
The Importance of CXCL1 in the Physiological State and in Noncancer Diseases of the Oral Cavity and Abdominal Organs

 , ,
, ,  , , , and
, , , and
Abstract
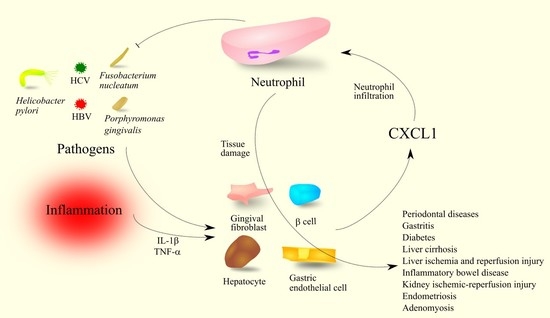
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Comments on Research Methodology
3. Gastrointestinal System and Organs Associated with Digestion
3.1. Periodontal Diseases and Periodontitis
3.2. Sjögren Syndrome
3.3. Helicobacter Pylori Infection, Gastritis and Peptic Ulcer Disease
- Virulence factor CagA in gastric epithelial cells [44,49,51]. This factor activates the Ras→mitogen-activated protein kinase kinase (MEK)→extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) pathway [52]. The latter kinase activates nuclear factor κB (NF-κB), which increases the expression of chemokines that are ligands for CXCR2. NF-κB activation by CagA also involves transforming growth factor-beta-activated kinase 1 (TAK1) [53]. At the same time, it seems that NF-κB activation may only indirectly affect the expression of CXCR2 ligands. More specifically, NF-κB activation increases interleukin-32 (IL-32) levels in the cytoplasm, which increases the expression of CXCR2 ligands [51];
- TLR2 activation on gastric epithelial cells by H. pylori [45]. Increased expression of CXCR2 ligands is independent of CapA and toll-like receptor 4 (TLR4) but dependent on TLR2 activation. This is followed by activation of EGFR, ERK MAPK, c-Jun N-terminal kinase (JNK) MAPK and signal transducer of activation (STAT) signaling pathways [45], which increase the expression of CXCR2 ligands. Studies on neutrophils have shown that HP-NAP increases CXCL8/IL-8 secretion via TLR2 activation [54], which indicates that this virulence factor may be involved in increased CXCL1 expression in gastric epithelial cells independent of CagA;
- NF-κB activation by H. pylori secreted TNF-α inducing protein (Tip-α) [55]. This mechanism was demonstrated on mouse gastric cancer MGT-40 cells.
3.4. Diabetes
3.5. The Physiology of Liver
3.6. Liver Diseases, Cirrhosis
3.7. Liver Ischemia and Reperfusion Injury
3.8. CXCL1 as an Acute-Phase Protein (APP)
3.9. Inflammatory Bowel Disease: Crohn’s Disease and Ulcerative Colitis
3.10. Ileum and Contraction Amplitude
3.11. Obesity and Overweight
4. Kidney
Kidney Transplantation and Ischemic-Reperfusion Injury
5. Reproductive System, Developmental Biology, Placenta, Pregnancy, Birth
5.1. Involvement of CXCL1 in Human Fetal Development and Placental Function
5.2. Endometriosis and Adenomyosis
6. CXCL1 as a Therapeutic Target
7. Perspectives for Further Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Fairbrother, W.J.; Reilly, D.; Colby, T.J.; Hesselgesser, J.; Horuk, R. The solution structure of melanoma growth stimulating activity. J. Mol. Biol. 1994, 242, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Gąssowska-Dobrowolska, M.; Wójcik, J.; Szatkowska, I.; Barczak, K.; Chlubek, M.; Baranowska-Bosiacka, I. The Importance of CXCL1 in Physiology and Noncancerous Diseases of Bone, Bone Marrow, Muscle and the Nervous System. Int. J. Mol. Sci. 2022, 23, 4205. [Google Scholar] [CrossRef] [PubMed]
- Zagorski, J.; DeLarco, J.E. Rat CINC (cytokine-induced neutrophil chemoattractant) is the homolog of the human GRO proteins but is encoded by a single gene. Biochem. Biophys. Res. Commun. 1993, 190, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Shibata, F.; Konishi, K.; Nakagawa, H. Identification of a common receptor for three types of rat cytokine-induced neutrophil chemoattractants (CINCs). Cytokine 2000, 12, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Bozic, C.R.; Kolakowski, L.F., Jr.; Gerard, N.P.; Garcia-Rodriguez, C.; von Uexkull-Guldenband, C.; Conklyn, M.J.; Breslow, R.; Showell, H.J.; Gerard, C. Expression and biologic characterization of the murine chemokine KC. J. Immunol. 1995, 154, 6048–6057. [Google Scholar] [PubMed]
- Shea-Donohue, T.; Thomas, K.; Cody, M.J.; Zhao, A.; Detolla, L.J.; Kopydlowski, K.M.; Fukata, M.; Lira, S.A.; Vogel, S.N. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immune 2008, 14, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, B.; Schumacher, C.; von Tscharner, V.; Clark-Lewis, I.; Baggiolini, M. Neutrophil-activating peptide 2 and gro/melanoma growth-stimulatory activity interact with neutrophil-activating peptide 1/interleukin 8 receptors on human neutrophils. J. Biol. Chem. 1991, 266, 10666–10671. [Google Scholar] [CrossRef]
- Loetscher, P.; Seitz, M.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Both interleukin-8 receptors independently mediate chemotaxis. Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GRO alpha and NAP-2. FEBS Lett. 1994, 341, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, S.K.; Murphy, P.M. The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J. Biol. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Patera, A.C.; Pong-Kennedy, A.; Deno, G.; Gonsiorek, W.; Manfra, D.J.; Vassileva, G.; Zeng, M.; Jackson, C.; Sullivan, L.; et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 2007, 282, 11658–11666. [Google Scholar] [CrossRef] [Green Version]
- Moser, B.; Clark-Lewis, I.; Zwahlen, R.; Baggiolini, M. Neutrophil-activating properties of the melanoma growth-stimulatory activity. J. Exp. Med. 1990, 171, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Kupper, R.W.; Dewald, B.; Jakobs, K.H.; Baggiolini, M.; Gierschik, P. G-protein activation by interleukin 8 and related cytokines in human neutrophil plasma membranes. Biochem. J. 1992, 282, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Li, M.; Spangler, L.C.; Spear, C.; Veenstra, M.; Darnall, L.; Chang, C.; Cotleur, A.C.; Ransohoff, R.M. Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis 2013, 51, 587–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomiyama, H.; Mera, A.; Ohneda, O.; Miura, R.; Suda, T.; Yoshie, O. Organization of the chemokine genes in the human and mouse major clusters of CC and CXC chemokines: Diversification between the two species. Genes Immun. 2001, 2, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomiyama, H.; Osada, N.; Yoshie, O. Systematic classification of vertebrate chemokines based on conserved synteny and evolutionary history. Genes Cells 2013, 18, 1–16. [Google Scholar] [CrossRef]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef]
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global Prevalence of Periodontal Disease and Lack of Its Surveillance. Sci. World J. 2020, 2020, 2146160. [Google Scholar] [CrossRef] [PubMed]
- Rath-Deschner, B.; Memmert, S.; Damanaki, A.; Nokhbehsaim, M.; Eick, S.; Cirelli, J.A.; Götz, W.; Deschner, J.; Jäger, A.; Nogueira, A.V.B. CXCL1, CCL2, and CCL5 modulation by microbial and biomechanical signals in periodontal cells and tissues-in vitro and in vivo studies. Clin. Oral Investig. 2020, 24, 3661–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikama, Y.; Kudo, Y.; Ishimaru, N.; Funaki, M. Possible Involvement of Palmitate in Pathogenesis of Periodontitis. J. Cell Physiol. 2015, 230, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, T.R.; Ge, S.; Bartold, P.M. Compromised inflammatory cytokine response to P. gingivalis LPS by fibroblasts from inflamed human gingiva. Clin. Oral Investig. 2018, 22, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara-Hirata, C.; Yamashiro, K.; Yamamoto, T.; Aoyagi, H.; Ideguchi, H.; Kawamura, M.; Suzuki, R.; Ono, M.; Wake, H.; Nishibori, M.; et al. Anti-HMGB1 Neutralizing Antibody Attenuates Periodontal Inflammation and Bone Resorption in a Murine Periodontitis Model. Infect. Immun. 2018, 86, e00111–e00118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Vieyra, R.; Rosales, C.; Uribe-Querol, E. Neutrophil Functions in Periodontal Homeostasis. J. Immunol. Res. 2016, 2016, 1396106. [Google Scholar] [CrossRef]
- Zenobia, C.; Luo, X.L.; Hashim, A.; Abe, T.; Jin, L.; Chang, Y.; Jin, Z.C.; Sun, J.X.; Hajishengallis, G.; Curtis, M.A.; et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. 2013, 15, 1419–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, A.; Irie, K.; Hashim, A.; Leroux, B.G.; Chang, A.M.; Curtis, M.A.; Darveau, R.P. Site-Specific Neutrophil Migration and CXCL2 Expression in Periodontal Tissue. J. Dent. Res. 2016, 95, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicu, E.A.; Rijkschroeff, P.; Wartewig, E.; Nazmi, K.; Loos, B.G. Characterization of oral polymorphonuclear neutrophils in periodontitis patients: A case-control study. BMC Oral Health 2018, 18, 149. [Google Scholar] [CrossRef]
- Valerio, M.S.; Herbert, B.A.; Basilakos, D.S.; Browne, C.; Yu, H.; Kirkwood, K.L. Critical role of MKP-1 in lipopolysaccharide-induced osteoclast formation through CXCL1 and CXCL2. Cytokine 2015, 71, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Govey, P.M.; Jacobs, J.M.; Tilton, S.C.; Loiselle, A.E.; Zhang, Y.; Freeman, W.M.; Waters, K.M.; Karin, N.J.; Donahue, H.J. Integrative transcriptomic and proteomic analysis of osteocytic cells exposed to fluid flow reveals novel mechano-sensitive signaling pathways. J. Biomech. 2014, 47, 1838–1845. [Google Scholar] [CrossRef] [Green Version]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin. Exp. Metastasis 2015, 32, 353–368. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Baldini, C.; Bootsma, H.; Bowman, S.J.; Jonsson, R.; Mariette, X.; Sivils, K.; Theander, E.; Tzioufas, A.; Ramos-Casals, M. Sjögren syndrome. Nat. Rev. Dis. Primers 2016, 2, 16047. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M.; De Lucro, R.; Ribatti, D. A potential role of the GRO-α/CXCR2 system in Sjögren’s syndrome: Regulatory effects of pro-inflammatory cytokines. Histochem Cell. Biol. 2013, 139, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M.; De Lucro, R.; Ribatti, D. GRO-α/CXCR2 system and ADAM17 correlated expression in Sjögren’s syndrome. Inflammation 2013, 36, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; D’Amore, M.; Lofrumento, D.D. The metalloproteinase ADAM17 and the epidermal growth factor receptor (EGFR) signaling drive the inflammatory epithelial response in Sjögren’s syndrome. Clin. Exp. Med. 2015, 15, 215–225. [Google Scholar] [CrossRef]
- Dunn, B.E.; Cohen, H.; Blaser, M.J. Helicobacter pylori. Clin. Microbiol. Rev. 1997, 10, 720–741. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Brito, B.B.; da Silva, F.A.F.; Soares, A.S.; Pereira, V.A.; Santos, M.L.C.; Sampaio, M.M.; Neves, P.H.M.; de Melo, F.F. Pathogenesis and clinical management of Helicobacter pylori gastric infection. World J. Gastroenterol. 2019, 25, 5578–5589. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Lacy, D.B.; Ohi, M.D. The Helicobacter pylori Cag Type IV Secretion System. Trends Microbiol. 2020, 28, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Umit, H.; Tezel, A.; Bukavaz, S.; Unsal, G.; Otkun, M.; Soylu, A.R.; Tucer, D.; Otkun, M.; Bilgi, S. The relationship between virulence factors of Helicobacter pylori and severity of gastritis in infected patients. Dig. Dis. Sci. 2009, 54, 103–110. [Google Scholar] [CrossRef]
- Salimzadeh, L.; Bagheri, N.; Zamanzad, B.; Azadegan-Dehkordi, F.; Rahimian, G.; Hashemzadeh-Chaleshtori, M.; Rafieian-Kopaei, M.; Sanei, M.H.; Shirzad, H. Frequency of virulence factors in Helicobacter pylori-infected patients with gastritis. Microb. Pathog. 2015, 80, 67–72. [Google Scholar] [CrossRef]
- Fu, H.W. Helicobacter pylori neutrophil-activating protein: From molecular pathogenesis to clinical applications. World J. Gastroenterol. 2014, 20, 5294–5301. [Google Scholar] [CrossRef]
- Suzuki, H.; Mori, M.; Sakaguchi, A.A.; Suzuki, M.; Miura, S.; Ishii, H. Enhanced levels of C-X-C chemokine, human GROalpha, in Helicobacter pylori-associated gastric disease. J. Gastroenterol. Hepatol. 1998, 13, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Tanahashi, T.; Kashima, K.; Imanishi, J. Chemokines in the gastric mucosa in Helicobacter pylori infection. Gut 1998, 42, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero, R.L.; Avé, P.; Ndiaye, D.; Bambou, J.C.; Huerre, M.R.; Philpott, D.J.; Mémet, S. NF-kappaB activation during acute Helicobacter pylori infection in mice. Infect. Immun. 2008, 76, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, P.; Paris, I.; Garcia, M.; Tran, C.T.; Cremniter, J.; Garnier, M.; Faure, J.P.; Barthes, T.; Boneca, I.G.; Morel, F.; et al. Chemokines and antimicrobial peptides have a cag-dependent early response to Helicobacter pylori infection in primary human gastric epithelial cells. Infect. Immun. 2014, 82, 2881–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.T.; Garcia, M.; Garnier, M.; Burucoa, C.; Bodet, C. Inflammatory signaling pathways induced by Helicobacter pylori in primary human gastric epithelial cells. Innate Immune 2017, 23, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, M.; Schmausser, B.; Scheller, K.; Toksoy, A.; Kraus, M.; Menzel, T.; Müller-Hermelink, H.K.; Gillitzer, R. CXC chemokines Gro(alpha)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin. Exp. Immunol. 2000, 122, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.C.; Weems, M.N.; Allen, L.H. Cutting Edge: Helicobacter pylori Induces Nuclear Hypersegmentation and Subtype Differentiation of Human Neutrophils In Vitro. J. Immunol. 2017, 198, 1793–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuper, T.; Frauenlob, T.; Sarajlic, M.; Posselt, G.; Wessler, S.; Horejs-Hoeck, J. TLR2, TLR4 and TLR10 Shape the Cytokine and Chemokine Release of H. pylori-Infected Human DCs. Int. J. Mol. Sci. 2020, 21, 3897. [Google Scholar] [CrossRef] [PubMed]
- Sieveking, D.; Mitchell, H.M.; Day, A.S. Gastric epithelial cell CXC chemokine secretion following Helicobacter pylori infection in vitro. J. Gastroenterol. Hepatol. 2004, 19, 982–987. [Google Scholar] [CrossRef]
- Sierra, J.C.; Asim, M.; Verriere, T.G.; Piazuelo, M.B.; Suarez, G.; Romero-Gallo, J.; Delgado, A.G.; Wroblewski, L.E.; Barry, D.P.; Peek, R.M., Jr.; et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut 2018, 67, 1247–1260. [Google Scholar] [CrossRef]
- Sakitani, K.; Hirata, Y.; Hayakawa, Y.; Serizawa, T.; Nakata, W.; Takahashi, R.; Kinoshita, H.; Sakamoto, K.; Nakagawa, H.; Akanuma, M.; et al. Role of interleukin-32 in Helicobacter pylori-induced gastric inflammation. Infect. Immun. 2012, 80, 3795–3803. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.; Kwok, T.; Hartig, R.; König, W.; Backert, S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [Green Version]
- Lamb, A.; Yang, X.D.; Tsang, Y.H.; Li, J.D.; Higashi, H.; Hatakeyama, M.; Peek, R.M.; Blanke, S.R.; Chen, L.F. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009, 10, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.H.; Hong, Z.W.; Chen, C.C.; Chang, H.W.; Fu, H.W. Helicobacter pylori Neutrophil-Activating Protein Directly Interacts with and Activates Toll-like Receptor 2 to Induce the Secretion of Interleukin-8 from Neutrophils and ATRA-Induced Differentiated HL-60 Cells. Int. J. Mol. Sci. 2021, 22, 11560. [Google Scholar] [CrossRef]
- Kuzuhara, T.; Suganuma, M.; Kurusu, M.; Fujiki, H. Helicobacter pylori-secreting protein Tipalpha is a potent inducer of chemokine gene expressions in stomach cancer cells. J. Cancer Res. Clin. Oncol. 2007, 133, 287–296. [Google Scholar] [CrossRef]
- Cai, Q.; Shi, P.; Yuan, Y.; Peng, J.; Ou, X.; Zhou, W.; Li, J.; Su, T.; Lin, L.; Cai, S.; et al. Inflammation-Associated Senescence Promotes Helicobacter pylori-Induced Atrophic Gastritis. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 857–880. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xie, C.; Lu, N.H. p53, a potential predictor of Helicobacter pylori infection-associated gastric carcinogenesis? Oncotarget 2016, 7, 66276–66286. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 2020, 69, 1582–1591. [Google Scholar] [CrossRef]
- Teng, Y.S.; Liu, Y.G.; Chen, X.H.; Wang, T.T.; Cheng, P.; Lv, Y.P.; Kong, H.; Mao, F.Y.; Hao, C.J.; Yang, S.M.; et al. Decreased IL-17RB expression impairs CD11b+CD11c- myeloid cell accumulation in gastric mucosa and host defense during the early-phase of Helicobacter pylori infection. Cell Death Dis. 2019, 10, 79. [Google Scholar] [CrossRef]
- Sun, D.; Novotny, M.; Bulek, K.; Liu, C.; Li, X.; Hamilton, T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 2011, 12, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Herjan, T.; Yao, P.; Qian, W.; Li, X.; Liu, C.; Bulek, K.; Sun, D.; Yang, W.P.; Zhu, J.; He, A.; et al. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J. Immunol. 2013, 191, 640–649. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.H.; Huang, S.T.; Yang, S.F.; Li, C.J.; Lin, H.W.; Weng, B.C.; Yang, S.M.; Huang, S.C.; Wu, J.C.; Chang, Y.C.; et al. Hepatoma-derived growth factor participates in Helicobacter Pylori-induced neutrophils recruitment, gastritis and gastric carcinogenesis. Oncogene 2019, 38, 6461–6477. [Google Scholar] [CrossRef]
- Polenghi, A.; Bossi, F.; Fischetti, F.; Durigutto, P.; Cabrelle, A.; Tamassia, N.; Cassatella, M.A.; Montecucco, C.; Tedesco, F.; de Bernard, M. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J. Immunol. 2007, 178, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Brisslert, M.; Enarsson, K.; Lundin, S.; Karlsson, A.; Kusters, J.G.; Svennerholm, A.M.; Backert, S.; Quiding-Järbrink, M. Helicobacter pylori induce neutrophil transendothelial migration: Role of the bacterial HP-NAP. FEMS Microbiol. Lett. 2005, 249, 95–103. [Google Scholar] [CrossRef]
- Cappon, A.; Babolin, C.; Segat, D.; Cancian, L.; Amedei, A.; Calzetti, F.; Cassatella, M.A.; D’Elios, M.M.; de Bernard, M. Helicobacter pylori-derived neutrophil-activating protein increases the lifespan of monocytes and neutrophils. Cell Microbiol. 2010, 12, 754–764. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.Q.; Wang, Z.H.; Liao, J.X.; Chen, X.Y.; Liu, W.Z.; Xiao, S.D.; Lu, H. Predictive value of neutrophil infiltration as a marker of Helicobacter pylori infection. World J. Gastroenterol. 2012, 18, 5101–5105. [Google Scholar] [CrossRef]
- Pérez-Figueroa, E.; Torres, J.; Sánchez-Zauco, N.; Contreras-Ramos, A.; Alvarez-Arellano, L.; Maldonado-Bernal, C. Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immune 2016, 22, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Jang, A.R.; Kang, M.J.; Shin, J.I.; Kwon, S.W.; Park, J.Y.; Ahn, J.H.; Lee, T.S.; Kim, D.Y.; Choi, B.G.; Seo, M.W.; et al. Unveiling the Crucial Role of Type IV Secretion System and Motility of Helicobacter pylori in IL-1β Production via NLRP3 Inflammasome Activation in Neutrophils. Front. Immunol. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Nishioka, H.; Baesso, I.; Semenzato, G.; Trentin, L.; Rappuoli, R.; Del Giudice, G.; Montecucco, C. The neutrophil-activating protein of Helicobacter pylori (HP-NAP) activates the MAPK pathway in human neutrophils. Eur. J. Immunol. 2003, 33, 840–849. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Hart, P.A.; Bellin, M.D.; Andersen, D.K.; Bradley, D.; Cruz-Monserrate, Z.; Forsmark, C.E.; Goodarzi, M.O.; Habtezion, A.; Korc, M.; Kudva, Y.C.; et al. Consortium for the Study of Chronic Pancreatitis, Diabetes, and Pancreatic Cancer(CPDPC). Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol. Hepatol. 2016, 1, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, P.; Diabetes in Pregnancy Working Group; Maternal Medicine Clinical Study Group; Royal College of Obstetricians and Gynaecologists, UK. Gestational diabetes: Opportunities for improving maternal and child health. Lancet Diabetes Endocrinol. 2020, 8, 793–800. [Google Scholar] [CrossRef]
- Hakimizadeh, E.; Shamsizadeh, A.; Nazari, M.; Arababadi, M.K.; Rezaeian, M.; Vazirinejad, R.; Jamali, Z.; Poor, N.M.; Khorramdelazad, H.; Darakhshan, S.; et al. Increased circulating levels of CXC chemokines is correlated with duration and complications of the disease in type-1 diabetes: A study on Iranian diabetic patients. Clin. Lab. 2013, 59, 531–537. [Google Scholar] [CrossRef]
- Takahashi, K.; Ohara, M.; Sasai, T.; Homma, H.; Nagasawa, K.; Takahashi, T.; Yamashina, M.; Ishii, M.; Fujiwara, F.; Kajiwara, T.; et al. Serum CXCL1 concentrations are elevated in type 1 diabetes mellitus, possibly reflecting activity of anti-islet autoimmune activity. Diabetes Metab. Res. Rev. 2011, 27, 830–833. [Google Scholar] [CrossRef]
- Diana, J.; Lehuen, A. Macrophages and β-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol. Med. 2014, 6, 1090–1104. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [Green Version]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [Green Version]
- Garciafigueroa, Y.; Phillips, B.E.; Engman, C.; Trucco, M.; Giannoukakis, N. Neutrophil-Associated Inflammatory Changes in the Pre-Diabetic Pancreas of Early-Age NOD Mice. Front. Endocrinol. 2021, 12, 565981. [Google Scholar] [CrossRef]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Sajadi, S.M.; Khoramdelazad, H.; Hassanshahi, G.; Rafatpanah, H.; Hosseini, J.; Mahmoodi, M.; Arababadi, M.K.; Derakhshan, R.; Hasheminasabzavareh, R.; Hosseini-Zijoud, S.M.; et al. Plasma levels of CXCL1 (GRO-alpha) and CXCL10 (IP-10) are elevated in type 2 diabetic patients: Evidence for the involvement of inflammation and angiogenesis/angiostasis in this disease state. Clin. Lab. 2013, 59, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Nunemaker, C.S.; Chung, H.G.; Verrilli, G.M.; Corbin, K.L.; Upadhye, A.; Sharma, P.R. Increased serum CXCL1 and CXCL5 are linked to obesity, hyperglycemia, and impaired islet function. J. Endocrinol. 2014, 222, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebuffat, S.A.; Sidot, E.; Guzman, C.; Azay-Milhau, J.; Jover, B.; Lajoix, A.D.; Peraldi-Roux, S. Adipose tissue derived-factors impaired pancreatic β-cell function in diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3378–3387. [Google Scholar] [CrossRef]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Citro, A.; Cantarelli, E.; Maffi, P.; Nano, R.; Melzi, R.; Mercalli, A.; Dugnani, E.; Sordi, V.; Magistretti, P.; Daffonchio, L.; et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J. Clin. Investig. 2012, 122, 3647–3651. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.J.; Weinberg, A.; Zammit, N.W.; Walters, S.N.; Hawthorne, W.J.; Loudovaris, T.; Thomas, H.; Kay, T.; Gunton, J.E.; Alexander, S.I.; et al. Human islets express a marked proinflammatory molecular signature prior to transplantation. Cell Transpl. 2012, 21, 2063–2078. [Google Scholar] [CrossRef] [Green Version]
- Darakhshan, S.; Fatehi, A.; Hassanshahi, G.; Mahmoodi, S.; Hashemi, M.S.; Karimabad, M.N. Serum concentration of angiogenic (CXCL1, CXCL12) and angiostasis (CXCL9, CXCL10) CXC chemokines are differentially altered in normal and gestational diabetes mellitus associated pregnancies. J. Diabetes Metab. Disord. 2019, 18, 371–378. [Google Scholar] [CrossRef]
- Glaser, N.; Chu, S.; Hung, B.; Fernandez, L.; Wulff, H.; Tancredi, D.; Odonnell, M.E. Acute and chronic neuroinflammation is triggered by diabetic ketoacidosis in a rat model. BMJ Open Diabetes Res. Care 2020, 8, e001793. [Google Scholar]
- Bigorgne, A.E.; John, B.; Ebrahimkhani, M.R.; Shimizu-Albergine, M.; Campbell, J.S.; Crispe, I.N. TLR4-Dependent Secretion by Hepatic Stellate Cells of the Neutrophil-Chemoattractant CXCL1 Mediates Liver Response to Gut Microbiota. PLoS ONE 2016, 11, e0151063. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Non-alcoholic fatty liver disease. BMC Med. 2017, 15, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and Management of Alcoholic Liver Disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection: A Review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Gkouvatsos, K.; Pantopoulos, K. Chronic hepatitis C and liver fibrosis. World J. Gastroenterol. 2014, 20, 11033–11053. [Google Scholar] [CrossRef]
- Li, H.; Yan, L.; Shi, Y.; Lv, D.; Shang, J.; Bai, L.; Tang, H. Hepatitis B Virus Infection: Overview. Adv. Exp. Med. Biol. 2020, 1179, 1–16. [Google Scholar]
- Kim, C.W.; Chang, K.M. Hepatitis C virus: Virology and life cycle. Clin. Mol. Hepatol. 2013, 19, 17–25. [Google Scholar] [CrossRef]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Vansaun, M.N.; Mendonsa, A.M.; Lee Gorden, D. Hepatocellular proliferation correlates with inflammatory cell and cytokine changes in a murine model of nonalchoholic fatty liver disease. PLoS ONE 2013, 8, e73054. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Oh, J.H.; Chae, Y.J.; Lee, J.S.; Koh, D.H.; Kang, H.T. Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J. Gastroenterol. Hepatol. 2019, 34, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlquist, K.J.V.; Voth, L.C.; Fee, A.J.; Stoeckman, A.K. An Autocrine Role for CXCL1 in Progression of Hepatocellular Carcinoma. Anticancer Res. 2020, 40, 6075–6081. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Sun, Y.; Jiang, Z.; Du, K.; Xia, N.; Zhong, G. Key genes associated with non-alcoholic fatty liver disease and acute myocardial infarction. Med. Sci. Monit. 2020, 26, e922492. [Google Scholar] [CrossRef] [PubMed]
- Maltby, J.; Wright, S.; Bird, G.; Sheron, N. Chemokine levels in human liver homogenates: Associations between GRO alpha and histopathological evidence of alcoholic hepatitis. Hepatology 1996, 24, 1156–1160. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.D.; Ueda, H.; Park, K.; Flanders, K.C.; Lee, Y.I.; Jay, G.; Kim, S.J. Regulation of transforming growth factor-beta 1 expression by the hepatitis B virus (HBV) X transactivator. Role in HBV pathogenesis. J. Clin. Investig. 1996, 97, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; Ju, D.; Zhang, D.W.; Li, H.; Kong, L.M.; Guo, Y.; Li, C.; Wang, X.L.; Chen, Z.N.; Bian, H. Activation of TGF-β1-CD147 positive feedback loop in hepatic stellate cells promotes liver fibrosis. Sci. Rep. 2015, 5, 16552. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.P.; Ju, D.; Li, H.; Yuan, L.; Cui, J.; Luo, D.; Chen, Z.N.; Bian, H. CD147 Promotes CXCL1 Expression and Modulates Liver Fibrogenesis. Int. J. Mol. Sci. 2018, 19, 1145. [Google Scholar] [CrossRef] [Green Version]
- Nishitsuji, H.; Funami, K.; Shimizu, Y.; Ujino, S.; Sugiyama, K.; Seya, T.; Takaku, H.; Shimotohno, K. Hepatitis C virus infection induces inflammatory cytokines and chemokines mediated by the cross talk between hepatocytes and stellate cells. J. Virol. 2013, 87, 8169–8178. [Google Scholar] [CrossRef] [Green Version]
- Costantini, S.; Capone, F.; Guerriero, E.; Marfella, R.; Sorice, A.; Maio, P.; Di Stasio, M.; Paolisso, G.; Castello, G.; Colonna, G. Cytokinome profile of patients with type 2 diabetes and/or chronic hepatitis C infection. PLoS ONE 2012, 7, e39486. [Google Scholar]
- Asselah, T.; Bièche, I.; Laurendeau, I.; Paradis, V.; Vidaud, D.; Degott, C.; Martinot, M.; Bedossa, P.; Valla, D.; Vidaud, M.; et al. Liver gene expression signature of mild fibrosis in patients with chronic hepatitis C. Gastroenterology 2005, 129, 2064–2075. [Google Scholar] [CrossRef] [PubMed]
- Nischalke, H.D.; Berger, C.; Luda, C.; Müller, T.; Berg, T.; Coenen, M.; Krämer, B.; Körner, C.; Trebicka, J.; Grünhage, F.; et al. The CXCL1 rs4074 A allele is associated with enhanced CXCL1 responses to TLR2 ligands and predisposes to cirrhosis in HCV genotype 1-infected Caucasian patients. J. Hepatol. 2012, 56, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.; Talloen, W.; Tuefferd, M.; Darling, J.M.; Scholliers, A.; Fanning, G.; Fried, M.W.; Aerssens, J. Plasma levels of growth-related oncogene (CXCL1-3) associated with fibrosis and platelet counts in HCV-infected patients. Aliment. Pharmacol. Ther. 2015, 42, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, L.; Brenner, D.A.; Stefanovic, B. Direct hepatotoxic effect of KC chemokine in the liver without infiltration of neutrophils. Exp. Biol. Med. 2005, 230, 573–586. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 399–413. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Ceni, E.; Salzano, R.; Biondi, P.; Parola, M.; Galli, A.; Foschi, M.; Caligiuri, A.; Pinzani, M.; Surrenti, C. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: Role of nitric oxide. Hepatology 1997, 25, 361–367. [Google Scholar] [CrossRef]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Mosher, B.; Dean, R.; Harkema, J.; Remick, D.; Palma, J.; Crockett, E. Inhibition of Kupffer cells reduced CXC chemokine production and liver injury. J. Surg. Res. 2001, 99, 201–210. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, H.; Qiu, Z.; Gao, S.; Wang, Z.; Xiao, L. Gene microarray analysis of expression profiles in liver ischemia and reperfusion. Mol. Med. Rep. 2017, 16, 3299–3307. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, M.; Shimizu, H.; Mitsuhashi, N.; Ohtsuka, M.; Wakabayashi, Y.; Ito, H.; Kimura, F.; Nakagawa, K.; Yoshidome, H.; Shimizu, Y.; et al. Effect of cold-ischemia time on C-X-C chemokine expression and neutrophil accumulation in the graft liver after orthotopic liver transplantation in rats. Transplantation 2002, 73, 1730–1735. [Google Scholar] [CrossRef]
- Lentsch, A.B.; Yoshidome, H.; Cheadle, W.G.; Miller, F.N.; Edwards, M.J. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: Roles for macrophage inflammatory protein-2 and KC. Hepatology 1998, 27, 1172–1177. [Google Scholar] [CrossRef]
- Honda, M.; Takeichi, T.; Asonuma, K.; Tanaka, K.; Kusunoki, M.; Inomata, Y. Intravital imaging of neutrophil recruitment in hepatic ischemia-reperfusion injury in mice. Transplantation 2013, 95, 551–558. [Google Scholar] [CrossRef]
- Uchida, Y.; Freitas, M.C.; Zhao, D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. The inhibition of neutrophil elastase ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2009, 15, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Sosa, R.A.; Zarrinpar, A.; Rossetti, M.; Lassman, C.R.; Naini, B.V.; Datta, N.; Rao, P.; Harre, N.; Zheng, Y.; Spreafico, R.; et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight 2016, 1, e89679. [Google Scholar] [CrossRef] [Green Version]
- Pesonen, E.J.; Höckerstedt, K.; Mäkisalo, H.; Vuorte, J.; Jansson, S.E.; Orpana, A.; Karonen, S.L.; Repo, H. Transhepatic neutrophil and monocyte activation during clinical liver transplantation. Transplantation 2000, 69, 1458–1464. [Google Scholar] [CrossRef]
- Yoshidome, H.; Lentsch, A.B.; Cheadle, W.G.; Miller, F.N.; Edwards, M.J. Enhanced pulmonary expression of CXC chemokines during hepatic ischemia/reperfusion-induced lung injury in mice. J. Surg. Res. 1999, 81, 33–37. [Google Scholar] [CrossRef]
- Colletti, L.M.; Kunkel, S.L.; Walz, A.; Burdick, M.D.; Kunkel, R.G.; Wilke, C.A.; Strieter, R.M. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein. J. Clin. Investig. 1995, 95, 134–141. [Google Scholar] [CrossRef]
- Su, L.; Li, N.; Tang, H.; Lou, Z.; Chong, X.; Zhang, C.; Su, J.; Dong, X. Kupffer cell-derived TNF-α promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis. 2018, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.J.; Perry, V.H.; Pitossi, F.J.; Butchart, A.G.; Chertoff, M.; Waters, S.; Dempster, R.; Anthony, D.C. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am. J. Pathol. 2005, 166, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Yates, A.G.; Jogia, T.; Gillespie, E.R.; Couch, Y.; Ruitenberg, M.J.; Anthony, D.C. Acute IL-1RA treatment suppresses the peripheral and central inflammatory response to spinal cord injury. J. Neuroinflamm. 2021, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Hughes, P.M.; Iredale, J.P.; Wilcockson, D.C.; Waters, S.; Docagne, F.; Perry, V.H.; Anthony, D.C. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003, 17, 1168–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010, 207, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Cummings, C.J.; Martin, T.R.; Frevert, C.W.; Quan, J.M.; Wong, V.A.; Mongovin, S.M.; Hagen, T.R.; Steinberg, K.P.; Goodman, R.B. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J. Immunol. 1999, 162, 2341–2346. [Google Scholar]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Brandt, E.; Müller-Alouf, H.; Desreumaux, P.; Woerly, G.; Colombel, J.F.; Capron, M. Circulating growth-regulator oncogene alpha contributes to neutrophil priming and interleukin-8-directed mucosal recruitment into chronic lesions of patients with Crohn’s disease. Eur. Cytokine Netw. 1998, 9, 647–653. [Google Scholar]
- Mitsuyama, K.; Tsuruta, O.; Tomiyasu, N.; Takaki, K.; Suzuki, A.; Masuda, J.; Yamasaki, H.; Toyonaga, A.; Sata, M. Increased circulating concentrations of growth-related oncogene (GRO)-alpha in patients with inflammatory bowel disease. Dig. Dis. Sci. 2006, 51, 173–177. [Google Scholar] [CrossRef]
- Alzoghaibi, M.A.; Al-Mofleh, I.A.; Al-Jebreen, A.M. Neutrophil chemokines GCP-2 and GRO-alpha in patients with inflammatory bowel disease. J. Dig. Dis. 2008, 9, 144–148. [Google Scholar] [CrossRef]
- Boucher, G.; Paradis, A.; Chabot-Roy, G.; Coderre, L.; Hillhouse, E.E.; Bitton, A.; Des Rosiers, C.; Levings, M.K.; Schumm, L.P.; Lazarev, M.; et al. Serum Analyte Profiles Associated With Crohn’s Disease and Disease Location. Inflamm. Bowel Dis. 2022, 28, 9–20. [Google Scholar] [CrossRef]
- Bjerrum, J.T.; Nyberg, C.; Olsen, J.; Nielsen, O.H. Assessment of the validity of a multigene analysis in the diagnostics of inflammatory bowel disease. J. Intern. Med. 2014, 275, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, K.L.; Sartor, R.B.; Haskill, S. Cytokine messenger RNA profiles in inflammatory bowel disease mucosa detected by polymerase chain reaction amplification. Gastroenterology 1992, 103, 1587–1595. [Google Scholar] [CrossRef]
- Lawrance, I.C.; Fiocchi, C.; Chakravarti, S. Ulcerative colitis and Crohn’s disease: Distinctive gene expression profiles and novel susceptibility candidate genes. Hum. Mol. Genet. 2001, 10, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Xu, L.; Zhang, Z.; Wang, F.; Tang, X. Investigation of Potential Genetic Biomarkers and Molecular Mechanism of Ulcerative Colitis Utilizing Bioinformatics Analysis. Biomed. Res. Int. 2020, 2020, 4921387. [Google Scholar] [CrossRef] [Green Version]
- Arijs, I.; De Hertogh, G.; Machiels, K.; Van Steen, K.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Quintens, R.; Van Assche, G.; Vermeire, S.; et al. Mucosal gene expression of cell adhesion molecules, chemokines, and chemokine receptors in patients with inflammatory bowel disease before and after infliximab treatment. Am. J. Gastroenterol. 2011, 106, 748–761. [Google Scholar] [CrossRef]
- Imada, A.; Ina, K.; Shimada, M.; Yokoyama, T.; Yokoyama, Y.; Nishio, Y.; Yamaguchi, T.; Ando, T.; Kusugami, K. Coordinate upregulation of interleukin-8 and growth-related gene product-alpha is present in the colonic mucosa of inflammatory bowel. Scand. J. Gastroenterol. 2001, 36, 854–864. [Google Scholar] [CrossRef]
- Egesten, A.; Eliasson, M.; Olin, A.I.; Erjefält, J.S.; Bjartell, A.; Sangfelt, P.; Carlson, M. The proinflammatory CXC-chemokines GRO-alpha/CXCL1 and MIG/CXCL9 are concomitantly expressed in ulcerative colitis and decrease during treatment with topical corticosteroids. Int. J. Colorectal. Dis. 2007, 22, 1421–1427. [Google Scholar] [CrossRef]
- Selleri, S.; Palazzo, M.; Deola, S.; Wang, E.; Balsari, A.; Marincola, F.M.; Rumio, C. Induction of pro-inflammatory programs in enteroendocrine cells by the Toll-like receptor agonists flagellin and bacterial LPS. Int. Immunol. 2008, 20, 961–970. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Cheng, Y.S.; Yu, L.; Xu, Y.Y.; Wang, Y. Bioinformatics Analysis of Differentially Expressed Genes and Protein-Protein Interaction Networks Associated with Functional Pathways in Ulcerative Colitis. Med. Sci. Monit. 2021, 27, e927917. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Kong, Y.; Chen, N.; Peng, W.; Zi, R.; Jiang, M.; Zhu, J.; Wang, Y.; Yue, J.; Lv, J.; et al. Identification of Immune-Related Gene Signature and Prediction of CeRNA Network in Active Ulcerative Colitis. Front. Immunol. 2022, 13, 855645. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Corbi, M.; Scaldaferri, F.; Petito, V.; Graziani, C.; Castri, F.; Neri, M.; Gasbarrini, A.; Sgambato, A.; Papa, A. Characterization of mucosal cytokine profile in ulcerative colitis patients under conventional and anti-TNF-a treatment. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.; Goll, R.; Cui, G.; Husebekk, A.; Vonen, B.; Birketvedt, G.S.; Florholmen, J. Tissue levels of tumor necrosis factor-alpha correlates with grade of inflammation in untreated ulcerative colitis. Scand. J. Gastroenterol. 2007, 42, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Kerami, Z.; Duijvis, N.W.; Vogels, E.W.; van Dooren, F.H.; Moerland, P.D.; Te Velde, A.A. Effect of interleukin-17 on gene expression profile of fibroblasts from Crohn’s disease patients. J. Crohn’s Colitis 2014, 8, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Fang, M.; Wang, X.M.; Liu, W.Y.; Zheng, Y.J.; Wu, X.B.; Tao, R. Proinflammatory effects and molecular mechanisms of interleukin-17 in intestinal epithelial cell line HT-29. World J. Gastroenterol. 2014, 20, 17924–17931. [Google Scholar] [CrossRef]
- Friedrich, M.; Diegelmann, J.; Beigel, F.; Brand, S. IL-17A alone weakly affects the transcriptome of intestinal epithelial cells but strongly modulates the TNF-α-induced expression of inflammatory mediators and inflammatory bowel disease susceptibility genes. Inflamm. Bowel Dis. 2014, 20, 1502–1515. [Google Scholar] [CrossRef]
- Østvik, A.E.; Svendsen, T.D.; Granlund, A.V.B.; Doseth, B.; Skovdahl, H.K.; Bakke, I.; Thorsvik, S.; Afroz, W.; Walaas, G.A.; Mollnes, T.E.; et al. Intestinal Epithelial Cells Express Immunomodulatory ISG15 During Active Ulcerative Colitis and Crohn’s Disease. J. Crohn’s Colitis 2020, 14, 920–934. [Google Scholar] [CrossRef]
- Nishida, A.; Hidaka, K.; Kanda, T.; Imaeda, H.; Shioya, M.; Inatomi, O.; Bamba, S.; Kitoh, K.; Sugimoto, M.; Andoh, A. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 303–314. [Google Scholar] [CrossRef]
- Llopis, M.; Antolin, M.; Carol, M.; Borruel, N.; Casellas, F.; Martinez, C.; Espín-Basany, E.; Guarner, F.; Malagelada, J.R. Lactobacillus casei downregulates commensals’ inflammatory signals in Crohn’s disease mucosa. Inflamm. Bowel Dis. 2009, 15, 275–283. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Vijay-Kumar, M.; Dalmasso, G.; Yang, D.; Linden, J.; Wang, L.; Gewirtz, A.; Ravid, K.; Merlin, D.; Sitaraman, S.V. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 2008, 135, 861–8670. [Google Scholar] [CrossRef] [Green Version]
- Stadnyk, A.W.; Carrigan, S.O.; Otley, A.R. Neutrophil transintestinal epithelial migration to CXCR2 ligands is regulated by adenosine. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 414–421. [Google Scholar] [CrossRef]
- Owhashi, M.; Taoka, Y.; Ishii, K.; Nakazawa, S.; Uemura, H.; Kambara, H. Identification of a ubiquitin family protein as a novel neutrophil chemotactic factor. Biochem. Biophys. Res. Commun. 2003, 309, 533–539. [Google Scholar] [CrossRef]
- Buanne, P.; Di Carlo, E.; Caputi, L.; Brandolini, L.; Mosca, M.; Cattani, F.; Pellegrini, L.; Biordi, L.; Coletti, G.; Sorrentino, C.; et al. Crucial pathophysiological role of CXCR2 in experimental ulcerative colitis in mice. J. Leukoc. Biol. 2007, 82, 1239–1246. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Lourda, M.; Ideström, M.; Chen, P.; Olsson-Åkefeldt, S.; Forkel, M.; Gavhed, D.; Lindforss, U.; Mjösberg, J.; Henter, J.I.; et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut 2016, 65, 1632–1641. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar]
- Verstockt, B.; Van Assche, G.; Vermeire, S.; Ferrante, M. Biological therapy targeting the IL-23/IL-17 axis in inflammatory bowel disease. Expert Opin. Biol. Ther. 2017, 17, 31–47. [Google Scholar] [CrossRef]
- Nemoto, Y.; Kanai, T.; Tohda, S.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; Sakamoto, N.; Fukuda, T.; Miura, O.; et al. Negative feedback regulation of colitogenic CD4+ T cells by increased granulopoiesis. Inflamm. Bowel Dis. 2008, 14, 1491–1503. [Google Scholar] [CrossRef]
- Zhang, R.; Ito, S.; Nishio, N.; Cheng, Z.; Suzuki, H.; Isobe, K. Up-regulation of Gr1+CD11b+ population in spleen of dextran sulfate sodium administered mice works to repair colitis. Inflamm. Allergy Drug. Targets 2011, 10, 39–46. [Google Scholar] [CrossRef]
- Buchholz, B.M.; Shapiro, R.A.; Vodovotz, Y.; Billiar, T.R.; Sodhi, C.P.; Hackam, D.J.; Bauer, A.J. Myocyte TLR4 enhances enteric and systemic inflammation driving late murine endotoxic ileus. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G852–G862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, A.B.; Hetz, R.A.; Xue, H.; Aroom, K.R.; Bhattarai, D.; Johnson, E.; Bedi, S.; Cox, C.S., Jr.; Uray, K. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol. Motil. 2013, 25, 593-e463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docsa, T.; Bhattarai, D.; Sipos, A.; Wade, C.E.; Cox, C.S., Jr.; Uray, K. CXCL1 is upregulated during the development of ileus resulting in decreased intestinal contractile activity. Neurogastroenterol. Motil. 2020, 32, e13757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, E.; Ehala-Aleksejev, K.; Guiot, Y.; Detry, R.; Vandenhooft, A.; Brichard, S.M. Adipokines oversecreted by omental adipose tissue in human obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E656–E665. [Google Scholar] [CrossRef] [Green Version]
- Maury, E.; Brichard, S.M.; Pataky, Z.; Carpentier, A.; Golay, A.; Bobbioni-Harsch, E. Effect of obesity on growth-related oncogene factor-alpha, thrombopoietin, and tissue inhibitor metalloproteinase-1 serum levels. Obesity 2010, 18, 1503–1509. [Google Scholar] [CrossRef]
- Oliveira, M.C.; Menezes-Garcia, Z.; Henriques, M.C.; Soriani, F.M.; Pinho, V.; Faria, A.M.; Santiago, A.F.; Cara, D.C.; Souza, D.G.; Teixeira, M.M.; et al. Acute and sustained inflammation and metabolic dysfunction induced by high refined carbohydrate-containing diet in mice. Obesity 2013, 21, E396–E406. [Google Scholar] [CrossRef]
- Grigoryev, D.N.; Cheranova, D.I.; Heruth, D.P.; Huang, P.; Zhang, L.Q.; Rabb, H.; Ye, S.Q. Meta-analysis of molecular response of kidney to ischemia reperfusion injury for the identification of new candidate genes. BMC Nephrol. 2013, 14, 231. [Google Scholar] [CrossRef] [Green Version]
- Daemen, M.A.; de Vries, B.; van’t Veer, C.; Wolfs, T.G.; Buurman, W.A. Apoptosis and chemokine induction after renal ischemia-reperfusion. Transplantation 2001, 71, 1007–1011. [Google Scholar] [CrossRef]
- Thurman, J.M.; Lenderink, A.M.; Royer, P.A.; Coleman, K.E.; Zhou, J.; Lambris, J.D.; Nemenoff, R.A.; Quigg, R.J.; Holers, V.M. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J. Immunol. 2007, 178, 1819–1828. [Google Scholar] [CrossRef] [Green Version]
- de Vries, B.; Köhl, J.; Leclercq, W.K.; Wolfs, T.G.; van Bijnen, A.A.; Heeringa, P.; Buurman, W.A. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J. Immunol. 2003, 170, 3883–3889. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Fu, X.; Zhang, Q.W.; Remick, D.G.; Fairchild, R.L. Neutralization of Gro alpha and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am. J. Pathol. 2001, 159, 2137–2145. [Google Scholar] [CrossRef]
- Cugini, D.; Azzollini, N.; Gagliardini, E.; Cassis, P.; Bertini, R.; Colotta, F.; Noris, M.; Remuzzi, G.; Benigni, A. Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia/reperfusion. Kidney Int. 2005, 67, 1753–1761. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, N.; Fukazawa, N.; Nonomura, K.; Fairchild, R.L. Cathepsin g is required for sustained inflammation and tissue injury after reperfusion of ischemic kidneys. Am. J. Pathol. 2007, 170, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Chujo, K.; Ueki, M.; Asaga, T.; Taie, S. Atrial natriuretic peptide attenuates ischemia/reperfusion-induced renal injury by reducing neutrophil activation in rats. Tohoku J. Exp. Med. 2008, 215, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Fukuzawa, N.; Schenk, A.D.; Petro, M.; Nonomura, K.; Baldwin, W.M., 3rd; Fairchild, R.L. High renal ischemia temperature increases neutrophil chemoattractant production and tissue injury during reperfusion without an identifiable role for CD4 T cells in the injury. Transpl. Immunol. 2009, 22, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Hess, A.P.; Hamilton, A.E.; Talbi, S.; Dosiou, C.; Nyegaard, M.; Nayak, N.; Genbecev-Krtolica, O.; Mavrogianis, P.; Ferrer, K.; Kruessel, J.; et al. Decidual stromal cell response to paracrine signals from the trophoblast: Amplification of immune and angiogenic modulators. Biol. Reprod. 2007, 76, 102–117. [Google Scholar] [CrossRef] [Green Version]
- Baston-Büst, D.M.; Schanz, A.; Böddeker, S.J.; Altergot-Ahmad, O.; Krüssel, J.S.; Rein, D.; Hess, A.P. CXCL1 expression in human decidua in vitro is mediated via the MAPK signalling cascade. Cytokine 2013, 64, 79–85. [Google Scholar] [CrossRef]
- Ma, C.; Liu, G.; Liu, W.; Xu, W.; Li, H.; Piao, S.; Sui, Y.; Feng, W. CXCL1 stimulates decidual angiogenesis via the VEGF-A pathway during the first trimester of pregnancy. Mol. Cell Biochem. 2021, 476, 2989–2998. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, X.; Liu, Z.; Xu, H.; Wang, T.; He, L.; Zhao, A. CXCR2-Mediated Granulocytic Myeloid-Derived Suppressor Cells’ Functional Characterization and Their Role in Maternal Fetal Interface. DNA Cell Biol. 2016, 35, 358–365. [Google Scholar] [CrossRef]
- Jung, J.H.; Kang, K.W.; Kim, J.; Hong, S.C.; Park, Y.; Kim, B.S. CXCR2 Inhibition in Human Pluripotent Stem Cells Induces Predominant Differentiation to Mesoderm and Endoderm Through Repression of mTOR, β-Catenin, and hTERT Activities. Stem Cells Dev. 2016, 25, 1006–1019. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kang, K.W.; Kim, J.H.; Lee, B.H.; Jung, J.H.; Park, Y.; Hong, S.C.; Kim, B.S. CXCR2 Ligands and mTOR Activation Enhance Reprogramming of Human Somatic Cells to Pluripotent Stem Cells. Stem Cells Dev. 2020, 29, 119–132. [Google Scholar] [CrossRef]
- Krtolica, A.; Larocque, N.; Genbacev, O.; Ilic, D.; Coppe, J.P.; Patil, C.K.; Zdravkovic, T.; McMaster, M.; Campisi, J.; Fisher, S.J. GROα regulates human embryonic stem cell self-renewal or adoption of a neuronal fate. Differentiation 2011, 81, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Lee, S.J.; Kim, J.; Lee, S.; Sung, H.J.; An, J.; Park, Y.; Kim, B.S. CXCR2 and its related ligands play a novel role in supporting the pluripotency and proliferation of human pluripotent stem cells. Stem Cells Dev. 2015, 24, 948–961. [Google Scholar] [CrossRef] [Green Version]
- Królak-Olejnik, B.; Beck, B.; Olejnik, I. Umbilical serum concentrations of chemokines (RANTES and MGSA/GRO-alpha) in preterm and term neonates. Pediatr. Int. 2006, 48, 586–590. [Google Scholar] [CrossRef]
- Sullivan, S.E.; Staba, S.L.; Gersting, J.A.; Hutson, A.D.; Theriaque, D.; Christensen, R.D.; Calhoun, D.A. Circulating concentrations of chemokines in cord blood, neonates, and adults. Pediatr. Res. 2002, 51, 653–657. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.W.; van den Berg, H.A.; Moore, J.D.; Quenby, S.; Blanks, A.M. Assessment of myometrial transcriptome changes associated with spontaneous human labour by high-throughput RNA-seq. Exp. Physiol. 2014, 99, 510–524. [Google Scholar] [CrossRef]
- Saliba, J.; Coutaud, B.; Solovieva, V.; Lu, F.; Blank, V. Regulation of CXCL1 chemokine and CSF3 cytokine levels in myometrial cells by the MAFF transcription factor. J. Cell Mol. Med. 2019, 23, 2517–2525. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.G.; Lenk, E.E.; Lebovic, D.I.; Shu, Y.; Yu, J.; Taylor, R.N. Pathogenesis of endometriosis: Interaction between Endocrine and inflammatory pathways. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 50–60. [Google Scholar] [CrossRef]
- Vannuccini, S.; Petraglia, F. Recent advances in understanding and managing adenomyosis. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, V.H.; Arbib, N.; Schiff, E.; Goldenberg, M.; Seidman, D.S.; Soriano, D. Sonographic Signs of Adenomyosis Are Prevalent in Women Undergoing Surgery for Endometriosis and May Suggest a Higher Risk of Infertility. BioMed Res. Int. 2017, 2017, 8967803. [Google Scholar] [CrossRef]
- Oral, E.; Seli, E.; Bahtiyar, M.O.; Olive, D.L.; Arici, A. Growth-regulated alpha expression in the peritoneal environment with endometriosis. Obstet. Gynecol. 1996, 88, 1050–1056. [Google Scholar] [CrossRef]
- Takamura, M.; Osuga, Y.; Izumi, G.; Yoshino, O.; Koga, K.; Saito, A.; Hirata, T.; Hirota, Y.; Harada, M.; Hasegawa, A.; et al. Interleukin-17A is present in neutrophils in endometrioma and stimulates the secretion of growth-regulated oncogene-α (Gro-α) from endometrioma stromal cells. Fertil. Steril. 2012, 98, 1218–1224. [Google Scholar] [CrossRef]
- Lai, T.H.; Wu, P.H.; Wu, W.B. Involvement of NADPH oxidase and NF-κB activation in CXCL1 induction by vascular endothelial growth factor in human endometrial epithelial cells of patients with adenomyosis. J. Reprod. Immunol. 2016, 118, 61–69. [Google Scholar] [CrossRef]
- Pateisky, P.; Pils, D.; Kuessel, L.; Szabo, L.; Walch, K.; Obwegeser, R.; Wenzl, R.; Yotova, I. The Serum Levels of the Soluble Factors sCD40L and CXCL1 Are Not Indicative of Endometriosis. Biomed. Res. Int. 2016, 2016, 2857161. [Google Scholar] [CrossRef]
- Furuya, M.; Suyama, T.; Usui, H.; Kasuya, Y.; Nishiyama, M.; Tanaka, N.; Ishiwata, I.; Nagai, Y.; Shozu, M.; Kimura, S. Up-regulation of CXC chemokines and their receptors: Implications for proinflammatory microenvironments of ovarian carcinomas and endometriosis. Hum. Pathol. 2007, 38, 1676–1687. [Google Scholar] [CrossRef]
- Malhotra, N.; Karmakar, D.; Tripathi, V.; Luthra, K.; Kumar, S. Correlation of angiogenic cytokines-leptin and IL-8 in stage, type and presentation of endometriosis. Gynecol. Endocrinol. 2012, 28, 224–227. [Google Scholar] [CrossRef]
- Ahn, S.H.; Edwards, A.K.; Singh, S.S.; Young, S.L.; Lessey, B.A.; Tayade, C. IL-17A Contributes to the Pathogenesis of Endometriosis by Triggering Proinflammatory Cytokines and Angiogenic Growth Factors. J. Immunol. 2015, 195, 2591–2600. [Google Scholar] [CrossRef] [Green Version]
- Young, V.J.; Ahmad, S.F.; Brown, J.K.; Duncan, W.C.; Horne, A.W. Peritoneal VEGF-A expression is regulated by TGF-β1 through an ID1 pathway in women with endometriosis. Sci. Rep. 2015, 5, 16859. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, J.; Man, G.C.W.; Leung, K.T.; Liang, B.; Xiao, B.; Ma, X.; Huang, S.; Huang, H.; Hegde, V.L.; et al. MDSCs drive the process of endometriosis by enhancing angiogenesis and are a new potential therapeutic target. Eur. J. Immunol. 2018, 48, 1059–1073. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Li, B.; Wang, X.; Sun, K.; Guo, Y. Therapeutic inhibition of CXC chemokine receptor 2 by SB225002 attenuates LPS-induced acute lung injury in mice. Arch. Med. Sci. 2018, 14, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, A.; Rojewska, E.; Pawlik, K.; Kreiner, G.; Ciechanowska, A.; Makuch, W.; Nalepa, I.; Mika, J. Pharmacological Blockade of Spinal CXCL3/CXCR2 Signaling by NVP CXCR2 20, a Selective CXCR2 Antagonist, Reduces Neuropathic Pain Following Peripheral Nerve Injury. Front. Immunol. 2019, 10, 2198. [Google Scholar] [CrossRef] [Green Version]
- Miyake, M.; Furuya, H.; Onishi, S.; Hokutan, K.; Anai, S.; Chan, O.; Shi, S.; Fujimoto, K.; Goodison, S.; Cai, W.; et al. Monoclonal Antibody against CXCL1 (HL2401) as a Novel Agent in Suppressing IL6 Expression and Tumoral Growth. Theranostics 2019, 9, 853–867. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbecki, J.; Szatkowska, I.; Kupnicka, P.; Żwierełło, W.; Barczak, K.; Poziomkowska-Gęsicka, I.; Wójcik, J.; Chlubek, D.; Baranowska-Bosiacka, I. The Importance of CXCL1 in the Physiological State and in Noncancer Diseases of the Oral Cavity and Abdominal Organs. Int. J. Mol. Sci. 2022, 23, 7151. https://doi.org/10.3390/ijms23137151
Korbecki J, Szatkowska I, Kupnicka P, Żwierełło W, Barczak K, Poziomkowska-Gęsicka I, Wójcik J, Chlubek D, Baranowska-Bosiacka I. The Importance of CXCL1 in the Physiological State and in Noncancer Diseases of the Oral Cavity and Abdominal Organs. International Journal of Molecular Sciences. 2022; 23(13):7151. https://doi.org/10.3390/ijms23137151
Chicago/Turabian StyleKorbecki, Jan, Iwona Szatkowska, Patrycja Kupnicka, Wojciech Żwierełło, Katarzyna Barczak, Iwona Poziomkowska-Gęsicka, Jerzy Wójcik, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2022. "The Importance of CXCL1 in the Physiological State and in Noncancer Diseases of the Oral Cavity and Abdominal Organs" International Journal of Molecular Sciences 23, no. 13: 7151. https://doi.org/10.3390/ijms23137151