
An Integrated Multi-Omic Network Analysis Identifies Seizure-Associated Dysregulated Pathways in the GAERS Model of Absence Epilepsy

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
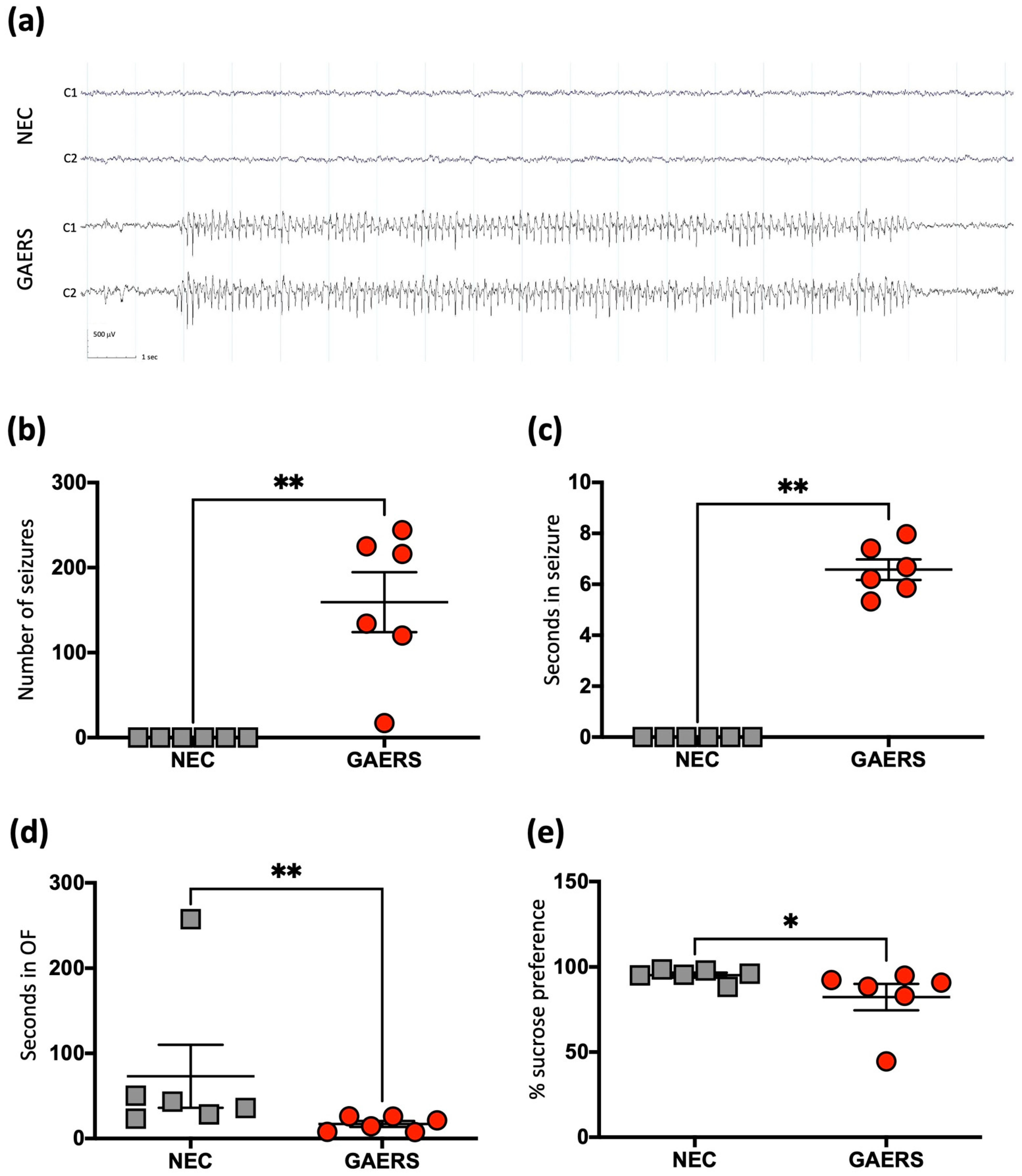
2.1. Behavioural Testing and Electroencephalography (EEG) Recordings Confirm Epileptic Phenotype in the GAERS Group
2.2. Proteomic Analysis Identifies Various Differentially Expressed Proteins in the GAERS Group
2.3. Metabolomic Analysis Identifies Differentially Abundant Metabolites and Significantly Enriched Metabolic Pathways in the GAERS Group
2.4. Modules with Varying Correlations to GAERS and Seizure Phenotype Identified in the Multi-Omic Networks from Somatosensory Cortex and Thalamus
2.5. Seizure-Associated Modules Show Significant Overlap in SCx and Thalamus
2.6. Quantitative Enrichment Analysis of the Seizure-Associated Modules Identifies Various Differentially Regulated Pathways
3. Discussion
4. Materials and Methods
4.1. EEG Electrode Implantation Surgery
4.2. EEG Acquisition and Analysis
4.3. Behavioural Tests
4.4. Statistical Analysis
4.5. Tissue Preparation
4.6. Proteomic Analysis Using LC-MS/MS
4.7. LC-MS Untargeted Metabolomic Analysis
4.8. Multi-Omic Data Integration and Weighted Gene Co-Expression Network Analysis (WGCNA)
4.9. Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Giussani, G.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Adib, M.G.; Agrawal, S.; Alahdab, F.; et al. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019, 18, 357–375. [Google Scholar] [CrossRef] [Green Version]
- World Health Organisation. Epilepsy: A Public Health Imperative; World Health Organisation: Geneva, Switzerland, 2019; pp. 1–171.
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, C.A.; Berkovic, S.F.; Petrou, S. Mechanisms of human inherited epilepsies. Prog. Neurobiol. 2009, 87, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Jackson, G.D.; Berkovic, S.F.; Petrou, S. New therapeutic opportunities in epilepsy: A genetic perspective. Pharmacol. Ther. 2010, 128, 274–280. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The genetics of epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 1–26. [Google Scholar] [CrossRef]
- Mullen, S.A.; Berkovic, S.F.; The IGC. Genetic generalized epilepsies. Epilepsia 2018, 59, 1148–1153. [Google Scholar] [CrossRef] [Green Version]
- Coenen, A.M.L.; Van Luijtelaar, E.L.J.M. Genetic Animal Models for Absence Epilepsy: A Review of the WAG/Rij Strain of Rats. Behav. Genet. 2003, 33, 635–655. [Google Scholar] [CrossRef]
- Crunelli, V.; Leresche, N. Childhood absence epilepsy: Genes, channels, neurons and networks. Nat. Rev. Neurosci. 2002, 3, 371–382. [Google Scholar] [CrossRef]
- Vergnes, M.; Marescaux, C.; Micheletti, G.; Reis, J.; Depaulis, A.; Rumbach, L.; Warter, J. Spontaneous paroxysmal electroclinical patterns in rat: A model of generalized non-convulsive epilepsy. Neurosci. Lett. 1982, 33, 97–101. [Google Scholar] [CrossRef]
- Powell, K.L.; Tang, H.; Ng, C.; Guillemain, I.; Dieuset, G.; Dezsi, G.; Çarçak, N.; Onat, F.; Martin, B.; O’Brien, T.J.; et al. Seizure expression, behavior, and brain morphology differences in colonies of genetic absence epilepsy rats from strasbourg. Epilepsia 2014, 55, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Marescaux, C.; Vergnes, M.; Depaulis, A. Genetic absence epilepsy in rats from Strasbourg—A review. J. Neural. Transm.-Gen. Sect. 1992, 35, 37–69. [Google Scholar]
- Marks, W.N.; Cavanagh, M.E.; Greba, Q.; Cain, S.M.; Snutch, T.P.; Howland, J.G. The Genetic Absence Epilepsy Rats from Strasbourg model of absence epilepsy exhibits alterations in fear conditioning and latent inhibition consistent with psychiatric comorbidities in humans. Eur. J. Neurosci. 2015, 43, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Danober, L.; Deransart, C.; Depaulis, A.; Vergnes, M.; Marescaux, C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog. Neurobiol. 1998, 55, 27–57. [Google Scholar] [CrossRef]
- Devinsky, O.; Schein, A.; Najjar, S. Epilepsy associated with systemic autoimmune disorders: Epilepsy and systemic autoimmune disorders. Epilepsy Curr. 2013, 13, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Depaulis, A.; Charpier, S. Pathophysiology of absence epilepsy: Insights from genetic models. Neurosci. Lett. 2018, 667, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Casillas-Espinosa, P.M.; Powell, K.L.; Zhu, M.; Campbell, C.R.; Maia, J.M.; Ren, Z.; Jones, N.C.; O’Brien, T.J.; Petrovski, S. Evaluating whole genome sequence data from the Genetic Absence Epilepsy Rat from Strasbourg and its related non-epileptic strain. PLoS ONE 2017, 12, e0179924. [Google Scholar] [CrossRef] [Green Version]
- Danış, Ö.; Demir, S.; Günel, A.; Aker, R.G.; Gülçebi, M.; Onat, F.; Ogan, A. Changes in intracellular protein expression in cortex, thalamus and hippocampus in a genetic rat model of absence epilepsy. Brain Res. Bull. 2011, 84, 381–388. [Google Scholar] [CrossRef]
- Yuce-Dursun, B.; Danis, O.; Demir, S.; Ogan, A.; Onat, F. Proteomic changes in the cortex membrane fraction of genetic absence epilepsy rats from Strasbourg. J. Integr. Neurosci. 2014, 13, 633–644. [Google Scholar] [CrossRef]
- Schadt, E.E. Molecular networks as sensors and drivers of common human diseases. Nature 2009, 461, 218–223. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horgan, R.P.; Kenny, L.C. ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Dejakaisaya, H.; Harutyunyan, A.; Kwan, P.; Jones, N.C. Altered metabolic pathways in a transgenic mouse model suggest mechanistic role of amyloid precursor protein overexpression in Alzheimer’s disease. Metabolomics 2021, 17, 42. [Google Scholar] [CrossRef]
- Harutyunyan, A.; Jones, N.C.; Kwan, P.; Anderson, A. Network preservation analysis reveals dysregulated synaptic modules and regulatory hubs shared between alzheimer’s disease and temporal lobe epilepsy. Front. Genet. 2022, 13, 821343. [Google Scholar] [CrossRef]
- Johnson, M.R.; Behmoaras, J.; Bottolo, L.; Krishnan, M.L.; Pernhorst, K.; Santoscoy, P.L.M.; Rossetti, T.; Speed, D.; Srivastava, P.K.; Chadeau-Hyam, M.; et al. Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat. Commun. 2015, 6, 6031. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.L.; Lukic, V.; Thorne, N.P.; Berkovic, S.F.; Scheffer, I.E.; Bahlo, M. Harnessing Gene Expression Networks to Prioritize Candidate Epileptic Encephalopathy Genes. PLoS ONE 2014, 9, e102079. [Google Scholar] [CrossRef] [Green Version]
- Snead, O.C. Basic mechanisms of generalized absence seizures. Ann. Neurol. 1995, 37, 146–157. [Google Scholar] [CrossRef]
- Chen, C.-H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.; Houten, S.M. The lysine degradation pathway: Subcellular compartmentalization and enzyme deficiencies. Mol. Genet. Metab. 2020, 131, 14–22. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.; Hartmann, H.; Jaggumantri, S.; Bok, L.A.; Cheng, B.; Connolly, M.; Coughlin, C.R., II; Das, A.M.; Gospe, S.M.J.; Jakobs, C.; et al. Lysine restricted diet for pyridoxine-dependent epilepsy: First evidence and future trials. Mol. Genet. Metab. 2012, 107, 335–344. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-S.; Tsai, C.-S. Enhanced lipid peroxidation in epileptics with null genotype of glutathione S-transferase M1 and intractable seizure. Jpn. J. Pharmacol. 2002, 90, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Smith, J.N.; Patel, M. Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [Green Version]
- Patel, M. A metabolic paradigm for epilepsy. Epilepsy Curr. 2018, 18, 318–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, M.C.; Horvath, S.; Geschwind, D.H. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc. Natl. Acad. Sci. USA 2006, 103, 17973–17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabasi, A.-L. Hierarchical Organization of Modularity in Metabolic Networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casillas-Espinosa, P.M.; Sargsyan, A.; Melkonian, D.; O’Brien, T.J. A universal automated tool for reliable detection of seizures in rodent models of acquired and genetic epilepsy. Epilepsia 2019, 60, 783–791. [Google Scholar] [CrossRef]
- Casillas-Espinosa, P.M.; Hicks, A.; Jeffreys, A.; Snutch, T.P.; O’Brien, T.J.; Powell, K.L. Z944, a novel selective T-type calcium channel antagonist delays the progression of seizures in the amygdala kindling model. PLoS ONE 2015, 10, e0130012. [Google Scholar] [CrossRef] [Green Version]
- Casillas-Espinosa, P.M.; Shultz, S.R.; Braine, E.L.; Jones, N.C.; Snutch, T.P.; Powell, K.L.; O’Brien, T.J. Disease-modifying effects of a novel T-type calcium channel antagonist, Z944, in a model of temporal lobe epilepsy. Prog. Neurobiol. 2019, 182, 101677. [Google Scholar] [CrossRef]
- Li, L.M.R.; Yuan, X.; Radfar, M.; Marendy, P.; Ni, W.; O’Brien, T.J.; Casillas-Espinosa, P. Spike wave discharges detection in animal models and epilepsy patients with genetic generalised epilepsy with absence seizures. Int. Epilepsy Congr. 2021, 62, 128–129. [Google Scholar]
- Jones, N.C.; Salzberg, M.R.; Kumar, G.; Couper, A.; Morris, M.; O’Brien, T. Elevated anxiety and depressive-like behavior in a rat model of genetic generalized epilepsy suggesting common causation. Exp. Neurol. 2008, 209, 254–260. [Google Scholar] [CrossRef]
- Johnstone, V.P.; Wright, D.K.; Wong, K.K.; O’Brien, T.J.; Rajan, R.; Shultz, S.R. Experimental Traumatic Brain Injury Results in Long-Term Recovery of Functional Responsiveness in Sensory Cortex but Persisting Structural Changes and Sensorimotor, Cognitive, and Emotional Deficits. J. Neurotrauma 2015, 32, 1333–1346. [Google Scholar] [CrossRef]
- Sarkisova, K.; Midzianovskaia, I.; Kulikov, M. Depressive-like behavioral alterations and c-fos expression in the dopaminergic brain regions in WAG/Rij rats with genetic absence epilepsy. Behav. Brain Res. 2003, 144, 211–226. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Stoessel, D.; Nowell, C.J.; Jones, A.J.; Ferrins, L.; Ellis, K.M.; Riley, J.; Rahmani, R.; Read, K.D.; McConville, M.J.; Avery, V.M.; et al. Metabolomics and lipidomics reveal perturbation of sphingolipid metabolism by a novel anti-trypanosomal 3-(oxazolo [4,5-b] pyridine-2-yl) anilide. Metabolomics 2016, 12, 126. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E.V. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography—Mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Jankevics, A.; Burgess, K.E.; Breitling, R.; Barrett, M.P. IDEOM: An excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics 2012, 28, 1048–1049. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Jankevics, A.; Jansen, R.C.; Swertz, M.A.; Breitling, R. PeakML/mzMatch: A File Format, Java Library, R Library, and Tool-Chain for Mass Spectrometry Data Analysis. Anal. Chem. 2011, 83, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jackson, J.; Edward, A. User’s Guide to Principal Components; Wiley: New York, NY, USA, 1991. [Google Scholar]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. Metaboanalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession ID | Protein | Description | Fold Change | FDR |
|---|---|---|---|---|
| Q6AZ33 | BLVRA | Biliverdin reductase A | −6.13954 | 1.02 × 10−10 |
| Q6P2A7 | FLOT | Flotillin | −2.05081 | 1.60 × 10−9 |
| G3V6T7 | PDIA4 | Protein disulfide isomerase family A, member 4 | 5.357599 | 2.55 × 10−9 |
| CAMKV | CAMKV | CaM kinase-like vesicle-associated | −6.52286 | 5.88 × 10−9 |
| A0A0G2JXT3 | FDPS | Farnesyl diphosphate synthase | −3.89937 | 5.88 × 10−9 |
| G3V983 | GSTM1 | Glutathione S-transferase mu 1 | 4.594248 | 1.78 × 10−8 |
| NIT2 | NIT2 | Nitrilase family, member 2 | −1.41015 | 1.78 × 10−8 |
| ALDH2 | ALDH2 | Aldehyde dehydrogenase 2 family member | 6.007907 | 3.39 × 10−8 |
| KAD1 | AK1 | Adenylate kinase isoenzyme 1 | 1.421165 | 2.03 × 10−7 |
| A0A0G2JSW3 | HBB | Haemoglobin subunit beta | 7.73431 | 3.46 × 10−7 |
| Accession ID | Protein | Description | Fold Change | FDR |
|---|---|---|---|---|
| ALDH2 | ALDH2 | Aldehyde dehydrogenase 2 family member | 5.39302723 | 7.34 × 10−13 |
| G3V6T7 | PDIA4 | Protein disulfide isomerase family A, member 4 | 4.98460727 | 5.74 × 10−12 |
| Q63011 | NA (fragment) | Zero beta-globin | −3.4885391 | 3.25 × 10−10 |
| CAMKV | CAMKV | CaM kinase-like vesicle-associated | −5.5727073 | 3.67 × 10−9 |
| A0A0G2JSW3 | HBB | Haemoglobin subunit beta | 8.40519465 | 3.75 × 10−9 |
| NIT2 | NIT2 | Nitrilase family, member 2 | −1.4379458 | 6.72 × 10−9 |
| A0A0G2JXT3 | FDPS | Farnesyl diphosphate synthase | −4.4110211 | 2.54 × 10−8 |
| KAD1 | AK1 | Adenylate kinase isoenzyme 1 | 1.49689657 | 4.63 × 10−8 |
| M0R544 | GAA | Glucosidase, alpha, acid | 3.66093389 | 4.65 × 10−8 |
| Q6AZ33 | BLVRA | Biliverdin reductase A | −6.0399292 | 4.65 × 10−8 |
| Somatosensory Cortex | ||
| Pathway | p-Value | FDR |
| Aminoacyl-tRNA biosynthesis | 1.38 × 10−6 | 0.000451 |
| ABC transporters | 7.02 × 10−6 | 0.001145 |
| Protein digestion and absorption | 4.85 × 10−5 | 0.005269 |
| Lysine degradation | 7.88 × 10−5 | 0.006423 |
| Glycine, serine and threonine metabolism | 0.000158 | 0.009242 |
| Arginine and proline metabolism | 0.00017 | 0.009242 |
| Amyotrophic lateral sclerosis (ALS) | 0.000314 | 0.013445 |
| Central carbon metabolism in cancer | 0.00033 | 0.013445 |
| Thalamus | ||
| Pathway | p-Value | FDR |
| Lysine degradation | 8.29 × 10−6 | 0.002703 |
| ABC transporters | 7.20 × 10−5 | 0.009727 |
| Aminoacyl-tRNA biosynthesis | 8.95 × 10−5 | 0.009727 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harutyunyan, A.; Chong, D.; Li, R.; Shah, A.D.; Ali, Z.; Huang, C.; Barlow, C.K.; Perucca, P.; O’Brien, T.J.; Jones, N.C.; et al. An Integrated Multi-Omic Network Analysis Identifies Seizure-Associated Dysregulated Pathways in the GAERS Model of Absence Epilepsy. Int. J. Mol. Sci. 2022, 23, 6063. https://doi.org/10.3390/ijms23116063
Harutyunyan A, Chong D, Li R, Shah AD, Ali Z, Huang C, Barlow CK, Perucca P, O’Brien TJ, Jones NC, et al. An Integrated Multi-Omic Network Analysis Identifies Seizure-Associated Dysregulated Pathways in the GAERS Model of Absence Epilepsy. International Journal of Molecular Sciences. 2022; 23(11):6063. https://doi.org/10.3390/ijms23116063
Chicago/Turabian StyleHarutyunyan, Anna, Debbie Chong, Rui Li, Anup D. Shah, Zahra Ali, Cheng Huang, Christopher K. Barlow, Piero Perucca, Terence J. O’Brien, Nigel C. Jones, and et al. 2022. "An Integrated Multi-Omic Network Analysis Identifies Seizure-Associated Dysregulated Pathways in the GAERS Model of Absence Epilepsy" International Journal of Molecular Sciences 23, no. 11: 6063. https://doi.org/10.3390/ijms23116063