
Arabinoxylan and Pectin Metabolism in Crohn’s Disease Microbiota: An In Silico Study


Abstract
:1. Introduction
2. Results and Discussion
2.1. MAGs Recovery
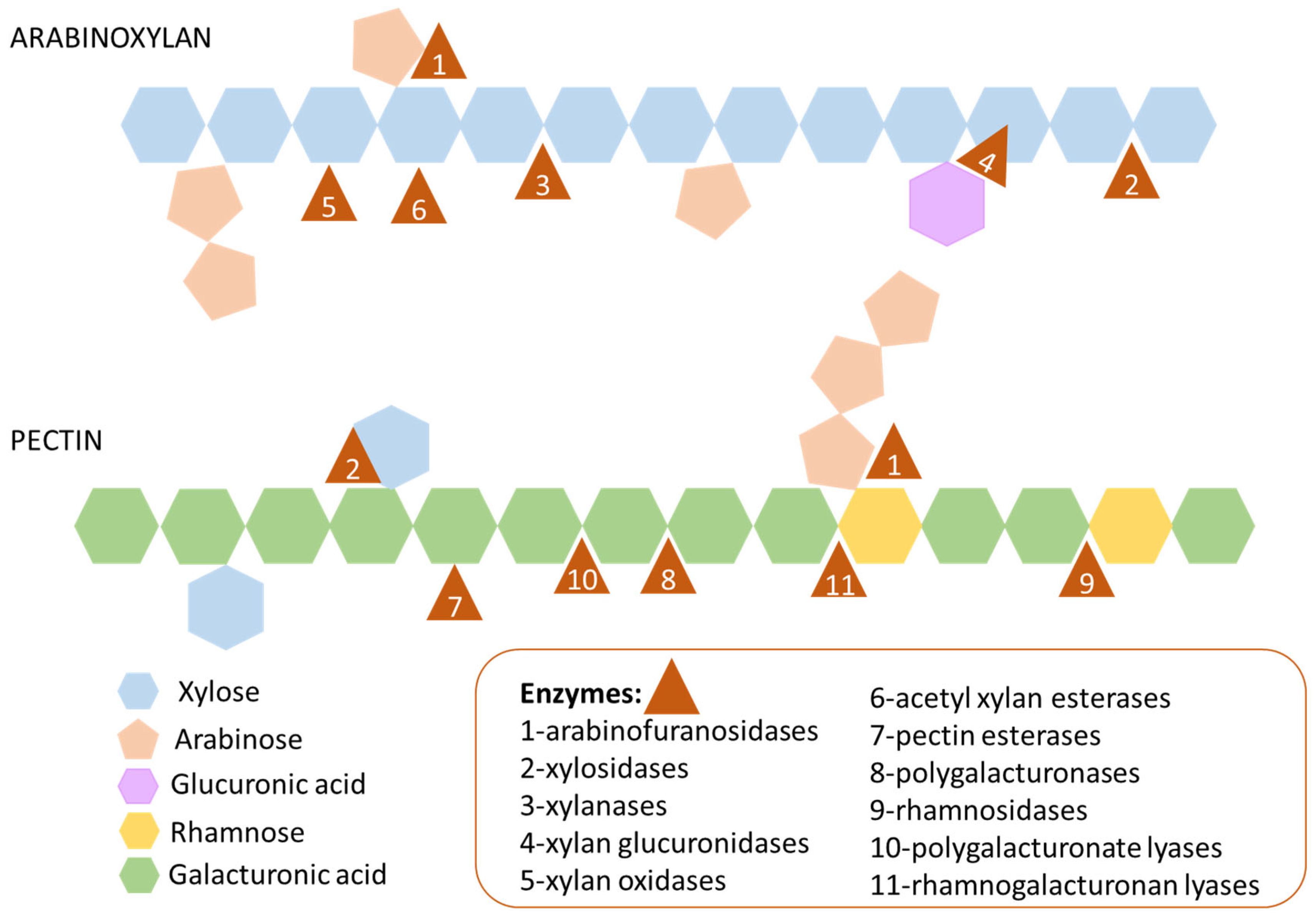
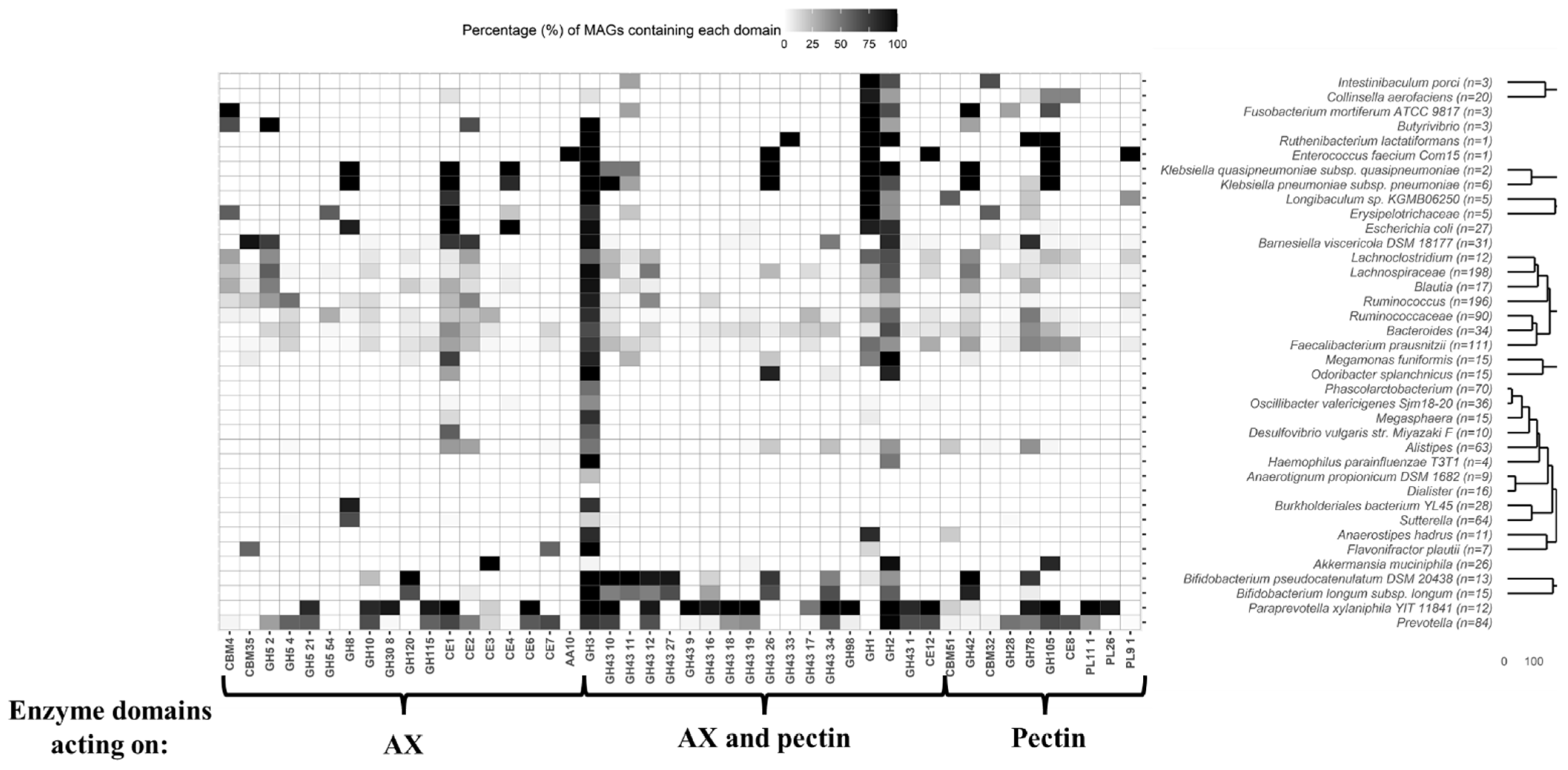
2.2. Study of MAGs Glycosidases Acting on Arabinoxylan and Pectin
2.3. Cross-Feeding between MAGs in the Presence of Arabinoxylan and Pectin
3. Materials and Methods
3.1. Metagenome Selection
3.2. Metagenome Assembly
3.3. In Silico Study of Arabinoxylan and Pectin Metabolism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.F.; Guan, X.X.; Tang, Y.J.; Sun, J.F.; Wang, X.K.; Wang, W.D.; Fan, J.M. Clinical effects and gut microbiota changes of using probiotics, prebiotics or synbiotics in inflammatory bowel disease: A systematic review and meta-analysis. Eur. J. Nutr. 2021, 60, 2855–2875. [Google Scholar] [CrossRef]
- Jairath, V.; Feagan, B.G. Global burden of inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2020, 5, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, D.; Saadah, O.; Mosli, M.; Edris, S.; Alhindi, R.; Bahieldin, A. Dysbiosis of gut microbiota in inflammatory bowel disease: Current therapies and potential for microbiota-modulating therapeutic approaches. Bosn. J. Basic Med. Sci. 2021, 21, 270. [Google Scholar] [CrossRef]
- Asnicar, F.; Berry, S.E.; Valdes, A.M.; Nguyen, L.H.; Piccinno, G.; Drew, D.A.; Leeming, E.; Gibson, R.; Le Roy, C.; Al Khatib, H.; et al. Microbiome connections with host metabolism and habitual diet from 1098 deeply phenotyped individuals. Nat. Med. 2021, 27, 321–332. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Kolmogorov, M.; Tseng, E.; Portik, D.M.; Korobeynikov, A.; Tolstoganov, I.; Uritskiy, G.; Liachko, I.; Sullivan, S.T.; Shin, S.B.; et al. Generating lineage-resolved, complete metagenome-assembled genomes from complex microbial communities. Nat. Biotechnol. 2022, 40, 711–719. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Belcour, A.; Frioux, C.; Aite, M.; Bretaudeau, A.; Hildebrand, F.; Siegel, A. Metage2Metabo, microbiota-scale metabolic complementarity for the identification of key species. Elife 2020, 9, e61968. [Google Scholar] [CrossRef]
- Sabater, C.; Ruiz, L.; Margolles, A. A Machine Learning Approach to Study Glycosidase Activities from Bifidobacterium. Microorganisms 2021, 9, 1034. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 2021, 22, 81. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef]
- Sugihara, K.; Kamada, N. Diet–Microbiota Interactions in Inflammatory Bowel Disease. Nutrients 2021, 13, 1533. [Google Scholar] [CrossRef]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The impact of western diet and nutrients on the microbiota and immune response at mucosal interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [Green Version]
- Sabater, C.; Calvete-Torre, I.; Villamiel, M.; Moreno, F.J.; Margolles, A.; Ruiz, L. Vegetable waste and by-products to feed a healthy gut microbiota: Current evidence, machine learning and computational tools to design novel microbiome-targeted foods. Trends Food Sci. Technol. 2021, 118, 399–417. [Google Scholar] [CrossRef]
- Lin, T.L.; Shu, C.C.; Lai, W.F.; Tzeng, C.M.; Lai, H.C.; Lu, C.C. Investiture of next generation probiotics on amelioration of diseases–Strains do matter. Med. Microecol. 2019, 1, 100002. [Google Scholar] [CrossRef]
- Lordan, C.; Thapa, D.; Ross, R.P.; Cotter, P.D. Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 2020, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Hadji, H.; Bouchemal, K. Advances in the treatment of inflammatory bowel disease: Focus on polysaccharide nanoparticulate drug delivery systems. Adv. Drug Deliv. Rev. 2022, 181, 114101. [Google Scholar] [CrossRef]
- Sabater, C.; Molina-Tijeras, J.A.; Vezza, T.; Corzo, N.; Montilla, A.; Utrilla, P. Intestinal anti-inflammatory effects of artichoke pectin and modified pectin fractions in the dextran sulfate sodium model of mice colitis. Artificial neural network modelling of inflammatory markers. Food Funct. 2019, 10, 7793–7805. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Masoodi, F.A.; Gani, A.; Gani, A.; Noor, N.; Fazli, A. Arabinoxylans. In Food Biopolymers: Structural, Functional and Nutraceutical Properties; Gani, A., Ashwar, B.A., Eds.; Springer: Cham, Switzerland, 2021; pp. 173–186. [Google Scholar] [CrossRef]
- Mathew, S.; Aronsson, A.; Karlsson, E.N.; Adlercreutz, P. Xylo-and arabinoxylooligosaccharides from wheat bran by endoxylanases, utilisation by probiotic bacteria, and structural studies of the enzymes. Appl. Microbiol. Biotechnol. 2018, 102, 3105–3120. [Google Scholar] [CrossRef]
- Tan, H.; Nie, S. Deciphering diet-gut microbiota-host interplay: Investigations of pectin. Trends Food Sci. Technol. 2020, 106, 171–181. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.; Xu, L.; Wang, Z.; Zhao, Y.; Chen, X. Effects of dietary fiber levels on cecal microbiota composition in geese. Asian Australas. J. Anim. Sci. 2018, 31, 1285. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The controversial role of human gut lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Breyner, N.M.; Michon, C.; de Sousa, C.S.; Vilas Boas, P.B.; Chain, F.; Azevedo, V.A.; Langella, P.; Chatel, J.M. Microbial anti-inflammatory molecule (MAM) from Faecalibacterium prausnitzii shows a protective effect on DNBS and DSS-induced colitis model in mice through inhibition of NF-κB pathway. Front. Microbiol. 2017, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Andoh, A.; Sugimoto, M. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in CD. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in CD revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Russell, W.R.; Quartieri, A.; Rossi, M.; Parkhill, J.; Walker, A.W.; Flint, H.J. Wheat bran promotes enrichment within the human colonic microbiota of butyrate-producing bacteria that release ferulic acid. Environ. Microbiol. 2016, 18, 2214–2225. [Google Scholar] [CrossRef] [Green Version]
- Zeybek, N.; Rastall, R.A.; Buyukkileci, A.O. Utilization of xylan-type polysaccharides in co-culture fermentations of Bifidobacterium and Bacteroides species. Carbohydr. Polym. 2020, 236, 116076. [Google Scholar] [CrossRef]
- Zhai, Q.; Feng, S.; Arjan, N.; Chen, W. A next generation probiotic, Akkermansia muciniphila. Crit. Rev. Food Sci. Nutr. 2019, 59, 3227–3236. [Google Scholar] [CrossRef]
- Zhu, K.; Mao, G.; Wu, D.; Yu, C.; Cheng, H.; Xiao, H.; Ye, X.; Linhardt, R.J.; Orfila, C.; Chen, S. Highly branched RG-I domain enrichment is indispensable for pectin mitigating against high-fat diet-induced obesity. J. Agric. Food Chem. 2020, 68, 8688–8701. [Google Scholar] [CrossRef]
- Lindsay, J.O.; Whelan, K.; Stagg, A.J.; Gobin, P.; Al-Hassi, H.O.; Rayment, N.; Kamm, M.A.; Kinight, S.C.; Forbes, A. Clinical, microbiological, and immunological effects of fructo-oligosaccharide in patients with Crohn’s disease. Gut 2006, 55, 348–355. [Google Scholar] [CrossRef]
- Wilson, B.; Whelan, K. Prebiotic inulin-type fructans and galacto-oligosaccharides: Definition, specificity, function, and application in gastrointestinal disorders. J. Gastroenterol. Hepatol. 2017, 32, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Kasmanas, J.C.; Bartholomäus, A.; Corrêa, F.B.; Tal, T.; Jehmlich, N.; Herberth, G.; von Bergen, M.; Stadler, P.F.; de Leon Ferreira de Carvalho, A.C.P.; da Rocha, U.N. HumanMetagenomeDB: A public repository of curated and standardized metadata for human metagenomes. Nucleic Acids Res. 2021, 49, D743–D750. [Google Scholar] [CrossRef]
- Fraser-Liggett, C. Metagenomic Analysis of the Structure and Function of the Human Gut Microbiota in CD. Nat. Preced. 2010, 1. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Vaughn, B.P.; Vatanen, T.; Allegretti, J.R.; Bai, A.; Xavier, R.J.; Korzenik, J.; Gevers, D.; Ting, A.; Robson, S.C.; Moss, A.C. Increased intestinal microbial diversity following fecal microbiota transplant for active CD. Inflamm. Bowel Dis. 2016, 22, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 38. [Google Scholar] [CrossRef]
- Džunková, M.; Low, S.J.; Daly, J.N.; Deng, L.; Rinke, C.; Hugenholtz, P. Defining the human gut host–phage network through single-cell viral tagging. Nat. Microbiol. 2019, 4, 2192–2203. [Google Scholar] [CrossRef]
- Fukuyama, J.; Rumker, L.; Sankaran, K.; Jeganathan, P.; Dethlefsen, L.; Relman, D.A.; Holmes, S.P. Multidomain analyses of a longitudinal human microbiome intestinal cleanout perturbation experiment. PLoS Comput. Biol. 2017, 13, e1005706. [Google Scholar] [CrossRef] [Green Version]
- Hiseni, P.; Rudi, K.; Wilson, R.C.; Hegge, F.T.; Snipen, L. HumGut: A comprehensive human gut prokaryotic genomes collection filtered by metagenome data. Microbiome 2021, 9, 165. [Google Scholar] [CrossRef]
- Minot, S.; Grunberg, S.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. USA 2012, 109, 3962–3966. [Google Scholar] [CrossRef] [Green Version]
- Nanjundappa, R.H.; Ronchi, F.; Wang, J.; Clemente-Casares, X.; Yamanouchi, J.; Umeshappa, C.S.; Yang, Y.; Blanco, J.; Bassolas-Molina, H.; Salas, A.; et al. A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell 2017, 171, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Petersen, L.M.; Bautista, E.J.; Nguyen, H.; Hanson, B.M.; Chen, L.; Lek, S.H.; Sodergren, E.; Weinstock, G.M. Community characteristics of the gut microbiomes of competitive cyclists. Microbiome 2017, 5, 98. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1. 0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.E.; Paladin, L.; Rai, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAGs Found in the Microbiota of Patients with Crohn’s Disease That were also Found in the Microbiota of Healthy Individuals | |
|---|---|
| Taxa | n |
| Lachnospiraceae | 174 |
| Alistipes | 113 |
| Faecalibacterium prausnitzii | 100 |
| Ruminococcus | 96 |
| Sutterella | 67 |
| Prevotella | 61 |
| [Eubacterium] eligens ATCC 27750 | 41 |
| Oscillibacter valericigenes Sjm18–20 | 39 |
| Bacteroides | 36 |
| Akkermansia muciniphila | 35 |
| Escherichia coli | 33 |
| Barnesiella viscericola DSM 18177 | 25 |
| Phascolarctobacterium | 23 |
| Acidaminococcus intestini RyC-MR95 | 22 |
| Blautia | 17 |
| Ruminococcaceae | 16 |
| Burkholderiales bacterium YL45 | 13 |
| Dialister | 13 |
| Flavonifractor plautii | 11 |
| Lachnoclostridium | 10 |
| Megasphaera | 6 |
| Paraprevotella xylaniphila YIT 11841 | 6 |
| Anaerostipes hadrus | 5 |
| Odoribacter splanchnicus | 5 |
| Anaerotignum propionicum DSM 1682 | 4 |
| Desulfovibrio vulgaris str. Miyazaki F | 4 |
| Clostridiales bacterium CCNA10 | 3 |
| Bifidobacterium longum subsp. longum | 2 |
| Bifidobacterium pseudocatenulatum DSM 20438 = JCM 1200 = LMG 10505 | 2 |
| Butyrivibrio | 2 |
| Haemophilus parainfluenzae T3T1 | 2 |
| Megamonas funiformis | 2 |
| Ruthenibacterium lactatiformans | 2 |
| Collinsella aerofaciens | 1 |
| Enterococcus faecium Com15 | 1 |
| Erysipelotrichaceae | 1 |
| Fusobacterium mortiferum ATCC 9817 | 1 |
| Intestinibaculum porci | 1 |
| Klebsiella pneumoniae subsp. pneumoniae | 1 |
| Klebsiella quasipneumoniae subsp. quasipneumoniae | 1 |
| Longibaculum sp. KGMB06250 | 1 |
| Total | 998 |
| MAGs Found in the Microbiota of Healthy Individuals That Were also Found in the Microbiota of Patients with Crohn’s Disease | |
|---|---|
| Taxa | n |
| Lachnospiraceae | 198 |
| Ruminococcus | 196 |
| Faecalibacterium prausnitzii | 111 |
| Ruminococcaceae | 90 |
| Prevotella | 84 |
| [Eubacterium] eligens ATCC 27750 | 79 |
| Phascolarctobacterium | 70 |
| Sutterella | 64 |
| Alistipes | 63 |
| Oscillibacter valericigenes Sjm18–20 | 36 |
| Bacteroides | 34 |
| Barnesiella viscericola DSM 18177 | 31 |
| Burkholderiales bacterium YL45 | 28 |
| Escherichia coli | 27 |
| Akkermansia muciniphila | 26 |
| Collinsella aerofaciens | 20 |
| Blautia | 17 |
| Dialister | 16 |
| Bifidobacterium longum subsp. longum | 15 |
| Megamonas funiformis | 15 |
| Megasphaera | 15 |
| Odoribacter splanchnicus | 15 |
| Bifidobacterium pseudocatenulatum DSM 20438 = JCM 1200 = LMG 10505 | 13 |
| Lachnoclostridium | 12 |
| Paraprevotella xylaniphila YIT 11841 | 12 |
| Anaerostipes hadrus | 11 |
| Desulfovibrio vulgaris str. Miyazaki F | 10 |
| Anaerotignum propionicum DSM 1682 | 9 |
| Flavonifractor plautii | 7 |
| Klebsiella pneumoniae subsp. pneumoniae | 6 |
| Erysipelotrichaceae | 5 |
| Longibaculum sp. KGMB06250 | 5 |
| Haemophilus parainfluenzae T3T1 | 4 |
| Acidaminococcus intestini RyC-MR95 | 3 |
| Butyrivibrio | 3 |
| Fusobacterium mortiferum ATCC 9817 | 3 |
| Intestinibaculum porci | 3 |
| Klebsiella quasipneumoniae subsp. quasipneumoniae | 2 |
| Clostridiales bacterium CCNA10 | 1 |
| Enterococcus faecium Com15 | 1 |
| Ruthenibacterium lactatiformans | 1 |
| Total | 1361 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabater, C.; Calvete-Torre, I.; Ruiz, L.; Margolles, A. Arabinoxylan and Pectin Metabolism in Crohn’s Disease Microbiota: An In Silico Study. Int. J. Mol. Sci. 2022, 23, 7093. https://doi.org/10.3390/ijms23137093
Sabater C, Calvete-Torre I, Ruiz L, Margolles A. Arabinoxylan and Pectin Metabolism in Crohn’s Disease Microbiota: An In Silico Study. International Journal of Molecular Sciences. 2022; 23(13):7093. https://doi.org/10.3390/ijms23137093
Chicago/Turabian StyleSabater, Carlos, Inés Calvete-Torre, Lorena Ruiz, and Abelardo Margolles. 2022. "Arabinoxylan and Pectin Metabolism in Crohn’s Disease Microbiota: An In Silico Study" International Journal of Molecular Sciences 23, no. 13: 7093. https://doi.org/10.3390/ijms23137093