83 | 0.041409388 |
| COPII-mediated vesicle transport | 07/09 | 0.043591541 |
| Peroxisome | 35/78 | 0.043974351 |
| Antigen processing and presentation | 36/81 | 0.046768701 |
| Gene Symbol | Description | Log2 FC | FDR p-Value | ||
|---|---|---|---|---|---|
| LECs | VECs | LECs | VECs | ||
| Glycolysis | |||||
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | −0.04 | 0.14 | 0.9046 | 0.7075 |
| PGK1 | phosphoglycerate kinase 1 | −2.47 | 0.12 | 3.22 × 10−7 | 0.6417 |
| PKM | pyruvate kinase, muscle | −2.18 | 0.44 | 1.81 × 10−7 | 0.2076 |
| ENO1 | enolase 1 | −0.41 | 0.14 | 0.1924 | 0.7325 |
| One-carbon metabolism/nucleotide synthesis | |||||
| PHGDH | phosphoglycerate dehydrogenase | 1.5 | 1.04 | 1.88 × 10−5 | 0.007 |
| SHMT2 | serine hydroxymethyltransferase 2 | 0.84 | 0 | 0.0719 | 0.5407 |
| CAD | carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase | 0.09 | −0.12 | 0.7939 | 0.8057 |
| IMPDH2 | IMPdehydrogenase 2 | −2.13 | 0.53 | 1.19 × 10−6 | 0.4923 |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2 | 4.45 | 0.57 | 1.29 × 10−9 | 0.3519 |
| NME4 | NME/NM23 nucleoside diphosphate kinase 4 | −1.86 | 0.01 | 1.44 × 10−6 | 0.9645 |
| NME1-NME2; NME1; NME2 | NME1-NME2 readthrough; NME/NM23 nucleoside diphosphate kinase 1; NME/NM23 nucleoside diphosphate kinase 2 | −2.6 | −0.26 | 4.29 × 10−8 | 0.8329 |
| TYMS | thymidylate synthetase | −2.05 | −0.85 | 1.46 × 10−5 | 0.2409 |
| Urea/TCA cycle | |||||
| ODC1; SNORA80B | ornithine decarboxylase 1; small nucleolar RNA, H/ACA box 80B | −0.98 | −0.5 | 0.0031 | 0.2514 |
| SRM | spermidine synthase | −2.93 | 0.62 | 2.63 × 10−8 | 0.2849 |
| ASS1 | argininosuccinate synthase 1 | −0.065 | −0.44 | 0.0653 | 0.3289 |
| PYCR1 | pyrroline-5-carboxylate reductase 1 | −0.57 | 0.28 | 0.2041 | 0.6933 |
| ASNS | asparagine synthetase | 6.03 | 1.51 | 4.11 × 10−12 | 0.0008 |
| GOT1 | glutamic-oxaloacetic transaminase 1 | 0.83 | −0.09 | 0.0125 | 0.8934 |
| Oxidative Phosphorylation Genes | |||||
| COX7C | cytochrome c oxidase subunit VIIc | −3.24 | 0.15 | 5.83 × 10−10 | 0.8302 |
| COX5B | cytochrome c oxidase subunit Vb | −3.06 | 0.26 | 1.84 × 10−8 | 0.5633 |
| NDUFB7 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7 | −0.98 | −0.17 | 0.0177 | 0.9995 |
| COX7B | cytochrome c oxidase subunit VIIb | −5.03 | −0.27 | 2.98 × 10−9 | 0.8898 |
| ATP5G1 | ATP synthase, H+ transporting, mitochondrial Fo complex subunit C1 | −4.59 | −0.88 | 8.96 × 10−8 | 0.1972 |
| COX6B1 | cytochrome c oxidase subunit VIb polypeptide 1 | −2.01 | −0.43 | 3.26 × 10−7 | 0.3624 |
| ATP5D | ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit | −4.36 | 0.93 | 2.42 × 10−9 | 0.0774 |
| NDUFB6 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 6 | −1.55 | −0.59 | 0.0003 | 0.2409 |
| NDUFS3 | NADH dehydrogenase (ubiquinone) Fe-S protein 3 | −0.12 | 0.06 | 0.7068 | 0.6352 |
| NDUFS2 | NADH dehydrogenase (ubiquinone) Fe-S protein 2 | −1.91 | 0.23 | 0.0006 | 0.8406 |
| NDUFS1 | NADH dehydrogenase (ubiquinone) Fe-S protein 1 | −0.15 | 0.15 | 0.6976 | 0.78 |
| UQCRH | ubiquinol-cytochrome c reductase hinge protein | −2.62 | −0.04 | 7.26 × 10−8 | 0.8027 |
| UQCRFS1 | ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | −2.06 | −0.05 | 2.23 × 10−5 | 0.6411 |
| NDUFA7 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7 | −2.98 | −0.03 | 8.83 × 10−8 | 0.9319 |
| ATP6V0C | ATPase, H+ transporting, V0 subunit c | −0.33 | 0.36 | 0.3446 | 0.2515 |
| UQCR10 | ubiquinol-cytochrome c reductase, complex III subunit X | −3.1 | −0.53 | 4.90 × 10−9 | 0.165 |
| ATP5A1 | ATP synthase, H+ transporting, alpha subunit 1 | −3.07 | −0.02 | 1.84 × 10−8 | 0.6776 |
| ATP5B | ATP synthase, H+ transporting, beta polypeptide | −1.54 | 0.3 | 0.0002 | 0.5228 |
| NDUFA8 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 8 | −0.5 | 0.63 | 0.2039 | 0.1373 |
| COX5A | cytochrome c oxidase subunit V a | −2.88 | −0.19 | 2.15 × 10−9 | 0.7889 |
| ATP6AP1 | ATPase, H+ transporting, lysosomal accessory protein 1 | −1.65 | 0.67 | 1.12 × 10−5 | 0.0482 |
| SDHD | succinate dehydrogenase complex subunit D | −2.96 | −0.11 | 5.72 × 10−9 | 0.9527 |
| NDUFV1 | NADH dehydrogenase (ubiquinone) flavoprotein 1 | −1.15 | −0.08 | 0.0021 | 0.9782 |
| COX8A | cytochrome c oxidase subunit VIIIA | −5.88 | 0.29 | 1.82 × 10−10 | 0.8555 |
| UQCRC1 | ubiquinol-cytochrome c reductase core protein I | −1.26 | −0.06 | 0.0004 | 0.9944 |
| ATP5J2 | ATP synthase, H+ transporting, mitochondrial Fo complex subunit F2 | −4.11 | −0.16 | 1.71 × 10−10 | 0.7345 |
| COX7A2 | cytochrome c oxidase subunit VII a polypeptide 2 | −2.15 | 0.01 | 6.82 × 10−6 | 0.6142 |
| COX4I1 | cytochrome c oxidase subunit IV isoform 1 | −1.3 | −0.09 | 0.0001 | 0.7254 |
| UQCRB | ubiquinol-cytochrome c reductase binding protein | −0.73 | 0.01 | 0.0115 | 0.9482 |
| ATP5C1 | ATP synthase, H+ transporting, gamma polypeptide 1 | −2.13 | −0.11 | 1.84 × 10−6 | 0.5134 |
| ATP5G3 | ATP synthase, H+ transporting, subunit C3 | −1.67 | −0.43 | 1.02 × 10−5 | 0.3869 |
| ATP5F1 | ATP synthase, H+ transporting, subunit B1 | −2.3 | −0.05 | 8.61 × 10−8 | 0.7665 |
| COX6A1 | cytochrome c oxidase subunit VI a polypeptide 1 | −3.53 | −0.02 | 6.41 × 10−10 | 0.9707 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzarito, G.; Visentin, M.; Leeners, B.; Dubey, R.K. Transcriptomic and Functional Evidence for Differential Effects of MCF-7 Breast Cancer Cell-Secretome on Vascular and Lymphatic Endothelial Cell Growth. Int. J. Mol. Sci. 2022, 23, 7192. https://doi.org/10.3390/ijms23137192
Azzarito G, Visentin M, Leeners B, Dubey RK. Transcriptomic and Functional Evidence for Differential Effects of MCF-7 Breast Cancer Cell-Secretome on Vascular and Lymphatic Endothelial Cell Growth. International Journal of Molecular Sciences. 2022; 23(13):7192. https://doi.org/10.3390/ijms23137192
Chicago/Turabian StyleAzzarito, Giovanna, Michele Visentin, Brigitte Leeners, and Raghvendra K. Dubey. 2022. "Transcriptomic and Functional Evidence for Differential Effects of MCF-7 Breast Cancer Cell-Secretome on Vascular and Lymphatic Endothelial Cell Growth" International Journal of Molecular Sciences 23, no. 13: 7192. https://doi.org/10.3390/ijms23137192