
Breast Cancer Metastasis: Mechanisms and Therapeutic Implications

Abstract
:1. Introduction
2. EMT in Metastatic Breast Cancer
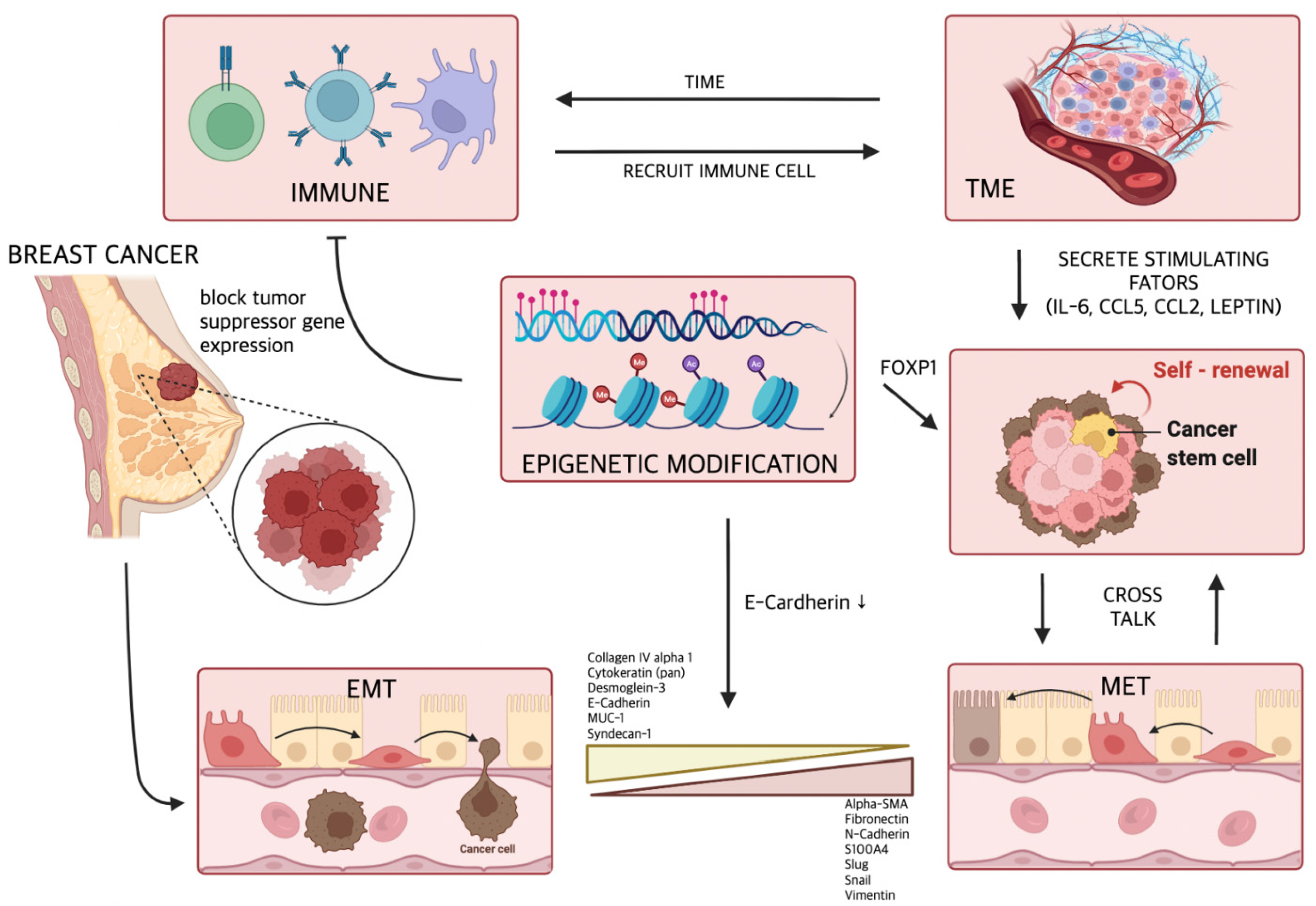
2.1. Relationship between EMT and Metastasis
2.2. EMT Transcription Factors (EMT-TFs) in Metastatic Breast Cancer
2.3. EMT-Mediated Cancer Stem Cells in Metastatic Breast Cancer
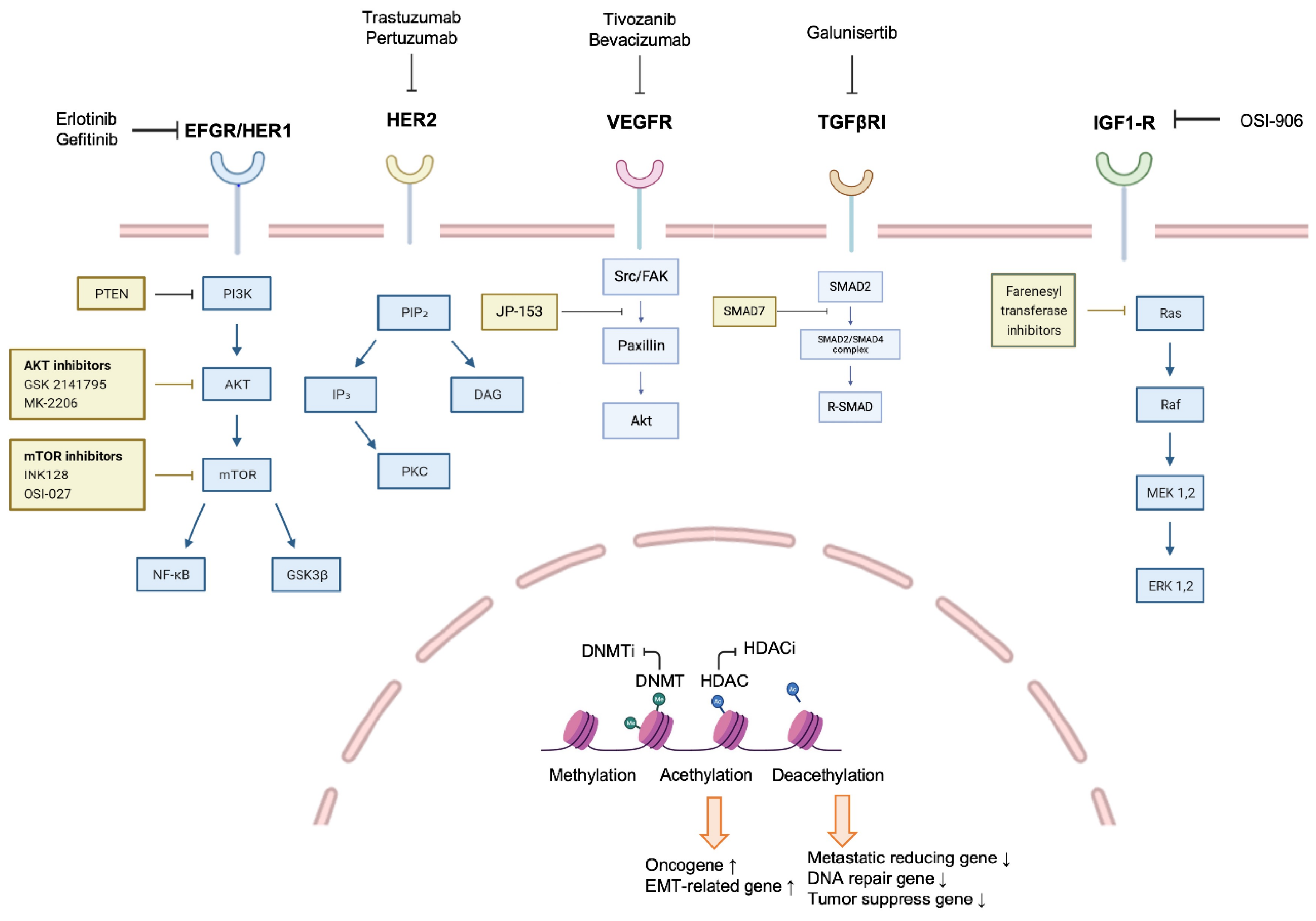
2.4. EMT-Targeted Therapy in Metastatic Breast Cancer
3. Tumor Microenvironment (TME) in Metastatic Breast Cancer
3.1. TME-Associated Cells in Metastatic Breast Cancer
3.2. Notch Signaling in the TME of Metastatic Breast Cancer
3.3. TME-Targeted Therapy in Metastatic Breast Cancer
4. Epigenetic Role in Metastatic Breast Cancer
4.1. Regulation of DNA Methylation in Metastatic Breast Cancer
4.2. Regulation of Histone Modification in Metastatic Breast Cancer
4.3. Epigenetic Change-Targeted Therapeutic Strategies for Metastatic Breast Cancer
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jin, X.; Mu, P. Targeting Breast Cancer Metastasis. Breast Cancer (Auckl.) 2015, 9 (Suppl. 1), 23–34. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef]
- Hicks, D.G.; Kulkarni, S. HER2+ breast cancer: Review of biologic relevance and optimal use of diagnostic tools. Am. J. Clin. Pathol. 2008, 129, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yu, D. Mechanisms of ErbB2-mediated paclitaxel resistance and trastuzumab-mediated paclitaxel sensitization in ErbB2-overexpressing breast cancers. Semin. Oncol. 2001, 28 (Suppl. 16), 12–17. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Andre, F.; Zielinski, C.C. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann. Oncol. 2012, 23 (Suppl. 6), vi46–vi51. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.; Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef]
- Helsby, N.; Yong, M.; Burns, K.; Findlay, M.; Porter, D. Cyclophosphamide bioactivation pharmacogenetics in breast cancer patients. Cancer Chemother. Pharmacol. 2021, 88, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Corso, G.; Frassoni, S.; Girardi, A.; De Camilli, E.; Montagna, E.; Intra, M.; Bottiglieri, L.; Margherita De Scalzi, A.; Fanianos, D.M.; Magnoni, F.; et al. Metaplastic breast cancer: Prognostic and therapeutic considerations. J. Surg. Oncol. 2021, 123, 61–70. [Google Scholar] [CrossRef]
- Mayer, E.L.; Scheulen, M.E.; Beckman, J.; Richly, H.; Duarte, A.; Cotreau, M.M.; Strahs, A.L.; Agarwal, S.; Steelman, L.; Winer, E.P.; et al. A Phase I dose-escalation study of the VEGFR inhibitor tivozanib hydrochloride with weekly paclitaxel in metastatic breast cancer. Breast Cancer Res. Treat. 2013, 140, 331–339. [Google Scholar] [CrossRef]
- Cortes, J.; Kim, S.B.; Chung, W.P.; Im, S.A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.M.; Petry, V.; Chung, C.F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Plosker, G.L.; Faulds, D. Epirubicin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs 1993, 45, 788–856. [Google Scholar] [CrossRef] [PubMed]
- van Denderen, B.J.; Thompson, E.W. Cancer: The to and fro of tumour spread. Nature 2013, 493, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol.-Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, R.J.; Palmieri, D.C.; Bronder, J.L.; Stark, A.M.; Steeg, P.S. Breast cancer metastasis to the central nervous system. Am. J. Pathol. 2005, 167, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Si, W.; Huang, W.; Zheng, Y.; Yang, Y.; Liu, X.; Shan, L.; Zhou, X.; Wang, Y.; Su, D.; Gao, J.; et al. Dysfunction of the Reciprocal Feedback Loop between GATA3- and ZEB2-Nucleated Repression Programs Contributes to Breast Cancer Metastasis. Cancer Cell 2015, 27, 822–836. [Google Scholar] [CrossRef] [Green Version]
- Siersbaek, R.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.P.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor alpha Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [Green Version]
- Marino, N.; Woditschka, S.; Reed, L.T.; Nakayama, J.; Mayer, M.; Wetzel, M.; Steeg, P.S. Breast cancer metastasis: Issues for the personalization of its prevention and treatment. Am. J. Pathol. 2013, 183, 1084–1095. [Google Scholar] [CrossRef] [Green Version]
- Foroni, C.; Broggini, M.; Generali, D.; Damia, G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat. Rev. 2012, 38, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, S.; Quader, S.; Cabral, H.; Ono, R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front. Pharmacol. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Imani, S.; Hosseinifard, H.; Cheng, J.; Wei, C.; Fu, J. Prognostic Value of EMT-inducing Transcription Factors (EMT-TFs) in Metastatic Breast Cancer: A Systematic Review and Meta-analysis. Sci. Rep. 2016, 6, 28587. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gu, L.N.; Shan, B.E.; Geng, C.Z.; Sang, M.X. Biomarkers for EMT and MET in breast cancer: An update (Review). Oncol. Lett. 2016, 12, 4869–4876. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bonisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhang, J.; Yarden, Y.; Fu, L. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct. Target. Ther. 2021, 6, 109. [Google Scholar] [CrossRef]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef]
- Badve, S.; Nakshatri, H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012, 13, e43–e48. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- da Silveira, W.A.; Palma, P.V.B.; Sicchieri, R.D.; Villacis, R.A.R.; Mandarano, L.R.M.; Oliveira, T.M.G.; Antonio, H.M.R.; Andrade, J.M.; Muglia, V.F.; Rogatto, S.R.; et al. Transcription Factor Networks derived from Breast Cancer Stem Cells control the immune response in the Basal subtype. Sci. Rep. 2017, 7, 2851. [Google Scholar] [CrossRef] [PubMed]
- Storci, G.; Bertoni, S.; De Carolis, S.; Papi, A.; Nati, M.; Ceccarelli, C.; Pirazzini, C.; Garagnani, P.; Ferrarini, A.; Buson, G.; et al. Slug/beta-catenin-dependent proinflammatory phenotype in hypoxic breast cancer stem cells. Am. J. Pathol. 2013, 183, 1688–1697. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.; Xia, Y.; Wu, Y.; Zhang, Z.; Wang, X.; Lu, L.; Dai, C.; Song, Y.; Xu, K.; Ji, W.; et al. Lin28B-high breast cancer cells promote immune suppression in the lung pre-metastatic niche via exosomes and support cancer progression. Nat. Commun. 2022, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, K.; Mendoza-Villanueva, D.; Sharan, S.; Summers, G.H.; Dobrolecki, L.E.; Lewis, M.T.; Sterneck, E. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene 2019, 38, 3765–3780. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Sterneck, E. The many faces of C/EBPdelta and their relevance for inflammation and cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Ballester, B.; Milara, J.; Cortijo, J. Pirfenidone anti-fibrotic effects are partially mediated by the inhibition of MUC1 bioactivation. Oncotarget 2020, 11, 1306–1320. [Google Scholar] [CrossRef] [Green Version]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-beta receptor type I inhibitor. Oncotarget 2018, 9, 6659–6677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER(+) Breast Cancer Cells. Clin. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E.; Skuli, N.; Cos, S.; Karpel-Massler, G.; Shiozawa, Y.; Goshen, R.; Halatsch, M.E. The ABC7 regimen: A new approach to metastatic breast cancer using seven common drugs to inhibit epithelial-to-mesenchymal transition and augment capecitabine efficacy. Breast Cancer Targets Ther. 2017, 9, 495–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Yan, Z.; Chen, W.; Wu, Y.; Han, J.; Guo, H.; Qiao, J. TET1 inhibits EMT of ovarian cancer cells through activating Wnt/beta-catenin signaling inhibitors DKK1 and SFRP2. Gynecol. Oncol. 2017, 147, 408–417. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Lu, X.; Wu, J.; Yu, K.; Ji, A.; Lu, W.; Wang, Z.; Wu, J.; et al. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/beta-catenin signaling. Sci. Rep. 2019, 9, 7539. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Yan, W.; Dai, Z.; Gao, X.; Ma, Y.; Xu, Q.; Jiang, J.; Zhang, S. Baicalein suppresses metastasis of breast cancer cells by inhibiting EMT via downregulation of SATB1 and Wnt/beta-catenin pathway. Drug Des. Dev. Ther. 2016, 10, 1419–1441. [Google Scholar] [CrossRef] [Green Version]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieu-Nosjean, M.C.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Sautes-Fridman, C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol. Rev. 2016, 271, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.; Martinez-Ordonez, A.; Fernandez-Garcia, B.; Gonzalez, L.O.; Cid, S.; Dominguez, F.; Perez-Fernandez, R.; Vizoso, F.J. Cancer-associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell. Oncol. (Dordr.) 2018, 41, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [Green Version]
- Picon-Ruiz, M.; Pan, C.; Drews-Elger, K.; Jang, K.; Besser, A.H.; Zhao, D.; Morata-Tarifa, C.; Kim, M.; Ince, T.A.; Azzam, D.J.; et al. Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b–Mediated Malignant Progression. Cancer Res. 2016, 76, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politi, K.; Feirt, N.; Kitajewski, J. Notch in mammary gland development and breast cancer. Semin. Cancer Biol. 2004, 14, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Reedijk, M. Notch Signaling and the Breast Cancer Microenvironment. Adv. Exp. Med. Biol. 2021, 1287, 183–200. [Google Scholar]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Yu, J.; Park, A.; Dubon, M.J.; Do, J.; Kim, Y.; Nam, D.; Noh, J.; Park, K.S. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 2019, 9, 11724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xie, Z.Y.; Guo, X.T.; Xiao, X.H.; Xiong, L.X. Notch and breast cancer metastasis: Current knowledge, new sights and targeted therapy. Oncol. Lett. 2019, 18, 2743–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoda, M.; Principe, S.; Jackson, H.W.; Luga, V.; Fang, H.; Molyneux, S.D.; Shao, Y.W.; Aiken, A.; Waterhouse, P.D.; Karamboulas, C.; et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat. Cell Biol. 2014, 16, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; Hao, L.; et al. Cross-talk between Notch and the Estrogen Receptor in Breast Cancer Suggests Novel Therapeutic Approaches. Cancer Res. 2008, 68, 5226–5235. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Cohen, B.; Goldvasser, P.; Berman, H.; Virtanen, C.; Reedijk, M. Plasminogen Activator uPA Is a Direct Transcriptional Target of the JAG1-Notch Receptor Signaling Pathway in Breast Cancer. Cancer Res. 2011, 71, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Sainson, R.C.; Williams, C.K.; Taylor, J.M.; Shi, W.; Li, J.L.; Harris, A.L. Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc. Res. 2008, 75, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Kitamura, T. Macrophage targeting: Opening new possibilities for cancer immunotherapy. Immunology 2018, 155, 285–293. [Google Scholar] [CrossRef]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.; van Vugt, M.; de Vries, E.G.E.; Schroder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw192. [Google Scholar] [CrossRef] [Green Version]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schafer, R.; Stanley, E.R.; Abraham, D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Xia, J.; Yao, Y.; Gong, D.W.; Shi, H.; Zhou, Q. Sulforaphane inhibits mammary adipogenesis by targeting adipose mesenchymal stem cells. Breast Cancer Res. Treat. 2013, 141, 317–324. [Google Scholar] [CrossRef]
- Locatelli, M.A.; Aftimos, P.; Dees, E.C.; LoRusso, P.M.; Pegram, M.D.; Awada, A.; Huang, B.; Cesari, R.; Jiang, Y.; Shaik, M.N.; et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget 2017, 8, 2320–2328. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Munster, P.N.; Hollebecque, A.; Argiles, G.; Dajani, O.; Cheng, J.D.; Wang, R.; Swift, A.; Tosolini, A.; Gupta, S. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur. J. Cancer 2015, 51, 1865–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, J.Y.; Wahler, J.; Das Gupta, S.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Suh, N. HES1-mediated inhibition of Notch1 signaling by a Gemini vitamin D analog leads to decreased CD44(+)/CD24(-/low) tumor-initiating subpopulation in basal-like breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. 2020, 25, 1058–1109. [Google Scholar]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyko, F.; Brown, R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 2005, 97, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Lee, Z.Y.; Chuah, L.H.; Mai, C.W.; Ngai, S.C. Epigenetics in Metastatic Breast Cancer: Its Regulation and Implications in Diagnosis, Prognosis and Therapeutics. Curr. Cancer Drug Targets 2019, 19, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Huo, Q.; Yang, F.; Xie, N. Perspectives on the Role of Histone Modification in Breast Cancer Progression and the Advanced Technological Tools to Study Epigenetic Determinants of Metastasis. Front. Genet. 2020, 11, 603552. [Google Scholar] [CrossRef]
- Sarrio, D.; Moreno-Bueno, G.; Hardisson, D.; Sanchez-Estevez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Ennour-Idrissi, K.; Dragic, D.; Issa, E.; Michaud, A.; Chang, S.L.; Provencher, L.; Durocher, F.; Diorio, C. DNA Methylation and Breast Cancer Risk: An Epigenome-Wide Study of Normal Breast Tissue and Blood. Cancers 2020, 12, 3088. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Lu, J.; Huang, B.; Wang, Y.; Dong, M.; Fan, D.; Li, H.; Gao, Y.; Hou, P.; et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 2020, 27, 3226–3242. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Rawal, R.M. Genetic and Epigenetic Modulation of Drug Resistance in Cancer: Challenges and Opportunities. Curr. Drug Metab. 2019, 20, 1114–1131. [Google Scholar] [CrossRef] [PubMed]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef]
- Wang, S.M.; Dowhan, D.H.; Muscat, G.E.O. Epigenetic arginine methylation in breast cancer: Emerging therapeutic strategies. J. Mol. Endocrinol. 2019, 62, R223–R237. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandy, D.; Rajam, S.M.; Dutta, D. A three layered histone epigenetics in breast cancer metastasis. Cell Biosci. 2020, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Adler, A.S.; Segal, E.; Chang, H.Y. A transcriptional program mediating entry into cellular quiescence. PLoS Genet. 2007, 3, e91. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lin, C.; Wang, X.; Li, Q.; Li, Y.; Wang, M.; Zhao, Z.; Wu, X.; Shi, D.; Xiao, Y.; et al. Epigenetic silencing of SALL2 confers tamoxifen resistance in breast cancer. EMBO Mol. Med. 2019, 11, e10638. [Google Scholar] [CrossRef]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients with Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Primary/Metastasis | Drug Name | Subtype | Effect | Ref. |
|---|---|---|---|---|
| Primary | Paclitaxel | Taxanes | Inhibit the proliferation of cancer cells by promoting the polymerization of microtubules in cells | [11] |
| Belinostat | Histone deacetylase inhibitor (HDACi) | Epidrug; Treatment of peripheral T-cell lymphoma | [12] | |
| 5-azacitidine | DNA methyltransferase inhibitor (DNMTi) | Epidrug; Treatment of myelodysplastic syndrome | [12] | |
| Tocilizumab (actemra) | Monoclonal Ab | IL-6 inhibitors; Inhibit the formation of cancer stem cells in TNBC cells | [13] | |
| Metastasis | Doxorubicin | Anthracyclins | Interfering with direct apoptotic effects and DNA local isomerase II action | [14] |
| Cyclophosphamide | Alkylating agents | Changes in active or inactive metabolites in the body and anticancer effects of phosphoramide mustard | [15] | |
| Vorinostat | Histone deacetylase inhibitor (HDACi) | Epidrug; Treatment of cutaneous T-cell lymphoma | [12] | |
| Zemetostat | Histone methyltransferase inhibitor (HMTi, EZH1i) | Epidrug; Treatment of epithelioid sarcoma, relapsed or refractory follicular lymphoma | [16] | |
| Atezolizumab +Paclitaxel | Monoclonal Ab | Prolongs progression-free survival in patients with PD-L1-positive breast cancer; Inhibition of metastatic TNBC targeting PD-L1 | [17] | |
| Methotrexate | Antimetabolites | Inhibit nucleotide synthesis; increase AICAR concentration | [18] | |
| Tivozanib | Tyrosine kinase inhibitor (TKI) | Inhibitors of VEGF/VEGFR; Inhibits angiogenesis | [19] | |
| Trastuzumab | Monoclonal antibody | Targets the HER2/neu receptor on cancer cells | [20] | |
| Lapatinib | Tyrosine kinase inhibitor (TKI) | Signal transduction inhibitors of epidermal growth factor receptor (EGFR) and human epidermal receptor type 2 (HER2) | [21] | |
| Palbociclib | Antineoplastic agents | CDK4/6 inhibitor; prevent cells from moving from the G1 to the S cell cycle phase during division | [22] | |
| Pertuzumab | Monoclonal antibody | Targets the HER2/neu receptor; induce antibody-dependent cell-mediated cytotoxicity (ADCC) | [23] | |
| Epirubicin | Anthracyclines | An epimer of doxorubicin; blocks the synthesis of nucleic acids and proteins by inserting planar rings between nucleotide base pairs | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Kim, D.; Ko, S.; Kim, A.; Mo, K.; Yoon, H. Breast Cancer Metastasis: Mechanisms and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 6806. https://doi.org/10.3390/ijms23126806
Park M, Kim D, Ko S, Kim A, Mo K, Yoon H. Breast Cancer Metastasis: Mechanisms and Therapeutic Implications. International Journal of Molecular Sciences. 2022; 23(12):6806. https://doi.org/10.3390/ijms23126806
Chicago/Turabian StylePark, Misung, Dohee Kim, Sunghyub Ko, Ayoung Kim, Kyumin Mo, and Hyunho Yoon. 2022. "Breast Cancer Metastasis: Mechanisms and Therapeutic Implications" International Journal of Molecular Sciences 23, no. 12: 6806. https://doi.org/10.3390/ijms23126806