
Galunisertib Exerts Antifibrotic Effects on TGF-β-Induced Fibroproliferative Dermal Fibroblasts

 , , and
, , and
Abstract
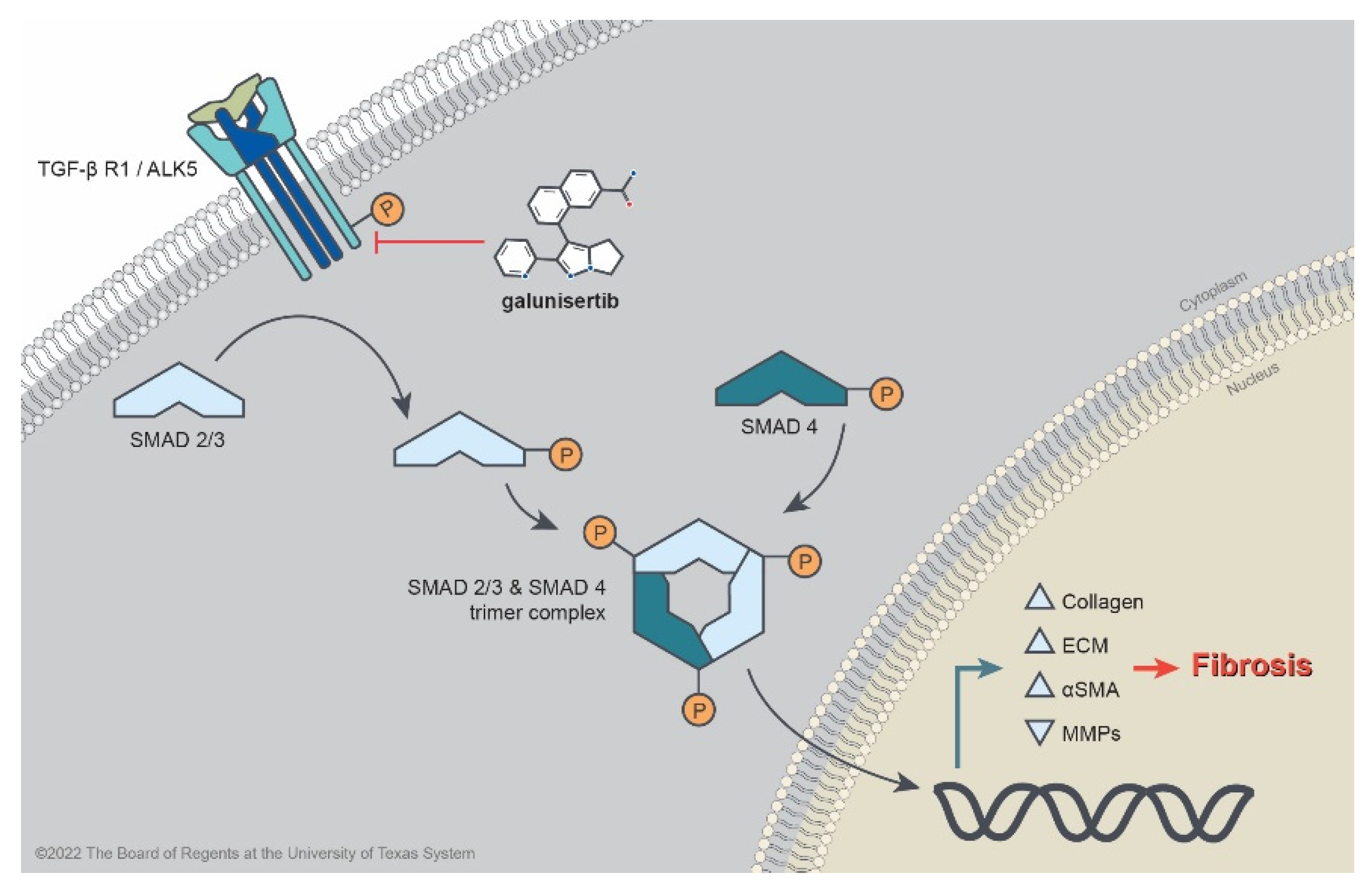
:1. Introduction
2. Results
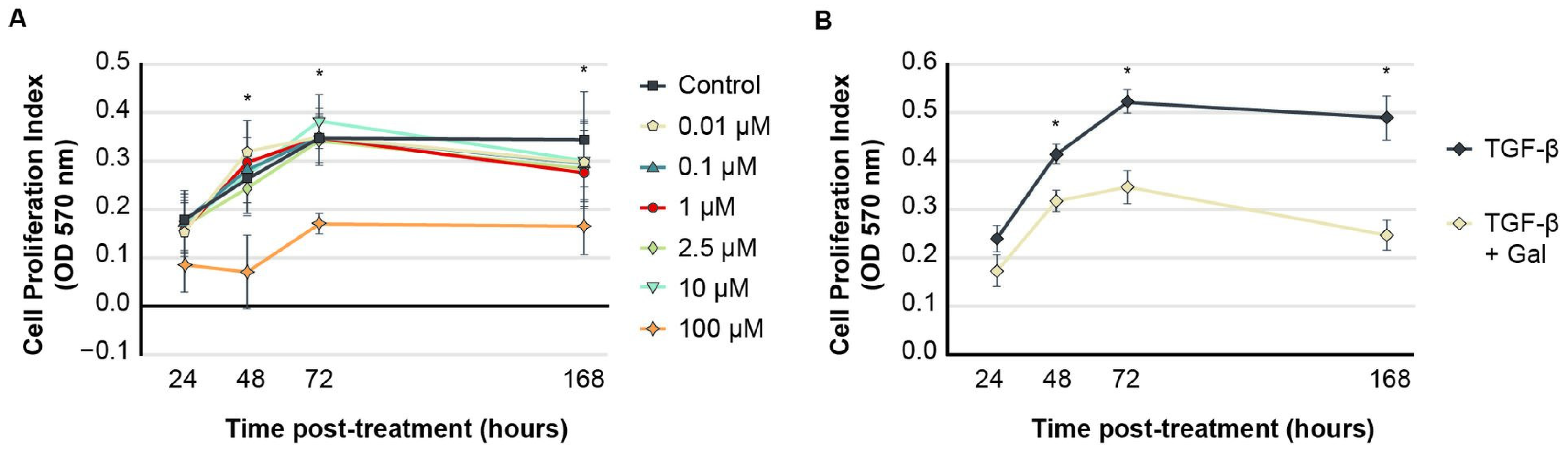
2.1. Galunisertib Normalizes Proliferation Indices in TGF-β-Induced Fibroblasts
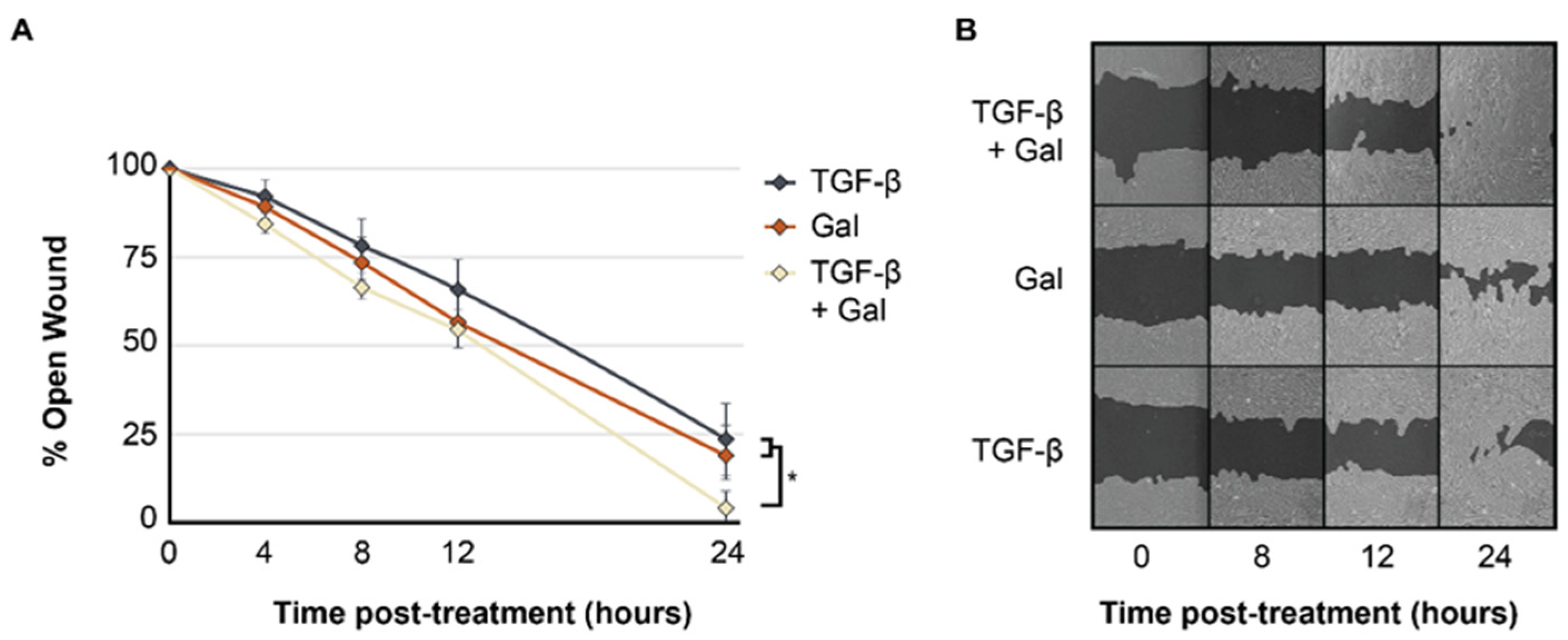
2.2. Galunisertib Expedites Rates of In Vitro Wound Closure in TGF-β-Induced Fibroblasts
2.3. Galunisertib Reduces the Expression of Fibrotic Genes and Promotes the Expression of Antifibrotic Genes in TGF-β-Induced Fibroblasts
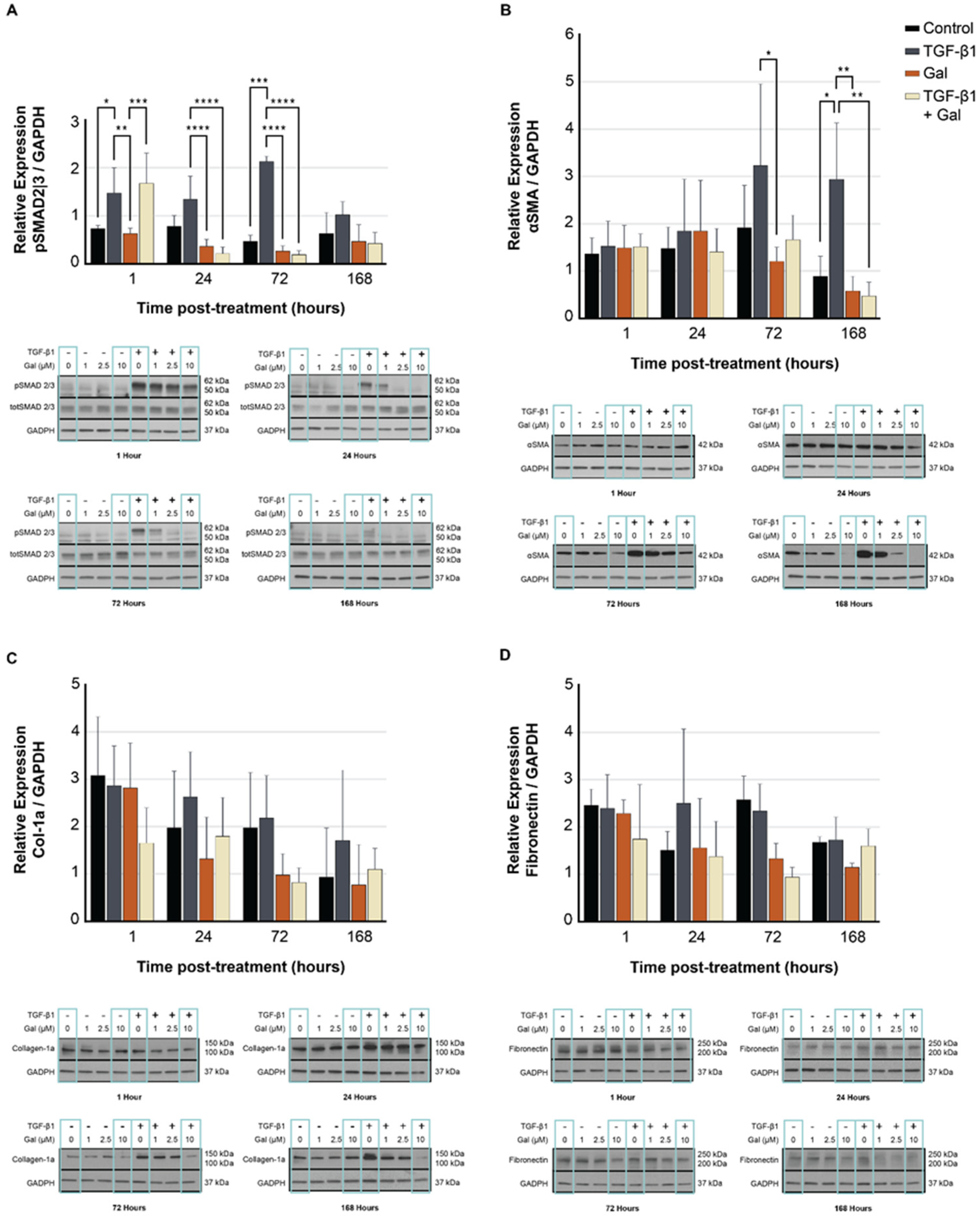
2.4. Galunisertib Diminishes the Production of Fibrotic Proteins in TGF-β-Treated Fibroblasts
3. Materials and Methods
3.1. Reagent and Cell Preparation
3.2. MTT Cell Proliferation Assay
3.3. In Vitro Model of Wound Healing (Scratch Assay)
3.4. Reverse Transcription-Quantitative PCR
3.5. Western Blot
3.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bochaton-Piallat, M.L.; Gabbiani, G.; Hinz, B. The myofibroblast in wound healing and fibrosis: Answered and unanswered questions. F1000Research 2016, 5, F1000 Faculty Rev-1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnson, K.W.; McLean, S.; Di Guglielmo, G.M.; Philip, A. Dynamics of transforming growth factor beta signaling in wound healing and scarring. Adv. Wound Care 2013, 2, 195–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-beta signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chua, C.H.; Wu, X.-L.; Wang, D.-R.; Yin, D.-M.; Cui, L.; Cao, Y.-L.; Longaker, M.T. Inhibiting scar formation in rat cutaneous wounds by blocking TGF-beta signaling. Zhonghua Yi Xue Za Zhi 2003, 83, 31–36. [Google Scholar]
- Bock, O.; Yu, H.; Zitron, S.; Bayat, A.; Ferguson, M.W.; Mrowietz, U. Studies of transforming growth factors beta 1-3 and their receptors I and II in fibroblast of keloids and hypertrophic scars. Acta Derm. Venereol. 2005, 85, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, L.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Raktoe, R.S.; Rietveld, M.H.; Out-Luiting, J.J.; Kruithof-de Julio, M.; van Zuijlen, P.P.; van Doorn, R.; El Ghalbzouri, A. Exon skipping of TGFbetaRI affects signalling and ECM expression in hypertrophic scar-derived fibroblasts. Scars Burn. Heal. 2020, 6, 2059513120908857. [Google Scholar]
- Santini, V.; Valcárcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II study of the ALK5 inhibitor galunisertib in very low-, low-, and intermediate-risk myelodysplastic syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralising antibody to TGF-beta 1,2 reduces cutaneous scarring in adult rodents. J. Cell Sci. 1994, 107 Pt 5, 1137–1157. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X.F.; Wang, Z.C.; Lou, D.; Fang, Q.Q.; Hu, Y.Y.; Zhao, W.Y.; Zhang, L.Y.; Wu, L.H.; Tan, W.Q. Current potential therapeutic strategies targeting the TGF-beta/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. Biomed. Pharmacother. 2020, 129, 110287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shan, S.; Wang, J.; Cheng, X.; Yi, B.; Zhou, J.; Li, Q. Galangin inhibits hypertrophic scar formation via ALK5/Smad2/3 signaling pathway. Mol. Cell Biochem. 2016, 413, 109–118, Erratum in Mol. Cell Biochem. 2020, 468, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Tredget, E.E.; Shankowsky, H.A.; Pannu, R.; Nedelec, B.; Iwashina, T.; Ghahary, A.; Taerum, T.V.; Scott, P.G. Transforming growth factor-beta in thermally injured patients with hypertrophic scars: Effects of interferon alpha-2b. Plast. Reconstr. Surg. 1998, 102, 1317–1328; discussion 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jang, Y.J. Recent understandings of biology, prophylaxis and treatment strategies for hypertrophic scars and keloids. Int. J. Mol. Sci. 2018, 19, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and hypertrophic scars: Pathophysiology, classification, and treatment. Dermatol. Surg. 2017, 43 (Suppl. 1), S3–S18. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Sun, P.; Li, D.; Dong, Z.; Chen, Z. Intralesional injection of botulinum toxin Type A compared with intralesional injection of corticosteroid for the treatment of hypertrophic scar and keloid: A systematic review and meta-analysis. Med. Sci. Monit. 2019, 25, 2950–2958. [Google Scholar] [CrossRef]
- Jones, M.E.; McLane, J.; Adenegan, R.; Lee, J.; Ganzer, C.A. Advancing keloid treatment: A novel multimodal approach to ear keloids. Dermatol. Surg. 2017, 43, 1164–1169. [Google Scholar] [CrossRef]
- O’Brien, L.; Jones, D.J. Silicone gel sheeting for preventing and treating hypertrophic and keloid scars. Cochrane Database Syst. Rev. 2013, 2013, CD003826. [Google Scholar] [CrossRef]
- Bian, F.; Render, J.; Ren, X.-D.; Chio, C.; Chan, K.; Boys, M.; Lala, D.S.; Pocalyko, D. An activin receptor-like kinase 5 inhibitor reduces collagen deposition in a rat dermal incision wound healing model. Plast. Reconstr. Surg. 2011, 128, 451e–459e. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Nokihara, H.; Yamada, Y.; Yamamoto, N.; Sunami, K.; Utsumi, H.; Asou, H.; TakahashI, O.; Ogasawara, K.; Gueorguieva, I.; et al. Phase 1 study of galunisertib, a TGF-beta receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1143–1152. [Google Scholar] [CrossRef]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-betaRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 study of galunisertib (TGF-beta1 receptor Type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Zhou, S.Z.; Cheng, X.Y.; Yi, B.; Shan, S.Z.; Wang, J.; Li, Q.F. Baicalein attenuates hypertrophic scar formation via inhibition of the transforming growth factor-beta/Smad2/3 signalling pathway. Br. J. Dermatol. 2016, 174, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [Green Version]
- El Ayadi, A.; Prasai, A.; Wang, Y.; Herndon, D.N.; Finnerty, C.C. b-Adrenergic receptor trafficking, degradation, and cell surface expression are altered in dermal fibroblasts from hypertrophic scars. J. Investig. Dermatol. 2018, 138, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012, 9, 671–675. [Google Scholar] [CrossRef]
- El Ayadi, A.; Jay, J.W.; Prasai, A. Current approaches targeting the wound healing phases to attenuate fibrosis and scarring. Int. J. Mol. Sci. 2020, 21, 1105. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.; Angeli, M.; Pastrello, C.; McQuaid, R.; Li, H.; Jurisicova, A.; Jurisica, I. Robust quantitative scratch assay. Bioinformatics 2016, 32, 1439–1440. [Google Scholar] [CrossRef] [Green Version]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Sada Pillay, N.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-beta inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharmacol. 2014, 77, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Crowe, M.J.; Doetschman, T.; Greenhalgh, D.G. Delayed wound healing in immunodeficient TGF-beta 1 knockout mice. J. Investig. Dermatol. 2000, 115, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Al-Attar, A.; Mess, S.; Thomassen, J.M.; Kauffman, C.L.; Davison, S.P. Keloid pathogenesis and treatment. Plast. Reconstr. Surg. 2006, 117, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, P.G.; Dodd, C.M.; Tredget, E.E.; Ghahary, A.; Rahemtulla, F. Chemical characterization and quantification of proteoglycans in human post-burn hypertrophic and mature scars. Clin. Sci. 1996, 90, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ezura, Y.; Chervoneva, I.; Robinson, P.S.; Beason, D.P.; Carine, E.T.; Soslowsky, L.J.; Iozzo, R.V.; Birk, D.E. Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J. Cell Biochem. 2006, 98, 1436–1449. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.J.; Liu, Y.; Zhang, X.; Li, Y.Y.; Xu, W.S. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns 2007, 33, 634–641. [Google Scholar] [CrossRef]
- Sayani, K.; Dodd, C.M.; Nedelec, B.; Shen, Y.J.; Ghahary, A.; Tredget, E.E.; Scott, P.P. Delayed appearance of decorin in healing burn scars. Histopathology 2000, 36, 262–272. [Google Scholar] [CrossRef]
- Hammad, S.; Cavalcanti, E.; Werle, J.; Caruso, M.L.; Dropmann, A.; Ignazzi, A.; Ebert, M.P.; Dooley, S.; Giannelli, G. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch. Toxicol. 2018, 92, 2297–2309. [Google Scholar] [CrossRef]
- Isaka, Y. Targeting TGF-beta signaling in kidney fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Morimoto, M.; Tajimi, M.; Inoue, K.; Benhadji, K.A.; Lahn, M.M.F.; Sakai, D. A phase 1b study of transforming growth factor-beta receptor I inhibitor galunisertib in combination with sorafenib in Japanese patients with unresectable hepatocellular carcinoma. Investig. New Drugs 2019, 37, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac safety of TGF-beta receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Treatment | ||||
|---|---|---|---|---|---|
| TGF-β (Comparison Group) | Galunisertib Only | TGF-β + Galunisertib | |||
| 1 h | |||||
| COL1A1 | 1.00 ± 0.04 | ↔ | 1.12 ± 0.12 | ↔ | 1.01 ± 0.02 |
| COL3A1 | 1.01 ± 0.01 | ↔ | 1.30 ± 0.22 | ↔ | 1.02 ± 0.07 |
| DCN | 1.08 ± 0.14 | ↔ | 1.18 ± 0.13 | ↔ | 0.91 ± 0.00 |
| ACTA2 | 1.02 ± 0.07 | ↔ | 1.04 ± 0.17 | ↔ | 0.80 ± 0.11 |
| CTGF | 1.55 ± 0.30 | ↓ | 1.10 ± 0.06 * | ↓ | 1.07 ± 0.18 * |
| FN1 | 1.01 ± 0.06 | ↔ | 1.28 ± 0.09 | ↔ | 0.78 ± 0.20 |
| MMP1 | 1.13 ± 0.16 | ↔ | 1.30 ± 0.13 | ↔ | 0.89 ± 0.02 |
| MMP13 | 1.11 ± 0.12 | ↔ | 1.23 ± 0.17 | ↔ | 1.08 ± 0.24 |
| Day 1 | |||||
| COL1A1 | 1.67 ± 0.12 | ↓ | 0.57 ± 0.07 * | ↓ | 0.69 ± 0.06 * |
| COL3A1 | 1.17 ± 0.07 | ↓ | 0.71 ± 0.17* | ↔ | 0.77 ± 0.23 |
| DCN | 0.38 ± 0.06 | ↔ | 0.79 ± 0.11 | ↔ | 0.79 ± 0.23 |
| ACTA2 | 6.33 ± 2.41 | ↓ | 0.81 ± 0.42 * | ↓ | 0.84 ± 0.51 * |
| CTGF | 1.72 ± 0.55 | ↓ | 0.06 ± 0.02 * | ↓ | 0.07 ± 0.03 * |
| FN1 | 2.93 ± 1.11 | ↓ | 0.76 ± 0.15 * | ↓ | 0.94 ± 0.28 * |
| MMP1 | 0.63 ± 0.06 | ↔ | 0.84 ± 0.17 | ↔ | 0.66 ± 0.13 |
| MMP13 | 0.85 ± 0.07 | ↔ | 0.78 ± 0.05 | ↔ | 0.79 ± 0.21 |
| Day 3 | |||||
| COL1A1 | 2.02 ± 0.16 | ↓ | 0.47 ± 0.05 * | ↓ | 0.48 ± 0.11 * |
| COL3A1 | 1.29 ± 0.17 | ↔ | 0.92 ± 0.32 | ↓ | 0.59 ± 0.12 * |
| DCN | 0.21 ± 0.01 | ↑ | 1.39 ± 0.09 * | ↑ | 1.00 ± 0.31 * |
| ACTA2 | 8.41 ± 2.60 | ↓ | 0.61 ± 0.27 * | ↓ | 0.44 ± 0.18 * |
| CTGF | 1.21 ± 0.15 | ↓ | 0.02 ± 0.01 * | ↓ | 0.01 ± 0.01 * |
| FN1 | 4.28 ± 2.60 | ↓ | 0.69 ± 0.13 * | ↓ | 0.55 ± 0.15 * |
| MMP1 | 0.57 ± 0.11 | ↔ | 2.90 ± 1.46 | ↔ | 1.17 ± 0.17 |
| MMP13 | 0.89 ± 0.11 | ↔ | 1.31 ± 0.25 | ↔ | 1.01 ± 0.13 |
| Day 7 | |||||
| COL1A1 | 1.24 ± 0.17 | ↓ | 0.35 ± 0.01 * | ↓ | 0.43 ± 0.03 * |
| COL3A1 | 0.86 ± 0.15 | ↔ | 1.13 ± 0.31 | ↔ | 1.18 ± 0.32 |
| DCN | 0.83 ± 0.23 | ↑ | 1.71 ± 0.55 * | ↑ | 1.66 ± 0.48 * |
| ACTA2 | 1.69 ± 0.74 | ↔ | 0.64 ± 0.17 | ↔ | 0.63 ± 0.32 |
| CTGF | 0.31 ± 0.10 | ↔ | No expression | ↔ | No expression |
| FN1 | 3.03 ± 0.88 | ↓ | 0.57 ± 0.22 * | ↓ | 0.61 ± 0.20 * |
| MMP1 | 1.54 ± 0.38 | ↑ | 11.33 ± 3.77 * | ↑ | 11.49 ± 2.91 * |
| MMP13 | 1.20 ± 0.11 | ↑ | 1.88 ± 0.76 * | ↔ | 1.50 ± 0.28 |
| Target Gene | Primers (5′-3′) Sense | Antisense |
|---|---|---|
| 18S | GGCCCTGTAATTGGAATGAGTC | CCAAGATCCAACTACGAGCTT |
| COL1A1 | GTCACCCACCGACCAAGAACC | AAGTCCAGGCTGTCCAGGGATG |
| COL3A1 | ATGCCCTACTGGTCCTCAGA | GGAACCAGGATGACCAGATG |
| DCN | CCTGATGACCGCGACTTCGAG | TTTGGCACTTTGTCCAGACCC |
| ACTA2 | GACGAAGCACAGAGCAAAAGAG | TGGTGATGATGCCATGTTCTATCG |
| CTGF | CGGCTTACCGACTGGAAGAC | CGTCGGTACATACTCCACAG |
| FN1 | GACTTCCTATGTGGTCGGAG | TGTCTTCAGCCACTGCATCC |
| MMP1 | CTGAACGGTGATGAAGCAGCC | AGTCCAAGAGAATGGCCGAG |
| MMP13 | CATTTGATGGGCCCTCTGGCCTGC | GTTTAGGGTTGGGGTCTTCATCTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, J.M.; Jay, J.W.; Wang, Y.; Joglar, A.A.; Prasai, A.; Palackic, A.; Wolf, S.E.; El Ayadi, A. Galunisertib Exerts Antifibrotic Effects on TGF-β-Induced Fibroproliferative Dermal Fibroblasts. Int. J. Mol. Sci. 2022, 23, 6689. https://doi.org/10.3390/ijms23126689
Peterson JM, Jay JW, Wang Y, Joglar AA, Prasai A, Palackic A, Wolf SE, El Ayadi A. Galunisertib Exerts Antifibrotic Effects on TGF-β-Induced Fibroproliferative Dermal Fibroblasts. International Journal of Molecular Sciences. 2022; 23(12):6689. https://doi.org/10.3390/ijms23126689
Chicago/Turabian StylePeterson, Joshua M., Jayson W. Jay, Ye Wang, Alejandro A. Joglar, Anesh Prasai, Alen Palackic, Steven E. Wolf, and Amina El Ayadi. 2022. "Galunisertib Exerts Antifibrotic Effects on TGF-β-Induced Fibroproliferative Dermal Fibroblasts" International Journal of Molecular Sciences 23, no. 12: 6689. https://doi.org/10.3390/ijms23126689