
Rlip76: An Unexplored Player in Neurodegeneration and Alzheimer’s Disease?

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
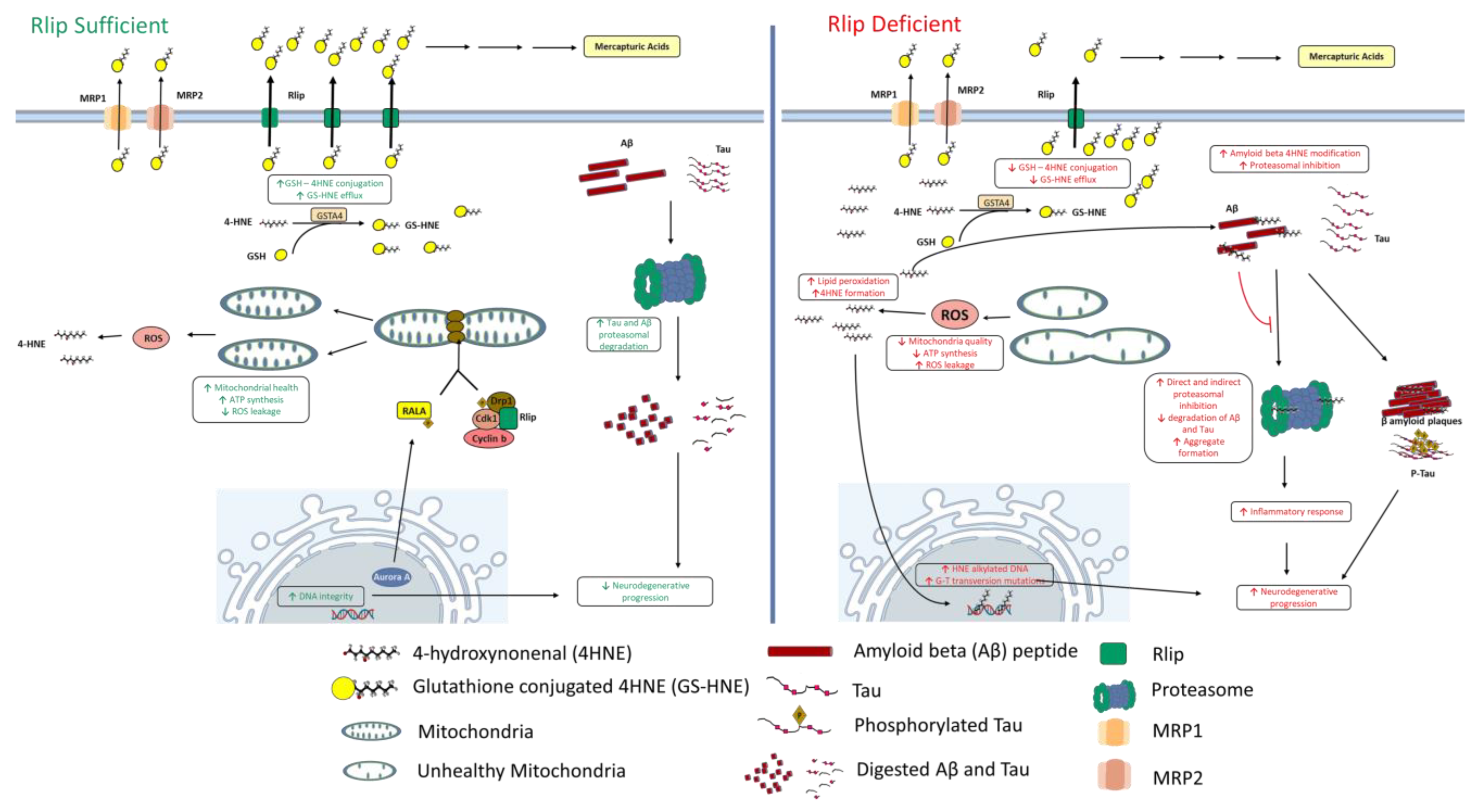
Abstract
:1. Introduction
2. Rlip Structure and Function
3. Stress Responsiveness
4. Energy Production: Rlip in Mitochondrial Biogenesis
5. 4-HNE, Oxidative Stress, and Neurodegeneration
6. Rlip in the Prevention of Oxidative Stress
7. Rlip in the Prevention of DNA Damage
8. Neurotransmission: Rlip in Endocytosis, Exocytosis and Receptor Regulation
9. Learning and Memory: Rlip and the Cytoskeleton in Axonal and Dendritic Remodeling
10. Rlip Gene Polymorphisms and Mechanistic Links
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Awasthi, S.; Singhal, S.S.; Sharma, R.; Zimniak, P.; Awasthi, Y.C. Transport of glutathione conjugates and chemotherapeutic drugs by RLIP76 (RALBP1): A novel link between G-protein and tyrosine kinase signaling and drug resistance. Int. J. Cancer 2003, 106, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sharma, A.; Sharma, R.; Patrick, B.; Singhal, S.S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: Role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003, 278, 41380–41388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Wickramarachchi, D.; Yadav, S.; Singhal, J.; Leake, K.; Vatsyayan, R.; Chaudhary, P.; Lelsani, P.; Suzuki, S.; Yang, S.; et al. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol. Cancer 2011, 10, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkind, J.M.; Nagababu, E.; Ramasamy, S.; Ravi, L.B. Hemoglobin redox reactions and oxidative stress. Redox Rep. 2003, 8, 234–237. [Google Scholar] [CrossRef]
- Miyanishi, K.; Tanaka, S.; Sakamoto, H.; Kato, J. The role of iron in hepatic inflammation and hepatocellular carcinoma. Free. Radic. Biol. Med. 2019, 133, 200–205. [Google Scholar] [CrossRef]
- Abrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxid. Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef]
- Saxena, M.; Singhal, S.S.; Awasthi, S.; Singh, S.V.; Labelle, E.F.; Zimniak, P.; Awasthi, Y.C. Dinitrophenyl S-glutathione ATPase purified from human muscle catalyzes ATP hydrolysis in the presence of leukotrienes. Arch. Biochem. Biophys. 1992, 298, 231–237. [Google Scholar] [CrossRef]
- Jullien-Flores, V.; Dorseuil, O.; Romero, F.; Letourneur, F.; Saragosti, S.; Berger, R.; Tavitian, A.; Gacon, G.; Camonis, J.H. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 1995, 270, 22473–22477. [Google Scholar] [CrossRef] [Green Version]
- Cantor, S.B.; Urano, T.; Feig, L.A. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol. Cell. Biol. 1995, 15, 4578–4584. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Weinberg, R.A. A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene 1995, 11, 2349–2355. [Google Scholar]
- Awasthi, S.; Cheng, J.; Singhal, S.S.; Saini, M.K.; Pandya, U.; Pikula, S.; Bandorowicz-Pikula, J.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 2000, 39, 9327–9334. [Google Scholar] [CrossRef] [PubMed]
- Fillatre, J.; Delacour, D.; Van Hove, L.; Bagarre, T.; Houssin, N.; Soulika, M.; Veitia, R.A.; Moreau, J. Dynamics of the subcellular localization of RalBP1/RLIP through the cell cycle: The role of targeting signals and of protein-protein interactions. FASEB J. 2012, 26, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Salgia, R.; Singhal, S.; Horne, D.; Awasthi, S. RLIP: An existential requirement for breast carcinogenesis. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Hindle, A.; Sawant, N.A.; George, M.; Vijayan, M.; Kshirsagar, S.; Morton, H.; Bunquin, L.E.; Palade, P.T.; Lawrence, J.J.; et al. RALBP1 in Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Cells 2021, 10, 3113. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Owen, D.; Mott, H.R. RLIP76: A Structural and Functional Triumvirate. Cancers 2021, 13, 2206. [Google Scholar] [CrossRef] [PubMed]
- Apken, L.H.; Oeckinghaus, A. The RAL signaling network: Cancer and beyond. Int. Rev. Cell. Mol. Biol. 2021, 361, 21–105. [Google Scholar] [CrossRef] [PubMed]
- Coon, B.G.; Burgner, J.; Camonis, J.H.; Aguilar, R.C. The epsin family of endocytic adaptors promotes fibrosarcoma migration and invasion. J. Biol. Chem. 2010, 285, 33073–33081. [Google Scholar] [CrossRef] [Green Version]
- Rosse, C.; L’Hoste, S.; Offner, N.; Picard, A.; Camonis, J. RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate epsin during the switch off of endocytosis in mitosis. J. Biol. Chem. 2003, 278, 30597–30604. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Wang, S.; Huang, Y.; Stamnes, M.; Chen, J.L. Roles of rho GTPases in intracellular transport and cellular transformation. Int. J. Mol. Sci. 2013, 14, 7089–7108. [Google Scholar] [CrossRef]
- Gentry, L.R.; Martin, T.D.; Reiner, D.J.; Der, C.J. Ral small GTPase signaling and oncogenesis: More than just 15minutes of fame. Biochim. Biophys. Acta. 2014, 1843, 2976–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell. Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Kim, M.H.; Seeburg, D.; Seo, J.; Verpelli, C.; Han, S.; Chung, H.S.; Ko, J.; Lee, H.W.; Kim, K.; et al. Regulated RalBP1 binding to RalA and PSD-95 controls AMPA receptor endocytosis and LTD. PLoS Biol. 2009, 7, e1000187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazat, K.; Harsat, M.; Goldshmid-Shagal, A.; Ehrlich, M.; Henis, Y.I. Dual effects of Ral-activated pathways on p27 localization and TGF-beta signaling. Mol. Biol. Cell. 2013, 24, 1812–1824. [Google Scholar] [CrossRef]
- Lebreton, S.; Boissel, L.; Iouzalen, N.; Moreau, J. RLIP mediates downstream signalling from RalB to the actin cytoskeleton during Xenopus early development. Mech. Dev. 2004, 121, 1481–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neel, N.F.; Rossman, K.L.; Martin, T.D.; Hayes, T.K.; Yeh, J.J.; Der, C.J. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol. Cell. Biol. 2012, 32, 1374–1386. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.; Warrington, A.; Taylor, K.A.; Svitkina, T. Structural organization of the actin cytoskeleton at sites of clathrin-mediated endocytosis. Curr. Biol. 2011, 21, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Hatch, A.L.; Ji, W.K.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol. Biol. Cell. 2016, 27, 3109–3121. [Google Scholar] [CrossRef]
- Abe, K.; Baba, K.; Huang, L.; Wei, K.T.; Okano, K.; Hosokawa, Y.; Inagaki, N. Mechanosensitive axon outgrowth mediated by L1-laminin clutch interface. Biophys. J. 2021, 120, 3566–3576. [Google Scholar] [CrossRef]
- Revach, O.Y.; Weiner, A.; Rechav, K.; Sabanay, I.; Livne, A.; Geiger, B. Mechanical interplay between invadopodia and the nucleus in cultured cancer cells. Sci. Rep. 2015, 5, 9466. [Google Scholar] [CrossRef]
- Hu, Y.; Mivechi, N.F. HSF-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J. Biol. Chem. 2003, 278, 17299–17306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaroni, A.; Paul, E.C. Cytocentrin is a Ral-binding protein involved in the assembly and function of the mitotic apparatus. J. Cell. Sci. 1999, 112, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Forth, S.; Kapoor, T.M. The mechanics of microtubule networks in cell division. J. Cell. Biol. 2017, 216, 1525–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Cheng, J.Z.; Singhal, S.S.; Pandya, U.; Sharma, R.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Functional reassembly of ATP-dependent xenobiotic transport by the N- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry 2001, 40, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Yadav, S.; Nagaprashantha, L.D.; Vatsyayan, R.; Singhal, S.S.; Awasthi, S. Targeting p53-null neuroblastomas through RLIP76. Cancer Prev. Res. 2011, 4, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Toma-Jonik, A.; Vydra, N.; Janus, P.; Widlak, W. Interplay between HSF1 and p53 signaling pathways in cancer initiation and progression: Non-oncogene and oncogene addiction. Cell. Oncol. 2019, 42, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Feldman, R.A.; Radhakrishnan, V.M.; Carey, S.; Martinez, J.D. Hsf1 is required for the nuclear translocation of p53 tumor suppressor. Neoplasia 2008, 10, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Yadav, S.; Drake, K.; Singhal, J.; Awasthi, S. Hsf-1 and POB1 induce drug sensitivity and apoptosis by inhibiting Ralbp1. J. Biol. Chem. 2008, 283, 19714–19729. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Tompkins, J.; Singhal, J.; Riggs, A.D.; Yadav, S.; Wu, X.; Singh, S.; Warden, C.; Liu, Z.; Wang, J.; et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 3918–3923. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Carna, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicine 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Bennani-Baiti, B.; Toegel, S.; Viernstein, H.; Urban, E.; Noe, C.R.; Bennani-Baiti, I.M. Inflammation Modulates RLIP76/RALBP1 Electrophile-Glutathione Conjugate Transporter and Housekeeping Genes in Human Blood-Brain Barrier Endothelial Cells. PLoS ONE 2015, 10, e0139101. [Google Scholar] [CrossRef] [Green Version]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Tiwari, M.N.; Biala, Y.; Yaari, Y. Regulation of Neuronal Na(+)/K(+)-ATPase by Specific Protein Kinases and Protein Phosphatases. J. Neurosci. 2019, 39, 5440–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Ryan, T.A. Glucose metabolism in nerve terminals. Curr. Opin. Neurobiol. 2017, 45, 156–161. [Google Scholar] [CrossRef]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mo.l Genet. 2017, 26, 1483–1496. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, D.F.; Counter, C.M. Breaking up is hard to do: RalA, mitochondrial fission and cancer. Small GTPases 2011, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Pollock, S.R.; Schinlever, A.R.; Rohani, A.; Kashatus, J.A.; Kashatus, D.F. RalA and RalB relocalization to depolarized mitochondria depends on clathrin-mediated endocytosis and facilitates TBK1 activation. PLoS ONE 2019, 14, e0214764. [Google Scholar] [CrossRef]
- Rockwell, P.; Yuan, H.; Magnusson, R.; Figueiredo-Pereira, M.E. Proteasome inhibition in neuronal cells induces a proinflammatory response manifested by upregulation of cyclooxygenase-2, its accumulation as ubiquitin conjugates, and production of the prostaglandin PGE(2). Arch. Biochem. Biophys. 2000, 374, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 5, S26–S36. [Google Scholar] [CrossRef] [PubMed]
- Shringarpure, R.; Grune, T.; Sitte, N.; Davies, K.J. 4-Hydroxynonenal-modified amyloid-beta peptide inhibits the proteasome: Possible importance in Alzheimer’s disease. Cell. Mol. Life Sci. 2000, 57, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Chesser, A.S.; Pritchard, S.M.; Johnson, G.V. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol 2013, 4, 122. [Google Scholar] [CrossRef] [Green Version]
- Piomboni, P.; Focarelli, R.; Stendardi, A.; Ferramosca, A.; Zara, V. The role of mitochondria in energy production for human sperm motility. Int. J. Androl. 2012, 35, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Gueraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell. Death Differ 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Bardag-Gorce, F.; Yuan, Q.X.; Li, J.; French, B.A.; Fang, C.; Ingelman-Sundberg, M.; French, S.W. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem. Biophys. Res. Commun. 2000, 279, 23–29. [Google Scholar] [CrossRef]
- Mattson, M.P.; Fu, W.; Waeg, G.; Uchida, K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. Neuroreport 1997, 8, 2275–2281. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell. Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.Z.; Sharma, R.; Yang, Y.; Singhal, S.S.; Sharma, A.; Saini, M.K.; Singh, S.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Accelerated metabolism and exclusion of 4-hydroxynonenal through induction of RLIP76 and hGST5.8 is an early adaptive response of cells to heat and oxidative stress. J. Biol. Chem. 2001, 276, 41213–41223. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Drake, K.; Nadkar, A.; Zajac, E.; Wickramarachchi, D.; Rowe, N.; Yacoub, A.; et al. RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 2005, 65, 6022–6028. [Google Scholar] [CrossRef] [Green Version]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48. [Google Scholar] [CrossRef] [Green Version]
- van ’t Erve, T.J.; Wagner, B.A.; Ryckman, K.K.; Raife, T.J.; Buettner, G.R. The concentration of glutathione in human erythrocytes is a heritable trait. Free. Radic. Biol. Med. 2013, 65, 742–749. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Zheng, R.; Po, I.; Mishin, V.; Black, A.T.; Heck, D.E.; Laskin, D.L.; Sinko, P.J.; Gerecke, D.R.; Gordon, M.K.; Laskin, J.D. The generation of 4-hydroxynonenal, an electrophilic lipid peroxidation end product, in rabbit cornea organ cultures treated with UVB light and nitrogen mustard. Toxicol Appl. Pharm. 2013, 272, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dygas, A.; Makowski, P.; Pikula, S. Is the glutathione conjugate of trans-4-hydroxy-2-nonenal transported by the multispecific organic anion transporting-ATPase of human erythrocytes? Acta. Biochim. Pol. 1998, 45, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renes, J.; de Vries, E.E.; Hooiveld, G.J.; Krikken, I.; Jansen, P.L.; Muller, M. Multidrug resistance protein MRP1 protects against the toxicity of the major lipid peroxidation product 4-hydroxynonenal. Biochem. J. 2000, 350, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug. Metab. Rev. 2011, 43, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA Damage to Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef] [Green Version]
- Coppede, F.; Migliore, L. DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 36–47. [Google Scholar] [CrossRef]
- Gackowski, D.; Rozalski, R.; Siomek, A.; Dziaman, T.; Nicpon, K.; Klimarczyk, M.; Araszkiewicz, A.; Olinski, R. Oxidative stress and oxidative DNA damage is characteristic for mixed Alzheimer disease/vascular dementia. J. Neurol. Sci. 2008, 266, 57–62. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Brocardo, P.S.; Christie, B.R. The role of oxidative stress in Huntington’s disease: Are antioxidants good therapeutic candidates? Curr. Drug. Targets 2014, 15, 454–468. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: A contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Deshpande, M.; Pang, H.; Stemmer, P.M.; Carruthers, N.J.; Shearn, C.T.; Backos, D.S.; Palaniyandi, S.S. 4-Hydroxy-2-nonenal attenuates 8-oxoguanine DNA glycosylase 1 activity. J. Cell. Biochem. 2020, 121, 4887–4897. [Google Scholar] [CrossRef] [PubMed]
- Smathers, R.L.; Fritz, K.S.; Galligan, J.J.; Shearn, C.T.; Reigan, P.; Marks, M.J.; Petersen, D.R. Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PLoS ONE 2012, 7, e38459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Neves, G.; Lagnado, L. The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J. Physiol. 1999, 515, 181–202. [Google Scholar] [CrossRef]
- Yao, C.K.; Liu, Y.T.; Lee, I.C.; Wang, Y.T.; Wu, P.Y. A Ca2+ channel differentially regulates Clathrin-mediated and activity-dependent bulk endocytosis. PLoS Biol. 2017, 15, e2000931. [Google Scholar] [CrossRef] [Green Version]
- Cousin, M.A. Integration of Synaptic Vesicle Cargo Retrieval with Endocytosis at Central Nerve Terminals. Front. Cell. Neurosci. 2017, 11, 234. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Chung, W.; Han, K.; Park, K.Y.; Kim, H.; Kim, E.; Kim, M.H. Down-regulation of RalBP1 expression reduces seizure threshold and synaptic inhibition in mice. Biochem. Biophys. Res. Commun. 2013, 433, 175–180. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, M.; Han, Y.; Zhang, H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 2020, 14, 660. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, M.; Pangalos, M.N.; Moss, S.J. Functional modulation of GABAB receptors by protein kinases and receptor trafficking. Adv. Pharm. 2010, 58, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Hanai, S.; Kishi, H.; Liu, Z.; Bao, Y.; Kikuchi, A.; Tsuchida, K.; Sugino, H. Regulation of endocytosis of activin type II receptors by a novel PDZ protein through Ral/Ral-binding protein 1-dependent pathway. J. Biol. Chem. 2002, 277, 19008–19018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, A.S.; Zheng, F.; Alzheimer, C. Activin Signaling in the Pathogenesis and Therapy of Neuropsychiatric Diseases. Front. Mol. Neurosci. 2016, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.; Bengtson, C.P.; Buchthal, B.; Bading, H. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin, A. Cell. Rep. 2015, 12, 1353–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, V.; Wang, Y.T. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: Implications for neuroprotective therapeutics for stroke. Neural Regen Res. 2016, 11, 1752–1753. [Google Scholar] [CrossRef]
- Cheng, F.; Li, X.; Li, Y.; Wang, C.; Wang, T.; Liu, G.; Baskys, A.; Ueda, K.; Chan, P.; Yu, S. alpha-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J. Neurochem. 2011, 119, 815–825. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Kidd, A.R., 3rd; Snider, J.L.; Martin, T.D.; Graboski, S.F.; Der, C.J.; Cox, A.D. Ras-related small GTPases RalA and RalB regulate cellular survival after ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Owens, C.; Chandra, N.; Popovic, K.; Conaway, M.; Theodorescu, D. RalBP1 is necessary for metastasis of human cancer cell lines. Neoplasia 2010, 12, 1003–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfinger, L.E.; Ptak, C.; Jeffery, E.D.; Shabanowitz, J.; Hunt, D.F.; Ginsberg, M.H. RLIP76 (RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J. Cell. Biol. 2006, 174, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Houssin, N.; Chikh, A.; Rynditch, A.; Van Hove, L.; Moreau, J. Recruitment of Cdc42 through the GAP domain of RLIP participates in remodeling of the actin cytoskeleton and is involved in Xenopus gastrulation. Dev. Biol. 2007, 312, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Olayioye, M.A.; Noll, B.; Hausser, A. Spatiotemporal Control of Intracellular Membrane Trafficking by Rho GTPases. Cells 2019, 8, 1478. [Google Scholar] [CrossRef] [Green Version]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell. Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.E.; Suter, D.M. An Integrated Cytoskeletal Model of Neurite Outgrowth. Front. Cell. Neurosci. 2018, 12, 447. [Google Scholar] [CrossRef]
- Nakajo, Y.; Miyamoto, S.; Nakano, Y.; Xue, J.H.; Hori, T.; Yanamoto, H. Genetic increase in brain-derived neurotrophic factor levels enhances learning and memory. Brain Res. 2008, 1241, 103–109. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The ’ins’ and ’outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell. Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Seano, G.; Primo, L. Podosomes and invadopodia: Tools to breach vascular basement membrane. Cell Cycle 2015, 14, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Brudvig, J.J.; Cain, J.T.; Sears, R.M.; Schmidt-Grimminger, G.G.; Wittchen, E.S.; Adler, K.B.; Ghashghaei, H.T.; Weimer, J.M. MARCKS regulates neuritogenesis and interacts with a CDC42 signaling network. Sci. Rep. 2018, 8, 13278. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Lalli, G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2010, 2, a001818. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leschziner, G.D.; Jorgensen, A.L.; Andrew, T.; Williamson, P.R.; Marson, A.G.; Coffey, A.J.; Middleditch, C.; Balding, D.J.; Rogers, J.; Bentley, D.R.; et al. The association between polymorphisms in RLIP76 and drug response in epilepsy. Pharmacogenomics 2007, 8, 1715–1722. [Google Scholar] [CrossRef]
- Lal, S.; Sutiman, N.; Ooi, L.L.; Wong, Z.W.; Wong, N.S.; Ang, P.C.S.; Chowbay, B. Pharmacogenetics of ABCB5, ABCC5 and RLIP76 and doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenom. J. 2017, 17, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Yadav, S.; Singhal, S.S.; Singhal, J.; Hindle, A.; Lee, J.; Cheedella, N.K.S.; Rehman, S.; Rahman, R.L.; Jones, C.; et al. Rlip Depletion Suppresses Growth of Breast Cancer. Cancers 2020, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Rodrigues, B.; Oue, H.; Banerjee, A.; Kanekiyo, T.; Singh, J. Dual functionalized liposome-mediated gene delivery across triple co-culture blood brain barrier model and specific in vivo neuronal transfection. J. Control. Release 2018, 286, 264–278. [Google Scholar] [CrossRef]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood-brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [Green Version]
- Nagabhushan Kalburgi, S.; Khan, N.N.; Gray, S.J. Recent gene therapy advancements for neurological diseases. Discov. Med. 2013, 15, 111–119. [Google Scholar]
- Li, T.Y.; Sleiman, M.B.; Li, H.; Gao, A.W.; Mottis, A.; Bachmann, A.M.; El Alam, G.; Li, X.; Goeminne, L.J.E.; Schoonjans, K.; et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 2021, 1, 165–178. [Google Scholar] [CrossRef]
- Liu, R.; Lei, J.X.; Luo, C.; Lan, X.; Chi, L.; Deng, P.; Lei, S.; Ghribi, O.; Liu, Q.Y. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehrawat, A.; Yadav, S.; Awasthi, Y.C.; Basu, A.; Warden, C.; Awasthi, S. P300 regulates the human RLIP76 promoter activity and gene expression. Biochem. Pharm. 2013, 85, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hindle, A.; Singh, S.P.; Pradeepkiran, J.A.; Bose, C.; Vijayan, M.; Kshirsagar, S.; Sawant, N.A.; Reddy, P.H. Rlip76: An Unexplored Player in Neurodegeneration and Alzheimer’s Disease? Int. J. Mol. Sci. 2022, 23, 6098. https://doi.org/10.3390/ijms23116098
Hindle A, Singh SP, Pradeepkiran JA, Bose C, Vijayan M, Kshirsagar S, Sawant NA, Reddy PH. Rlip76: An Unexplored Player in Neurodegeneration and Alzheimer’s Disease? International Journal of Molecular Sciences. 2022; 23(11):6098. https://doi.org/10.3390/ijms23116098
Chicago/Turabian StyleHindle, Ashly, Sharda P. Singh, Jangampalli Adi Pradeepkiran, Chhanda Bose, Murali Vijayan, Sudhir Kshirsagar, Neha A. Sawant, and P. Hemachandra Reddy. 2022. "Rlip76: An Unexplored Player in Neurodegeneration and Alzheimer’s Disease?" International Journal of Molecular Sciences 23, no. 11: 6098. https://doi.org/10.3390/ijms23116098