1. Introduction
Mitochondria play central roles in maintaining cellular metabolic homeostasis, cell survival, and cell death. They are also the so-called “powerhouses” of cells as they produce most of the cellular adenosine triphosphate (ATP). Mitochondrial homeostasis is maintained by a series of protective mechanisms, such as fission, fusion, and mitophagy, the degradation and removal of selectively damaged or dysfunctional mitochondria via autophagy. Mitophagy is mediated mainly by the Pink1 (Phosphatase-and-tensin-homolog-instigated putative kinase 1)/Parkin pathway [
1].
Pink1 (aka BRPK and PARK6) is a 63 kDa mitochondrial serine/threonine-protein kinase encoded by the
PINK1 gene that was first identified in 2001 as a target of phosphatase and tensin homolog (PTEN) in cancer cells [
2]. Although Pink1 was initially identified as a gene upregulated in cancer cells, the specific role of Pink1 in tumorgenesis has not been fully understood yet. Interestingly, the role of Pink1 has been widely studied in Parkinson’s disease since mutations in the
PINK1 gene were identified in 2004 as a cause of early-onset Parkinson’s disease [
3]. Parkinson’s disease (PD) is a long-term degenerative disorder of the central nervous system that mainly affects the motor system. It is a progressive disorder that is caused by the degeneration of nerve cells in the part of the brain called the substantia nigra, which controls movement. Currently, there is no cure for Parkinson’s disease. However, treatments are available to help reduce the main symptoms and maintain quality of life. One recent unconventional approach that is becoming more and more common is based on nutraceuticals, substances that are food or part of food that provide medical or health benefits. Several nutraceuticals from natural sources have been substantiated to provide neuroprotection in experimental models through multiple mechanistic pathways, i.e., mitochondrial dysfunction, neuroinflammation, oxidative stress, and protein misfolding. The intake of nutraceuticals or nutritional modifications is generally safe and can be combined with current common drug therapies to improve the patient’s quality of life and/or mitigate PD symptoms [
4,
5,
6].
Various studies have demonstrated numerous functions of Pink1, including regulating complex I activity and maintaining neuronal viability in response to stress, yet regulation of mitochondrial functions is considered to be one of the most critical ones. For example, it has been suggested that by regulating the enzymatic activity of complex I, Pink1 modulates the overall electron transport chain (ETC) capacity and, ultimately, the overall output levels of ATP [
7]. In healthy mitochondria, under physiological conditions, mitochondria have an optimal, relatively high mitochondrial membrane potential (ΔΨm), and under these conditions, Pink1 is continuously imported into the mitochondria, and the transmembrane segment of Pink1 is cleaved and degraded. However, under unhealthy conditions (e.g., oxidative stress), when mitochondria are depolarized due to damage, the degradation of Pink1 is reduced, thus promoting Pink1 accumulation on the outer mitochondrial membrane (OMM). Accumulation of Pink1 on the impaired mitochondria recruits Parkin, which induces the degradation of the damaged mitochondria via a selective form of autophagy, named mitophagy [
8,
9].
The current prevailing model by which Pink1 and Parkin promote mitophagy posits that upon mitochondrial damage, Pink1 import is blocked, and it accumulates on the OMM of depolarized mitochondria. Pink1 then undergoes autophosphorylation and phosphorylates ubiquitin, which stimulates the recruitment of Parkin to the mitochondrial surface. Next, Parkin is phosphorylated by Pink1, and these events stimulate the ubiquitin-ligase activity of Parkin, allowing it to ubiquitinate further OMM targets, resulting in an additional substrate for Pink1, promoting more Parkin recruitment that leads to the recruitment of ubiquitin adaptor proteins, which in turn promote engulfment of the depolarized mitochondria by autophagosomes. This pathway is governed by various post-transcriptional modifications such as phosphorylation and ubiquitination, mediated by Pink1 and Parkin, respectively [
10]. Although some aspects of Pink1 mitochondrial import and processing have been elucidated, several issues are still unclear. Herein, to better understand the mechanism by which Pink1 regulates mitochondrial morphology, we developed a linear peptide that targets Pink1, and based on this bioactive peptide, we engineered peptide-based targeted peptidomimetics (modified peptides), biomolecular probes, with various properties. These biomolecular probes are optimized for enhanced cell permeability, chemical crosslinking, and molecular imaging. Furthermore, these probes demonstrated improved bioactivity and superior stability. We suggest these biomolecular probes as novel and unique tools to further study the role of Pink1 under physiological and pathological conditions. In addition, we present a step-by-step guideline that can be used by most researchers and laboratories for the development of biomolecular probes based on identified naturally occurring and/or linear peptides for basic research studies as well as for therapeutic applications.
3. Discussion
Pink1 is a mitochondrially targeted serine/threonine kinase which controls the specific elimination of dysfunctional mitochondria. It accumulates on defective mitochondria, eliciting the translocation of Parkin from the cytosol to mediate the clearance of damaged mitochondria via mitophagy. Pink1 is involved in many biological metabolic processes, and there is much evidence that altered Pink1/Parkin related mitophagy may be involved in the pathogenesis of various human diseases. In addition, Pink1 is rapidly turned over under basal conditions, and its expression level is very low. While Pink1 has a critical role in physiological and pathological conditions, there are very limited tools to study its role in vitro and in vivo.
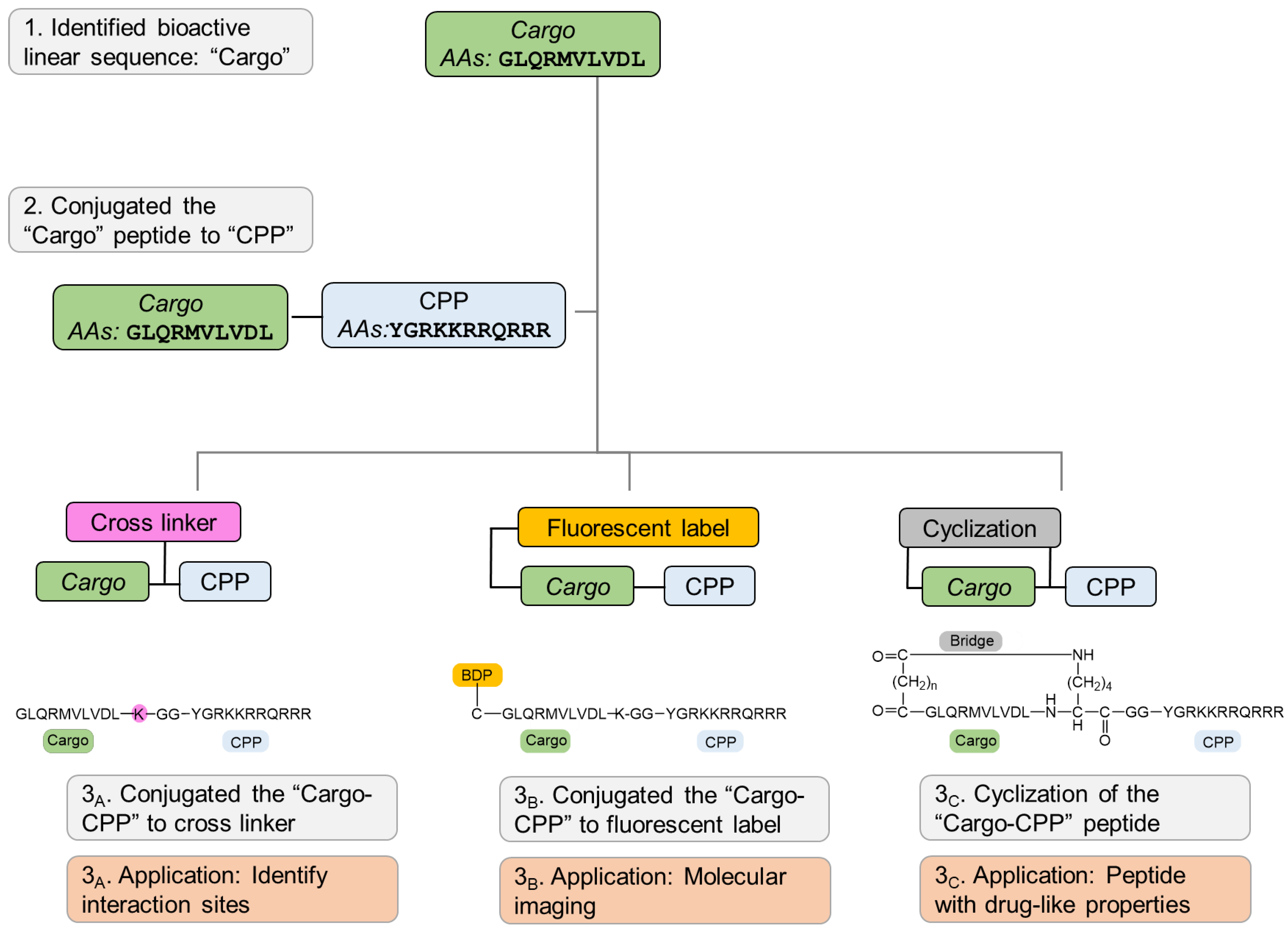
We developed a set of molecular probes to further study the role of Pink1 in health and disease (
Figure 8). Initially, we rationally designed a peptide that targets Pink1 protein as a tool to explore Pink1 functions, and we conjugated CPP to the bioactive peptide to deliver it inside the intact cells. Next, using cross linker fusion, we mapped the three-dimensional (3D) structure of the interaction site of the target protein with the peptide, which aligned with the docking prediction. In addition, using straightforward chemistry, we conjugated fluorescent dye to the bioactive peptide and demonstrated that it is colocalized with the target protein in cells. Finally, we used the backbone cyclization approach to overcome linear peptide limitation, mainly rapid proteolysis and inadequate membrane permeability, and developed peptidomimetics with increased stability and improved binding efficacy compared to their linear counterparts.
In summary, we present a step-by-step guideline to transform bioactive peptides into molecular probes, generating unique research tools for basic research and drug discovery. We demonstrate, using straightforward chemical synthesis approaches, how to optimize a bioactive linear peptide into designated probes. Modifications, such as attachment to cell-penetrating peptide, dye conjugation, cross-linker fusion, and cyclization, all result in a promising class of tools that can advance the revealing of basic research mechanisms and unknown signaling pathways, as well as for drug discovery in which peptides demonstrate extremely tight binding to their targets, high specificity, and low toxicity. The methods presented herein are general and common and can be used by many laboratories to custom identify bioactive peptides to be more specific and effective bioactive tools to advance basic research as well as therapeutic leads.
4. Materials and Methods
4.1. Sequence Alignments
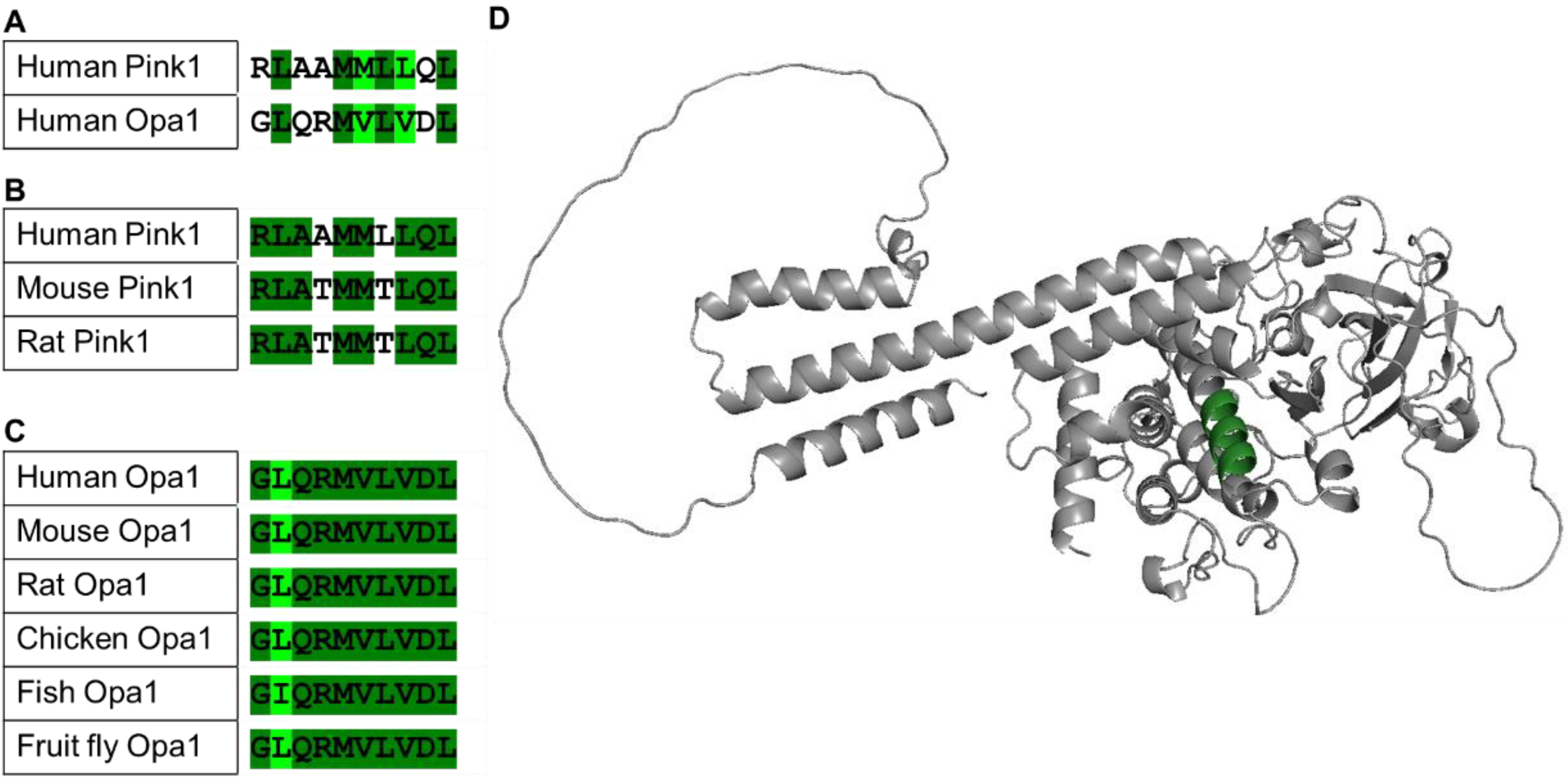
Sequences from different species were aligned using the LALIGN server (accessed on 1 April 2022), using Pink1 proteins (Homo sapiens (Q9BXM7), Mus musculus (Q99MQ3), Rattus norvegicus (B5DFG1)), Opa1 proteins (Homo sapiens (O60313), Mus musculus (P58281), Rattus norvegicus (Q2TA68), Gallus gallus (Q5F499), Danio rerio (Q5U3A7), and Drosophila melanogaster (A1Z9N0)).
4.2. Peptide Synthesis
In brief: Peptides were chemically synthesized using a fully automated peptide synthesizer (Syro I, Biotage, Uppsala, Sweden) on solid support by following the solid-phase peptide synthesis (SPPS) methodology [
48] with a fluorenylmethoxycarbonyl (Fmoc)/tert-Butyl (tBu) protocol. The cyclic peptides were synthesized with a modified lysine whose side chain was protected with N-methyltrityl (Mtt), a protection group that can be deprotected selectively using acid labile conditions [
40]. After completion of the synthesis of the linear peptide, an anhydride spacer was coupled to the N-terminal amino group and cyclization was performed using amide bonds between the moiety linker at the backbone N-terminus and an epsilon amino on the side chain of a lysine residue [
37]. The final cleavage and side chain deprotection were done manually. Peptides were analyzed by analytical reverse-phase high-pressure liquid chromatography (RP-HPLC) (1260 Infinity II LC System, Agilent, Santa Clara, CA, USA) and matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS) (autoflex
® maX, Bruker, Billerica, MA, USA) and purified by preparative RP-HPLC (1260 Infinity II LC System, Agilent, Santa Clara, CA, USA).
Further details: All commercially available solvents and reagents were used without further purification. Dichloromethane (DCM), Piperidine, Diethyl ether, N,N Diisopropylethylamine (DIEA), Trifluoroacetic acid (TFA), and Water (HPLC grade) were purchased from Bio-Lab (Jerusalem, Israel); Acetonitrile (ACN) (HPLC grade) was purchased from J.T. Baker (Poland); Acetic anhydride was purchased from Daejung (Gimhae-si, Korea); Triisopropylsilane (TIS), and Succinic anhydride were purchased from Acros organics (Branchburg, NJ, USA); Dithiothreitol (DTT) was purchased from Fisher Bioreagents (Ottawa, ON, Canada); Dimethylformamide (DMF) was purchased from Carlo Erba (Val De Reuil, France); Oxyma Pure was contributed by Luxembourg Bio Technologies Ltd. (Ness Ziona, Israel); N,N-Diisopropylcarbodiimide (DIC) was purchased from Angene International Limited (Nanjing, China); Glutaric anhydride was purchased from Alfa Aesar (Chaoyang, China); Benzotriazole-1-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBOP) was purchased from GL Biochem Ltd. (Shanghai, China); Fmoc Rink amide MBHA resin was purchased from AnaSpec (substitution 0.67 mmol/g, Fremont, CA, USA); Fmoc-protected amino acids were purchased from Ontores Biotechnologies (Hangzhou, China). Side chains of the amino acids used in the synthesis were protected as follows: tert-Butyloxycarbonyl (BOC) (Lys/Trp), tert-butyl (tBu) (Ser/Thr/Tyr/Glu), t-butyl ester (OtBu) (Asp), 2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) (Arg), 4-methyltrityl (Mtt) (Lys), and triphenylmethy (Trt) (Asn/Cys/Gln/His).
Peptides were chemically synthesized using a fully automated parallel peptide synthesizer, Syro I (Biotage, Uppsala, Sweden), on solid support following the fluorenylmethoxycarbonyl (Fmoc)/tert-Butyl (tBu) method. Fmoc deprotection was performed in two steps: 3 min and 12 min, both at 75 °C using piperidine (40%) in DMF solution. Coupling reactions were performed by repetition of the following cycle conditions: 45 min, 75 °C, with DIC (0.2 M) in DMF, Oxyma Pure (0.2 M) in DMF, and amino acid (0.2 M) in DMF. Whenever necessary, coupling and Fmoc deprotection steps were monitored using small cleavage. Anhydride coupling was carried out by the repetition of the following cycle conditions: 30 min at room temperature, using anhydride (10 eq)/ DIEA (10 eq)/ peptide (1 eq) in DMF. Mtt deprotection was carried out using TFA/TIS/DCM (1:5:94) and the reaction was done for 5 min, three times, at room temperature. Cyclization was performed manually in a chemical hood using DCM and PyBOP/DIEA (5:10) for 60 min at room temperature.
Peptide cleavage from the resin and deprotection of the amino acid side chains were carried out with a pre-cooled mixture of TFA/TIS/H2O solution (90:2.5:2.5 v/v/v) for three hours at room temperature. For peptides that have Cys in the sequence, the cleavage cocktail was TFA/TIS/H2O/DTT solution (94:1:5:50 v/v/v/(mg/mL)) and the reaction was done for three hours at room temperature. The resin was removed by filtration, and the solvents were evaporated by a stream of compressed air. The crude products were precipitated with diethyl ether, collected by centrifugation, dissolved in CH3CN/H2O (30:70) and lyophilized.
Small Cleavage. A small amount of resin was treated with a pre-cooled mixture of TFA/H2O/TIS (95:2.5:2.5) and the reaction was carried out for 60 min at room temperature. The resin was removed by filtration, and the solvents were evaporated by a stream of compressed air. The residue was dissolved in CH3CN/H2O (30:70). The filtrated solution was analyzed by HPLC and/or MS.
Products were analyzed by analytical reverse-phase high-pressure liquid chromatography (RP-HPLC) 1260 Infinity II LC System equipped with: G7129A 1260 vialsampler, G7111B 1260 quaternary pump, G7115A 1260 DAD (Diode Array Detector) WR, G1364C 1260 FC-AS, G1330B 1290 thermostat, from Agilent (Santa Clara, CA, USA) using a Luna 5 µm C18(2) 100 Å (250 × 4.6 mm) column (Phenomenex, Torrance, CA, USA) at 1 mL/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). A linear gradient of 5–95% B in 45 min was applied and the detection was at 214 nm and 254 nm. The synthesis products were purified using a Luna 5 µm C18(2) 100 Å (250 × 10 mm) column (Phenomenex, Torrance, CA, USA) at 4.7 mL/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). For separation, a linear gradient of 5–95% B in 45 min was applied and the detection was at 214 nm and 254 nm.
4.3. Conjugating Dye to the Peptide
In brief: Dye was covalently attached by the Sulphur of the Cys in the peptide with Tris buffer solution at room temperature.
BDP 630/650 maleimide was purchased from Lumiprobe (Hunt Valley, MD, USA); Tris HCl was purchased from Fisher Bioreagents (Shanghai, China); Tris base was purchased from Fisher Bioreagents (Pittsburgh, PA, USA). The peptide CVD-205 (CH
3-C(O)-CGLQRMVLVDLKGGYGRKKRRQRRR-NH
2) (
Figure S3) was dissolved in Tris buffer (100 mM (pH 7.4)) to 4 mM. The dye was dissolved in ACN to 3 mM. The dye solution was added to the peptide solution in equal volumes with proper shaking at 700 rounds per minute (rpm) overnight at room temperature.
4.4. Molecular Docking
The protein structure was input as the receptor molecule, and the peptides were used as the ligands in the PatchDock webserver [
28]. The final clustering was selected on the basis of the root-mean-square deviation (RMSD) value. The best protein–peptide complex was selected from among the top ten conformers based on energy scoring. This web tool was employed to obtain rigid protein–peptide docking.
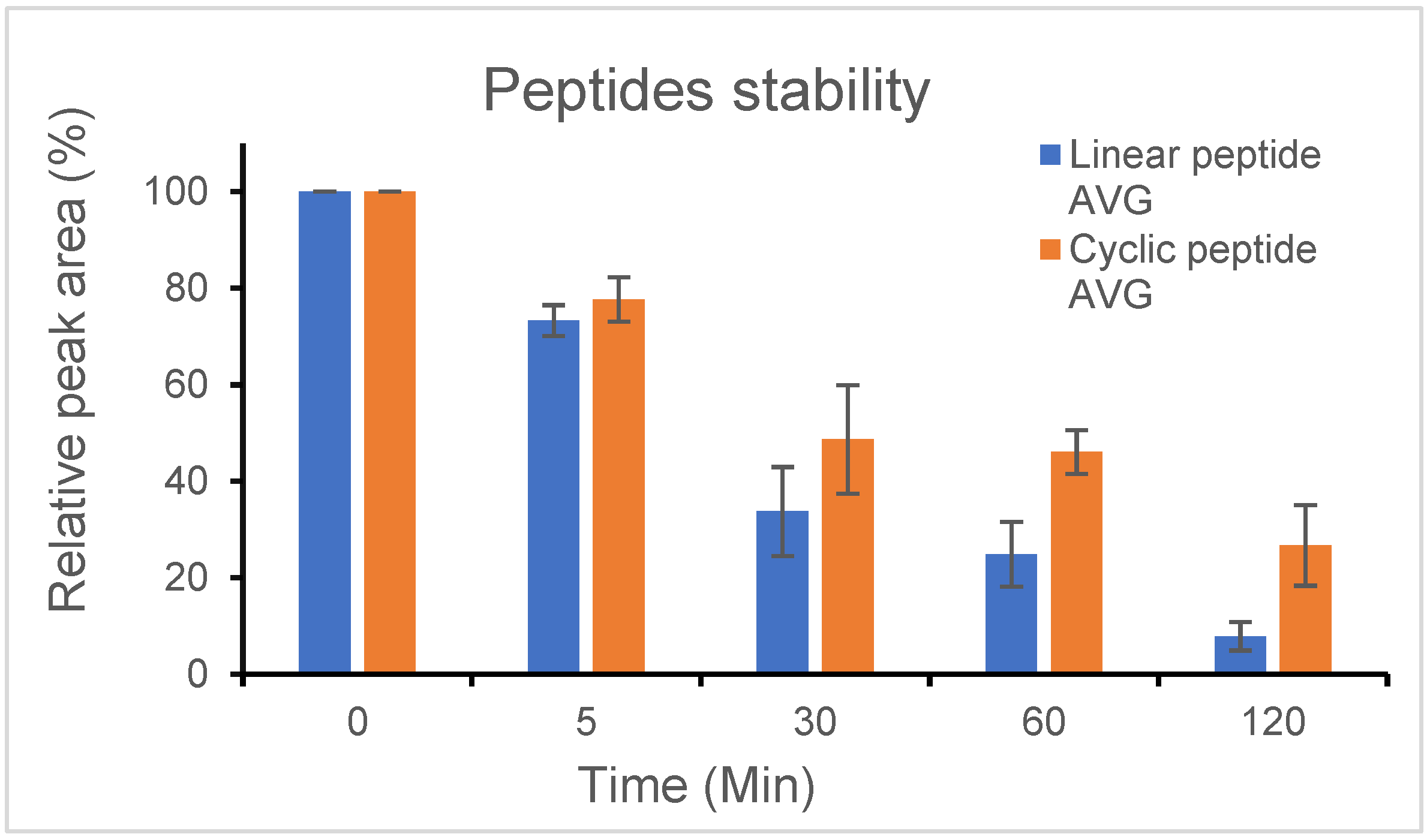
4.5. Stability Test of Linear and Cyclic Peptide
In brief: The stability studies of the peptides were performed by dissolving the peptides in Tris buffer and incubating them with trypsin solution at 37 °C using HPLC.
Further details: Trypsin from porcine pancreas was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Peptide (2 mg) was dissolved in Tris buffer (800 µL, 50 mM (pH 8.0)). The peptide solution was mixed with 1 μL of trypsin solution (1 mg/mL in 50 mM Tris buffer (pH 8.0)). The peptide was incubated at 37 °C and samples (90 μL) were taken at 0, 5, 30, 60, and 120 min. The samples were mixed with TFA/CH3CN/H2O (2%/5%/93%, 90 µL). The peptides were analyzed by analytical reverse-phase high-pressure liquid chromatography (RP-HPLC) 1260 Infinity II LC System from Agilent, (Santa Clara, CA, USA) using a Luna 5 μm C18(2) 100 Å (250 × 4.6 mm) column (Phenomenex, Torrance, CA, USA) at 1 mL/min. The solvent systems used were A (H2O with 0.1% TFA) and B (CH3CN with 0.1% TFA). A linear gradient of 5–50% B in 15 min was applied, and the detection was at 214 nm and 254 nm.
4.6. Protein Expression and Purification
4.6.1. Bacterial Protein Expression
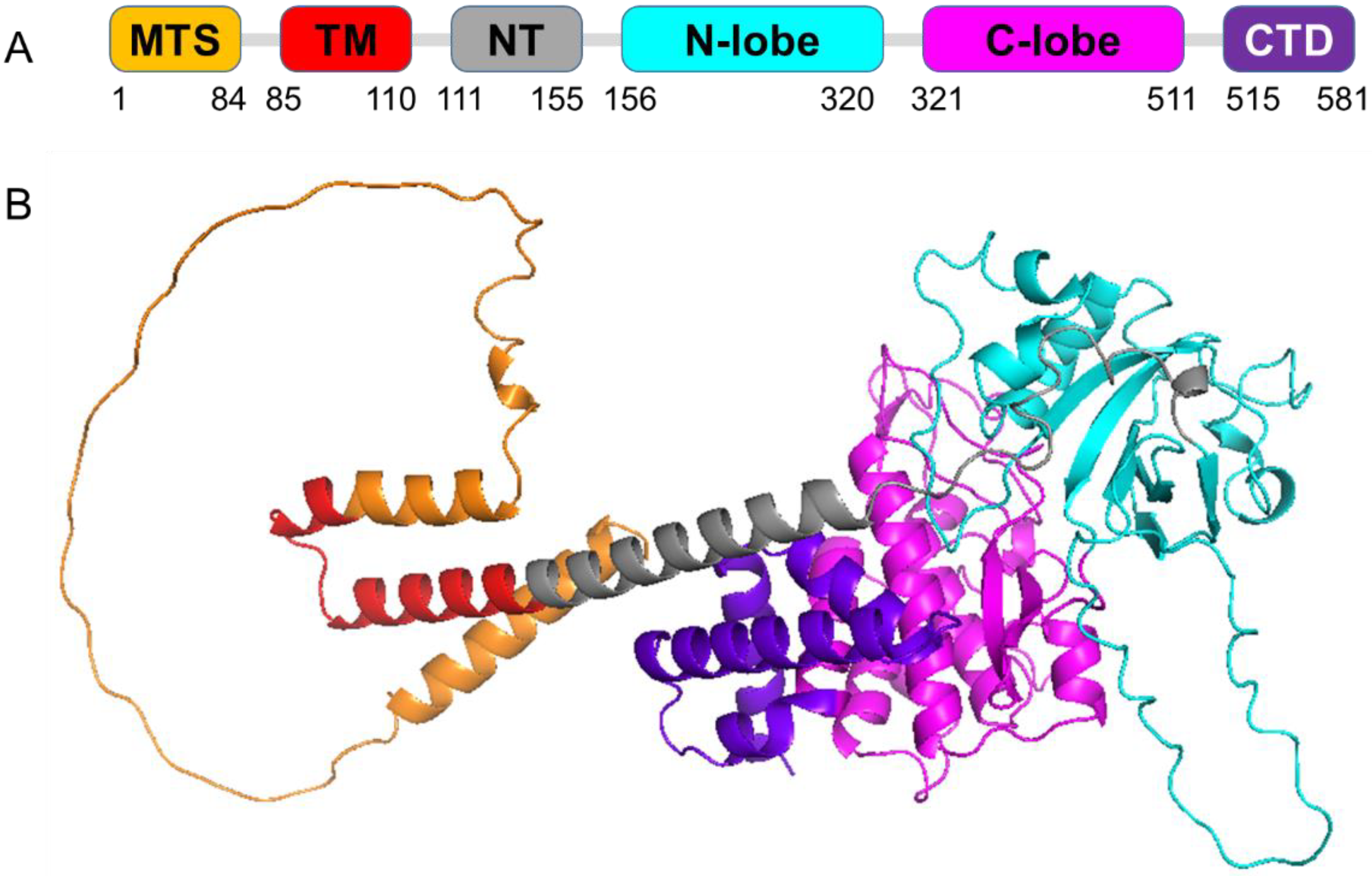
The two recombinant proteins, human Pink1 (hPink1) and Tribolium castaneum Pink1 (TcPink1), were expressed using the Escherichia coli (E. coli) Rosetta bacterial expression system. The hPink1 plasmid construct, pET-21a(+)/Maltose binding protein (MBP)-His-Pink1, consists of the partial AA (125–581) sequence of hPink1, which was purchased from Addgene (Watertown, MA, USA). The TcPink1 plasmid construct, pMal4c Pink1 (Tribolium castaneum) DU34701 consists of full-length AA sequence (1–570) was purchased from the MRC Protein Phosphorylation and Ubiquitylation Unit, Dundee University (Dundee, UK).
All constructs were transformed into E. coli XL1-Gold strain Stratagene (San Francisco, CA, USA) by using the conventional heat shock method, followed by cultivation and plasmid DNA purification using the QIAprep minispin kit (QIAGEN, Hilden, Germany). All plasmids were further sequenced and validated by Sanger sequencing (Macrogen via IDT). The plasmids were stored at −20 °C until use for protein expression.
For protein expression, the validated plasmid constructs of hPink1 and TcPink1 were transformed into E. coli Rosetta (DE3) strain cells (Novagen, Madison, WI, USA) by the heat shock method. The cells were plated on Lysogeny broth (LB)-agar supplemented with Ampicillin + Chloramphenicol and allowed to incubate overnight at 37 °C. A single colony was inoculated into 2 mL LB media supplemented with the appropriate antibiotics and glucose (0.2%) and incubated overnight at 37 °C with shaking at 200 rpm. Upon reaching OD600 of 0.5–0.8, the expressions of hPink1 and TcPink1 were induced in bacterial cells with isopropyl β-D-1-thiogalactopyranoside (IPTG) (0.25 mM), and the culture was incubated at 16 °C for 14–16 h with proper shaking at 200 rpm. Cells were then harvested by centrifugation (SL 16R, Thermo Fisher, Waltham, MA, USA) (20 min, 4000 rpm, 4 °C). The pellet was re-suspended in ice-cold lysis (L) buffer (L-MBP buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 5% (v/v) glycerol, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, 1 mM benzamidine, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and complete TM EDTA-free protease inhibitors (Roche, Basel, Switzerland)).
4.6.2. Protein Purification
After re-suspension in lysis buffer, cells were further lysed via EmulsiFlex®-C3, high pressure homogenizer (AVESTIN, Ottawa, ON, Canada) (15,000 psi, 10 min, 4 °C). To separate the soluble protein from the cell debris, the cell lysate was subjected to centrifugation in the Avanti J-E centrifuge system (Beckman Coulter, Brea, CA, USA) (30,000× g, 45 min, 4 °C). The soluble supernatant of the lysate was filtered and loaded onto an immobilized-amylose affinity chromatography column (Amylose resin, New England Bio Labs, Ipswich, MA, USA) using an automated fast liquid chromatography (FPLC) AKTA system (GE Healthcare, Chicago, IL, USA). The affinity column bound hPink1 or TcPink1 proteins were eluted from the column using the elution buffer containing maltose (12 mM).
The OD of eluted fractions were monitored in a nano-spectrophotometer at 280 nm and fractions containing hPink1 were pooled together and concentrated to 1.3 mg/mL in 3 mL using a spin concentrator (vivaspin 500, Sartorius, Goettingen, Germany), with the appropriate cut off (100 kDa). The sample was loaded onto size-exclusion chromatography (SEC) with a Superdex 200 prep grade (16/700) GL column (GE Healthcare, Chicago, IL, USA) using the FPLC AKTA system, preequilibrated with Tris-HCl buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1 mM EGTA, 5% (v/v) glycerol, 0.03% (v/v) Brij-35, and 0.1% (v/v) 2-mercaptoethanol).
Peak fractions were analyzed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (10%) and stained with Coomassie blue. The Western blot analysis for TcPink1 was done with a MBP mouse monoclonal antibody, sc-13564 (Santa Cruz Biotechnology, Dallas, TX, USA). To identify the recombinant hPink1, Western blot analysis was done with Pink1 rabbit monoclonal antibody, 6946S (Cell Signaling Technology, Beverly, MA, USA). Secondary antibodies, Anti-mouse DyLight™ 680, 5470S (Cell Signaling Technology, Beverly, MA, USA) and Anti-rabbit DyLight™ 800, 5151S (Cell Signaling Technology, Beverly, MA, USA), were used in the analysis that was done using Odyssey® CLx (LI COR Biosciences, Lincoln, NE, USA). Relevant fractions containing Pink1 were flash frozen in liquid nitrogen. Purified Pink1 proteins were stored at −80 °C in affinity chromatography elution buffer or in SEC elution buffer. Next, the proteins were dialyzed into the proper buffer as needed in each protocol.
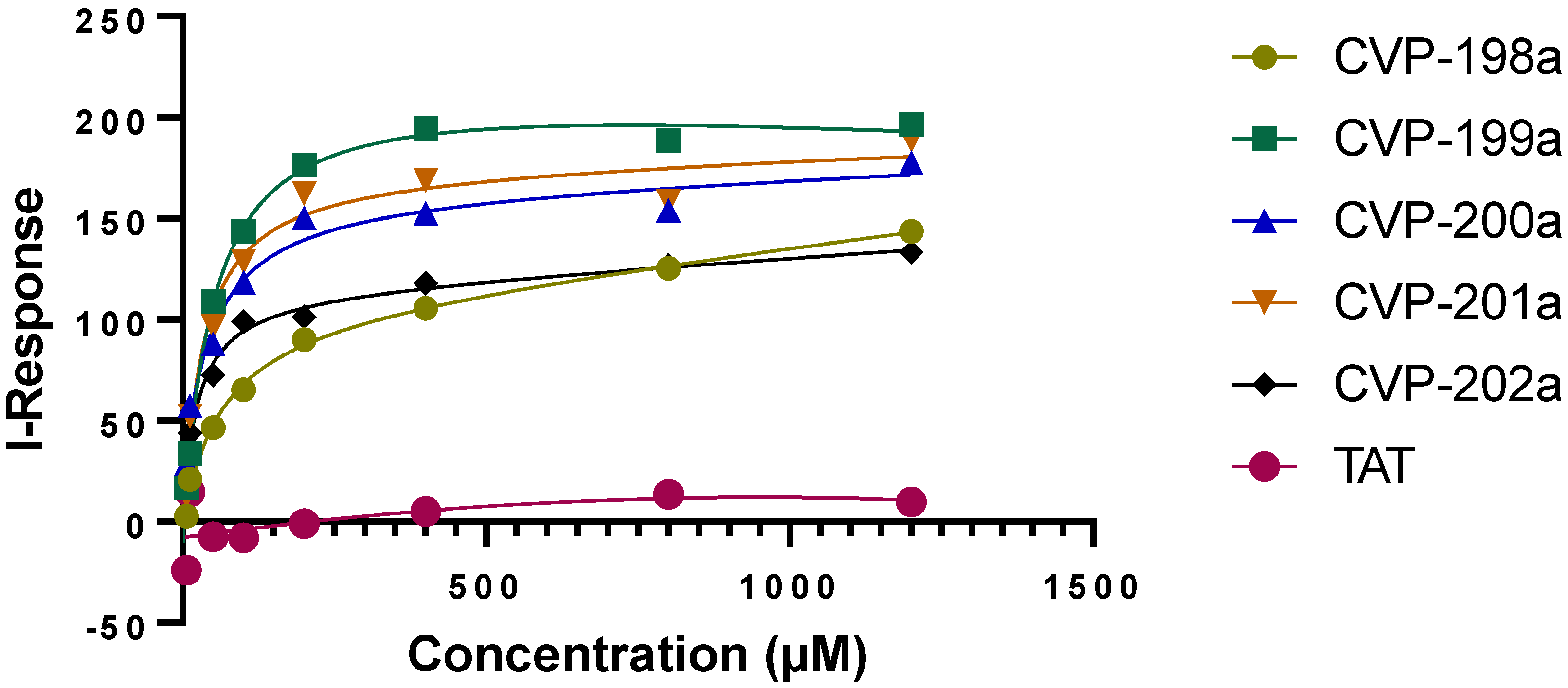
4.7. Peptide Binding to Protein, In Vitro
In brief: Protein was immobilized/cross-linked into the carboxyl group present on the activated graphene biosensor chip. The analyte is applied in solution to the chip and when an interaction occurs, an alteration in the current (I) is measured and recorded in real-time throughout the experiment. The entire cycle of the experiment starts with calibration using PBS (pH 7.4) to record the baseline equilibration response. Followed by the association step by adding analyte (50 µL) at the desired concentrations in PBS (pH 7.4). After the experiment, data at the concentration points of the analytes were exported from 3 transistors, averaged, and any background drift recorded in PBS was subtracted.
Further details: N-hydroxysulfosuccinimide sadium salt (Sulfo-NHS) was purchased from Biosynth Carbosynth (Compton, UK); 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC-HCl) was purchased from Alfa Aesar (Kandel, Germany); Quench 1 (3.9 mM amino-PEG5-alcohol in PBS (pH 7.4)) and Quench 2 (1 M ethanolamine (pH 8.5)) were purchased from Cardea (San Diego, CA, USA); 2-(N-morpholino)-ethanesulfonic acid (MES) was purchased from Sigma-Aldrich (Saint Louis, MO, USA); PBS X 10 (pH 7.0) was purchased from Hylabs (Rehovot, Israel).
Binding data of the peptides to immobilized protein in vitro was gathered using an AGILE Dev Kit label-free binding assay (Cardea, San Diego, CA, USA). Following the standard protocol from the manufacturer. The capture molecules were bound to the chip with a zero length linker (EDC/sulfo-NHS). EDC (2 mg) and sulfo-NHS (6 mg) were used in MES buffer (1 M (pH 6.0)) for 15 min to covalently attach the amine of the protein to the carboxyl on the chip. The protein solution (500 nM) was incubated with the chip for 15 min. Next, Quench 1 followed by Quench 2 were applied serially for 15 min each to quench remaining unoccupied binding sites on the chip. After a rinse in PBS, base-line current levels for the chip were recorded for at least 5 min. Next, the PBS was aspirated, and a droplet of the tested analyte (50 μL) was applied to the chip and the change in the sensor chip readout was recorded, after that the analyte was aspirated and the chip was rinsed with PBS. Additional measurements were performed using varying analyte concentrations. After data were gathered, the responses of the sensors on a single assay chip were averaged, and any background drift recorded in PBS was subtracted. A Hill fit plot was used to determine a KD. The KD values were calculated by GraghPad Prism 9, statistical analysis software. Data presented as mean ± SEM of all measurements. All samples were identical prior to allocation of treatments.
4.8. Crosslinking Study
4.8.1. Protein Cross-Linking
In brief: The recombinant protein and the peptide were incubated together with a crosslinker. The reaction was stopped with a quencher. The proteins were precipitated collected and trypsinased, the outcome was analysed by liquid chromatography mass spectrometry (LCMS).
Further details: Suberic acid bis(3-sulfo-N-hydroxysuccinimide ester) sodium salt (BS3), 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM), and iodoacetamide were purchased from Sigma-Aldrich (Saint Louis, MO, USA); Sequencing Grade Modified Trypsin (lyophilized) was purchased from Promega (Fitchburg, WI, USA); Empore™ SPE Disks matrix active group C18 was purchased from Merck (Darmstadt, Germany); 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) was purchased from Fisher Bioreagents (Pittsburgh, PA, USA); Dimethyl Sulfoxide (DMSO) was purchased from Bio-Lab (Jerusalem, Israel).
A mixture solution of purified protein and peptide was prepared in HEPES buffer (50 mM HEPES, 150 mM NaCl (pH 8.0)). Protein TcPink1’s final concentration was 10 μM and hPink1’s final concentration was 2 μM. The peptide was added at a 1:10 or 1:100 protein-to-peptide ratio, and the mixture incubated with agitation for 20 min at room temperature. For BS3 cross-linking, a solution of BS3 in DMSO (150 mM) was used, which was diluted with the proteins to a final BS3 concentration of 1 mM. The cross-linking reaction was incubated at room temperature for 30 min with agitation. The reaction was quenched by adding ammonium bicarbonate to a final concentration of 30 mM. For DMTMM cross-linking, a solution of DMTMM in HEPES buffer (100 mM) was used, which was diluted with the proteins to a final DMTMM concentration of 7 mM. The cross-linking reaction was incubated at room temperature for 30 min with agitation at 600 rpm. The reaction was quenched by adding ammonium bicarbonate (100 mM).
4.8.2. Mass Spectrometry
The proteins were precipitated in acetone at −80 °C for 1 h, followed by centrifugation at 13,000× g. The pellet was resuspended in urea (20 μL, 8 M) with DTT (10 mM). After 30 min, iodoacetamide was added to a final concentration of 25 mM and the alkylation reaction proceeded for 30 min in the dark. The urea was diluted by adding 250 μL digestion buffer (25 mM TRIS (pH 8.0); 10% acetonitrile), trypsin (Promega, Fitchburg, WI, USA) was added at a 1:100 protease-to-protein ratio, and the protein was digested overnight at 37 °C with agitation. Following digestion, the peptides were desalted on C18 stage-tips and eluted by 55% acetonitrile. The eluted peptides were dried in a SpeedVac, reconstituted in formic acid (0.1%), and measured in a mass spectrometer. The samples were analyzed by a 60 min 0–40% acetonitrile gradient on a liquid chromatography system (Acquity M UPLC, Waters, Milford, MA, USA) coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher, Waltham, MA, USA). The RAW data files from the mass spectrometer were converted into MGF format in a Proteome Discoverer (Thermo Fisher, Waltham, MA, USA), which was the input format for the analysis pipeline. The method parameters of the runs were as follows: data-dependent acquisition; full MS resolution, 70,000; MS1 AGC target, 1e6; MS1 maximum IT, 200 ms; scan range, 450–1800; dd-MS/MS resolution, 35,000; MS/MS AGC target, 2e5; MS2 maximum IT, 300 ms; loop count, top 12; isolation window, 1.1; fixed first mass, 130; HCD energy (NCE), 26; MS2 minimum AGC target, 800; charge exclusion: unassigned, 1, 2, 3, 8, >8; peptide match, off; exclude isotope, on; and dynamic exclusion, 45 s.
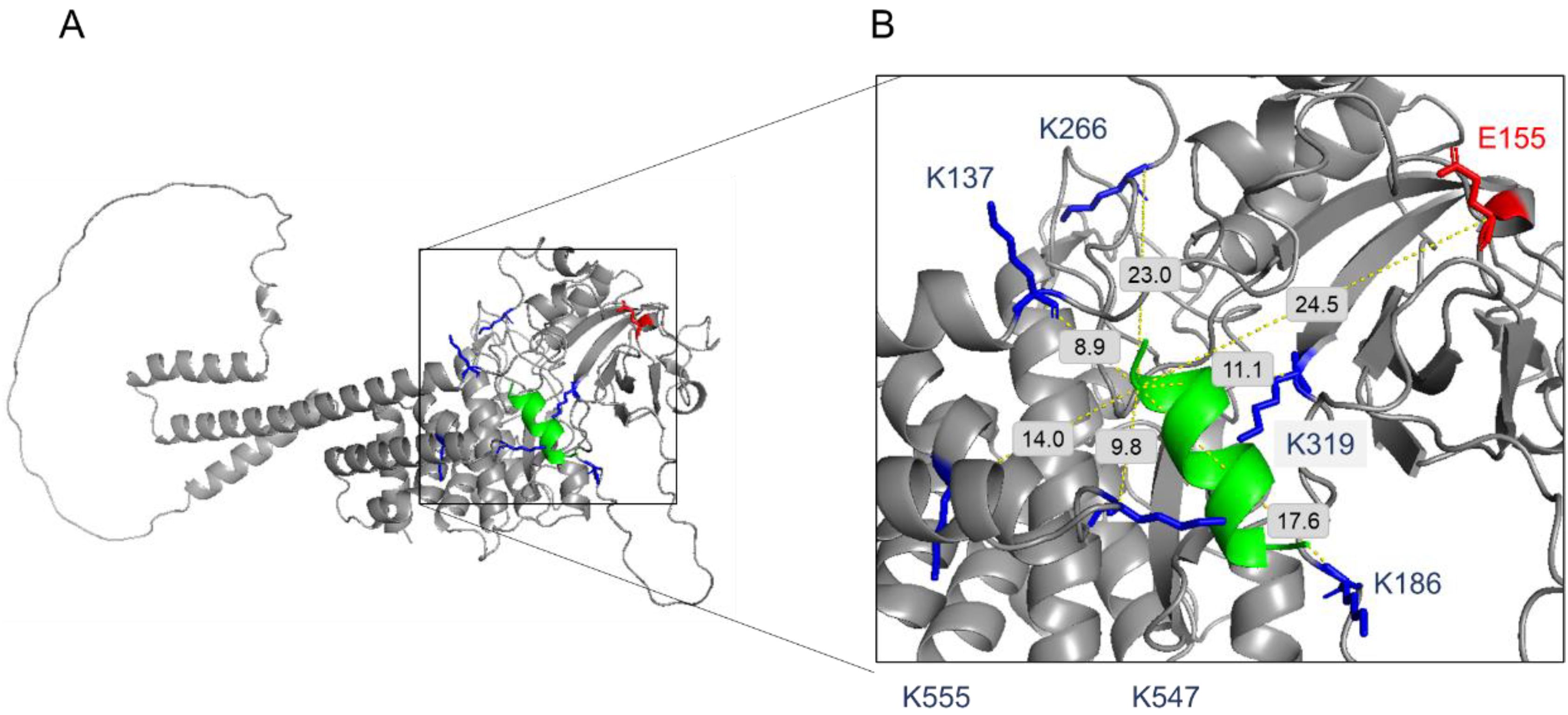
4.8.3. Identification of Cross-Links
We used an established search application [
49]. The sequence database included TcPink1, hPink1 and the peptide, CVP-198. The false discovery rate (FDR) was estimated from decoy-based analysis, which repeated the identification analysis 20 times with an erroneous cross-linker mass. For example, in the case of BS3 cross-linking, which has a true cross-linker mass of 138.0681 Da, erroneous cross-linker masses of 138.0681 * N/138 Da were used, where N = 160, 161, 162, …, 179. This led to bogus identifications with fragmentation scores that were generally much lower than the scores obtained with the correct cross-linker mass (see histograms in
Figures S9 and S10). For the identification of true cross-links, we set the threshold on the fragmentation score according to the desired FDR value. For example, a threshold of 0.65 on the fragmentation score of the BS3 cross-linked CVP-198 and hPink1 datasets, gave 75 cross-links above the threshold in the true analysis and a median of 1 cross-link in a typical decoy run (
Figure S10). We therefore estimate the corresponding FDR to be about 1 in 75, or ~2%. The final thresholds used: BS3 cross-linked mixtures, 0.65, and DMTMM cross-linked mixtures, 0.7.
4.9. Cells Study
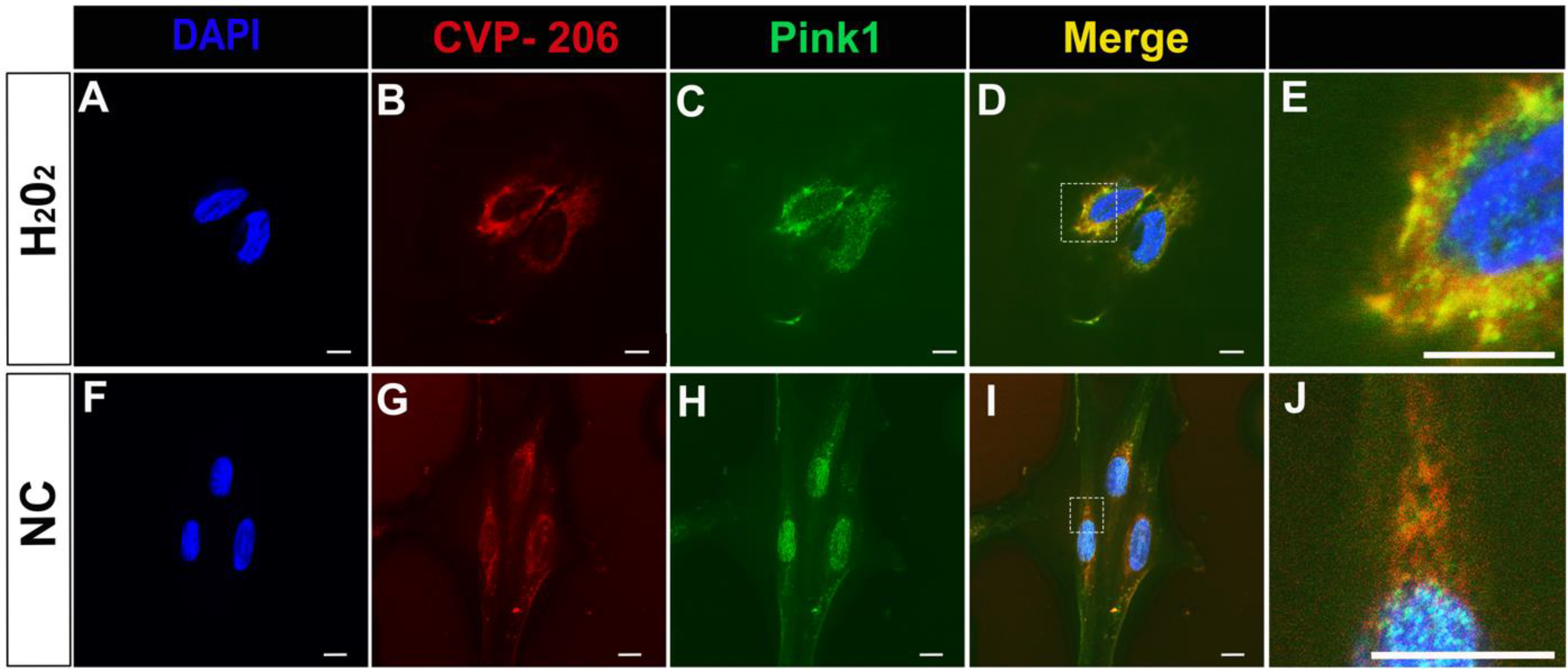
In brief: H9c2 cells were treated by hydrogen peroxide (H2O2) followed by the addition of dye conjugated peptide, and the cells were fixed and permeabilized. Immunocytochemistry was done with anti-Pink1 and a secondary antibody. The control experiment was done without the H2O2 stress. The Pink1 location on the mitochondria was checked by adding MitoTracker to the cells, which were fixed and permeabilized. The cells in all conditions were analyzed by a confocal microscope.
Further details: H9c2 cell line derived from embryonic rat heart tissue (CRL-1446) was purchased from the American Type Culture Collection (ATCC, Gaithersburg, MD, USA); Dulbecco’s modified Eagle’s high glucose medium (DMEM), fetal bovine serum (FBS), sodium pyruvate, penicillin–streptomycin (Pen–Strep) (10×) and combined antibiotic solutions were purchased from Biological Industries (Beit-Haemek, Israel); Sodium hydrogen carbonate was purchased from Daejung (Siheung-Si, Korea); Glass bottom dish 35 mm with 10 mm bottom well #1.5 glass were purchased from Cellvis (Mountain View, CA, USA); Hydrogen peroxide (30%) was purchased from Fisher Bioreagents (Pittsburgh, PA, USA); Formaldehyde solution phosphate buffered (4%) was purchased from Fisher Chemical (Loughborough, UK); MitoTracker Red CM-H2Xros was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
H9c2 cells were grown in standard DMEM containing FBS (10%), penicillin-streptomycin (100 U/mL–100 ug/mL), sodium pyruvate (1%), and sodium hydrogen carbonate (0.15%). Cells were plated in glass bottom 35 mm plates at a density of 10,000 cells per well and incubated overnight in a cell culture incubator supplied with 5% CO2 at 37 °C. A stock solution of the colored peptide CVP-206 (1 mM) was prepared in DMSO. Cells undergoing oxidative stress were incubated in serum-free DMEM media with H2O2 (1.6 mM) and incubated for 0.5 h in a cell culture incubator supplied with 5% CO2 at 37 °C. After washing with serum-free media, a peptide with dye, CVP-206 (1 µM) or MitoTracker Red CM-H2Xros (300 nM) in serum-free media, was applied to the cells and incubated for an additional 0.5 h in a cell culture incubator supplied with 5% CO2 at 37 °C. The cells were washed with PBS and fixed with paraformaldehyde (4%) in PBS (pH 7.3) for 15 min. Cells washed with PBS 0.2% Tween 20 and undergo permeabilization with PBS 0.3% Triton X-100 for 15 min. Next, cells were washed with PBS 0.2% Tween 20 and blocked with horse serum (1%) in PBS 0.2% Tween 20 for 1 h at room temperature. Anti-Pink1 antibody (AB-ab186303, Abcam, Cambridge, UK) 1:100 in horse serum (1%) in PBS 0.2% Tween 20 was applied to the cells for 1 h at room temperature. Next, Goat anti-Mouse IgG H&L (Alexa Fluor 488) preadsorbed (ab150117, Abcam, Cambridge, UK) 1:1000 in horse serum (1%) in PBS 0.2% Tween 20 was applied to the cells for 1 h at room temperature. The washes after each antibody were with PBS 0.2% Tween 20. The dish was sealed with a mounting medium with 4′,6-diamidino-2-phenylindole (DAPI)—aqueous (ab104139-20, Abcam, Cambridge, UK). Immunofluorescence images were acquired with Zeiss LSM780 inverted confocal microscope through a 63X objective (ZEISS International, Oberkochen, Germany).


 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
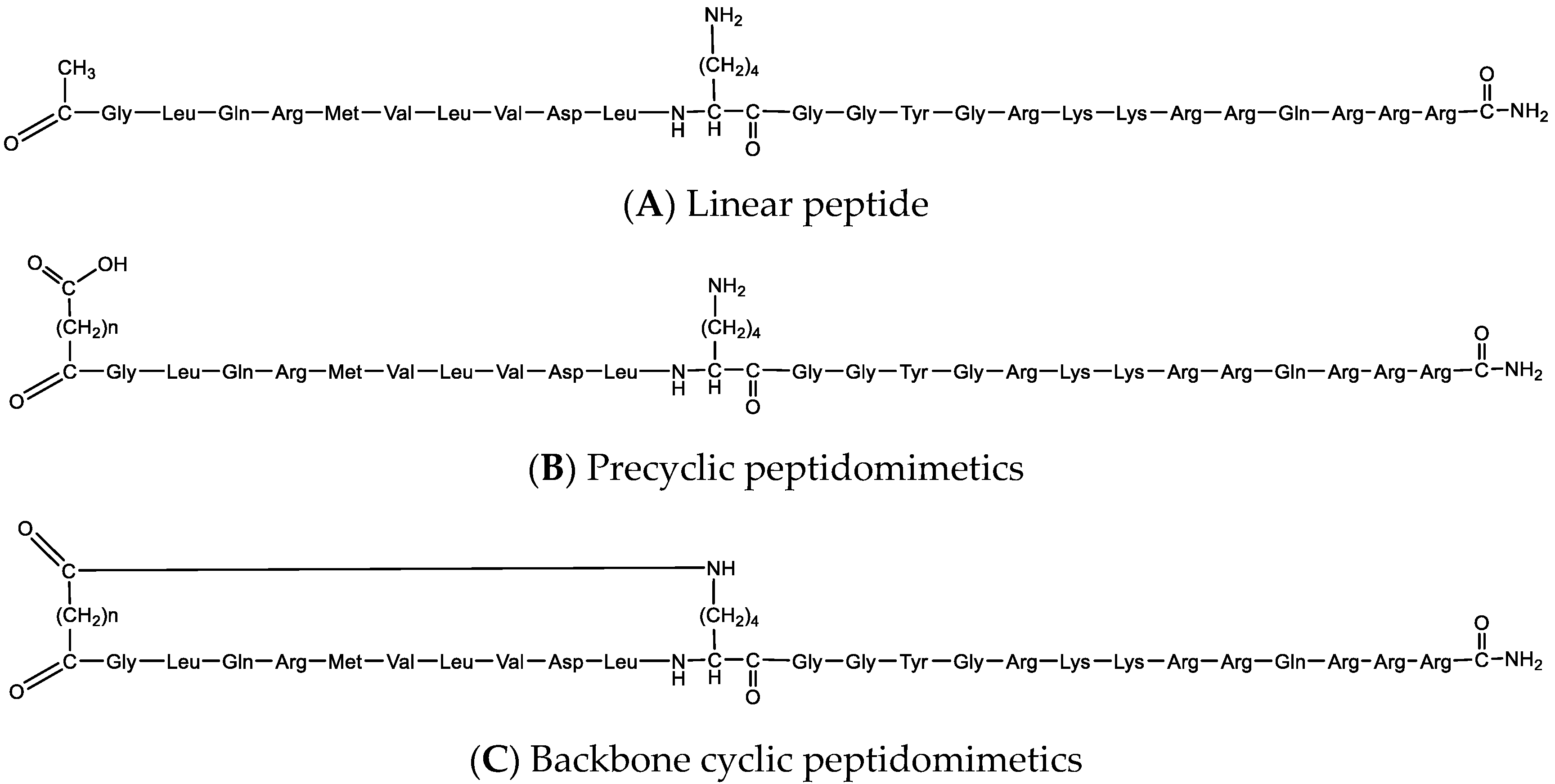{kind=link}
{kind=link}
{kind=link}
{kind=link}