
Mechanism of Ir(ppy)3 Guest Exciton Formation with the Exciplex-Forming TCTA:TPBI Cohost within a Phosphorescent Organic Light-Emitting Diode Environment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory and Computational Details
2.1. Förster Theory
2.2. Computational Details
3. Results and Discussion
3.1. Trimer Model Systems
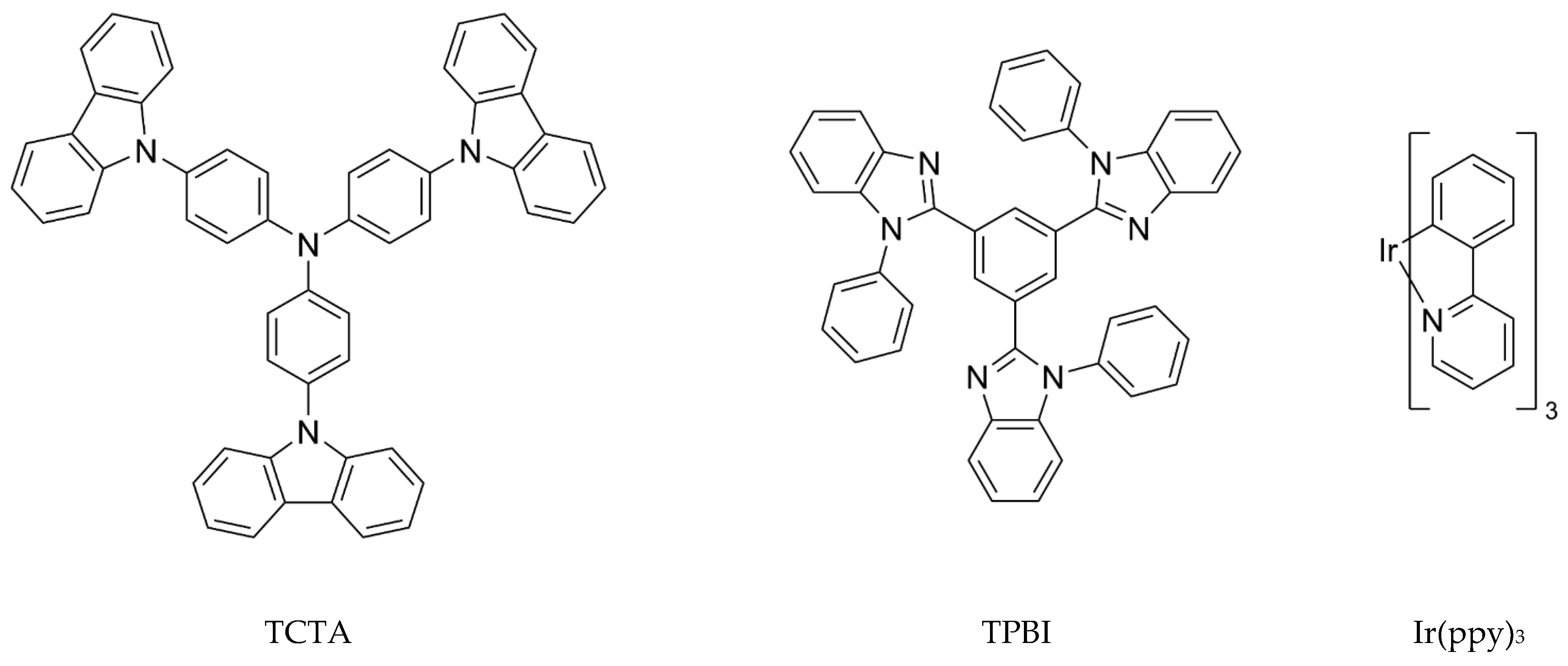
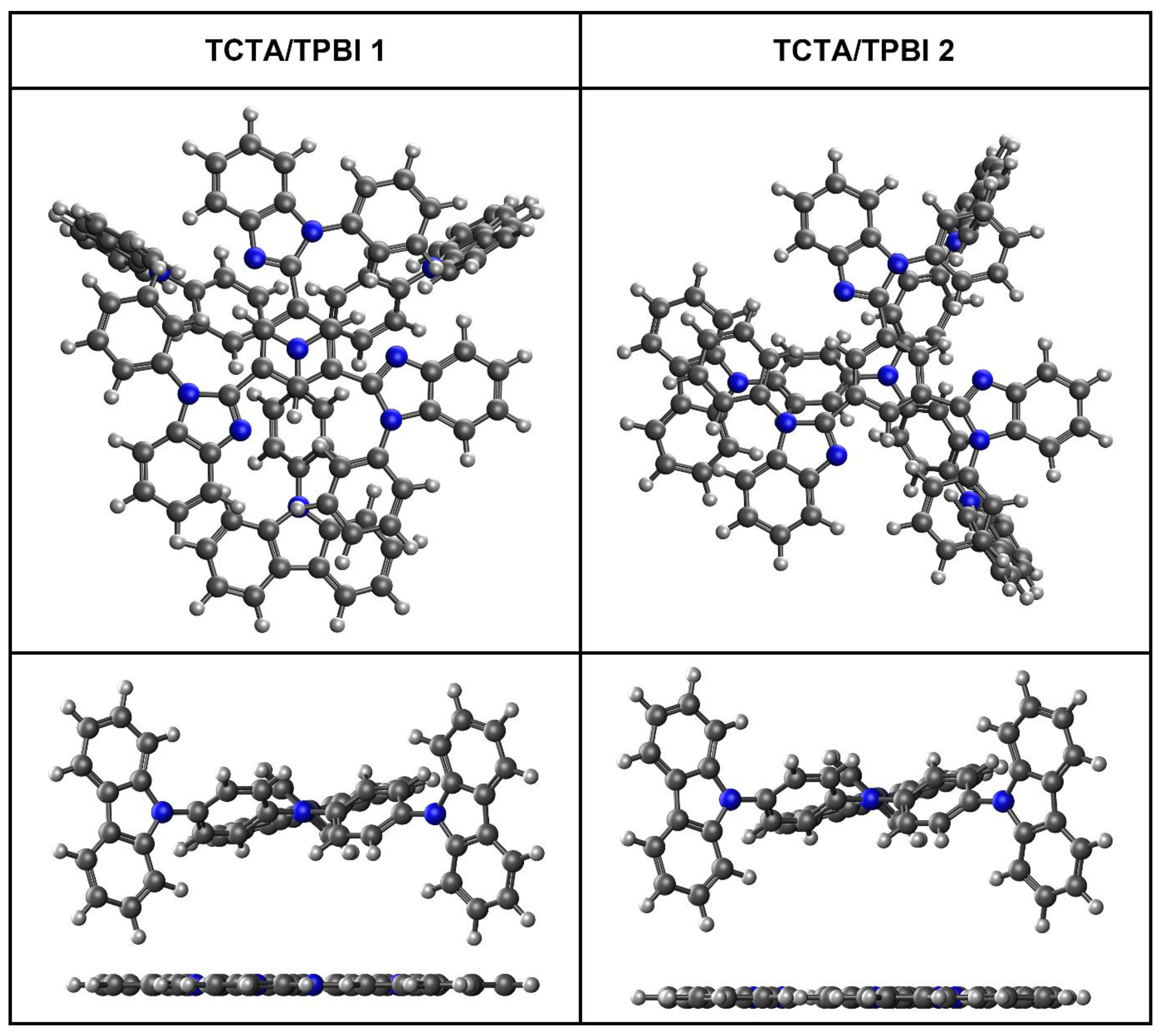
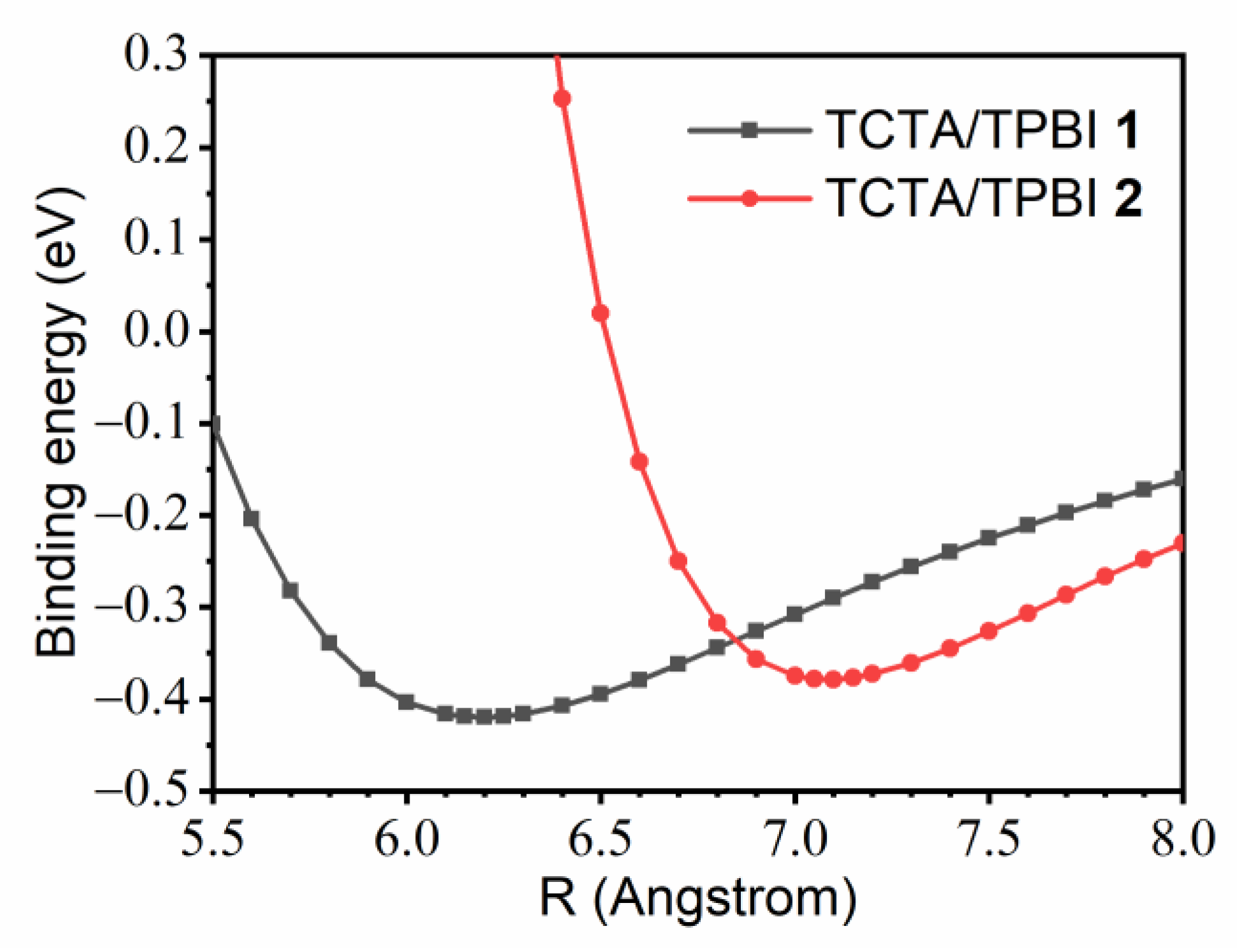
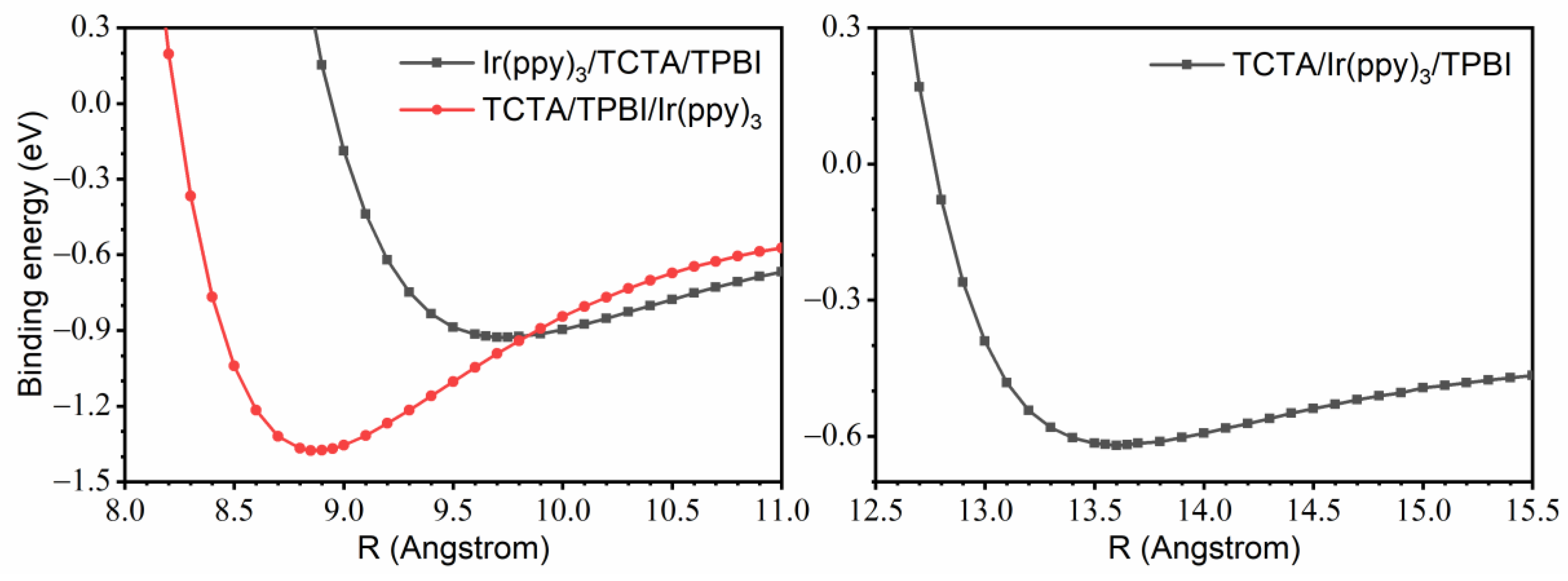
3.1.1. Construction of TCTA/TPBI Complex
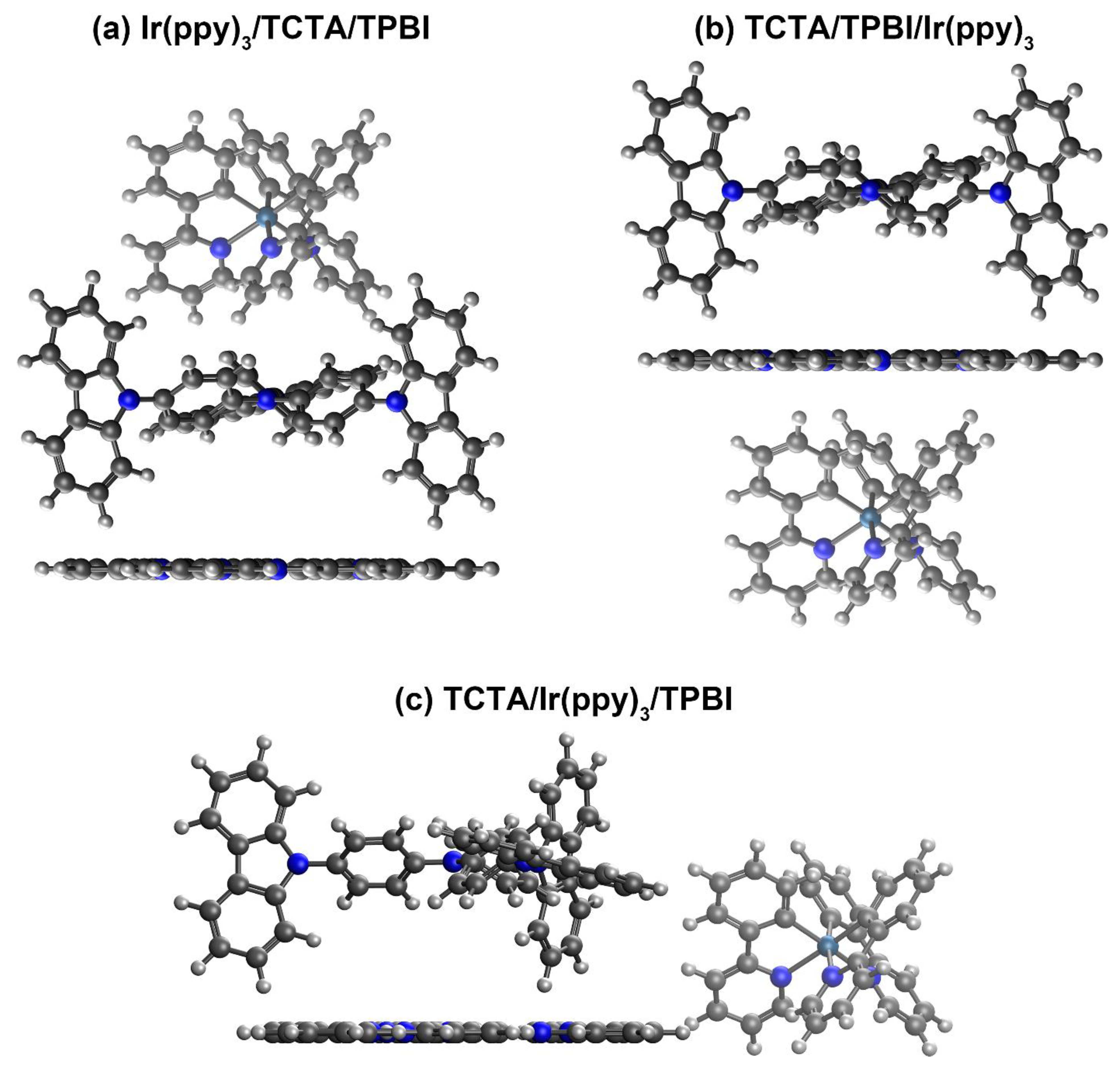
3.1.2. Construction of Trimer Model Systems
3.2. Excited States of Trimer Model Systems
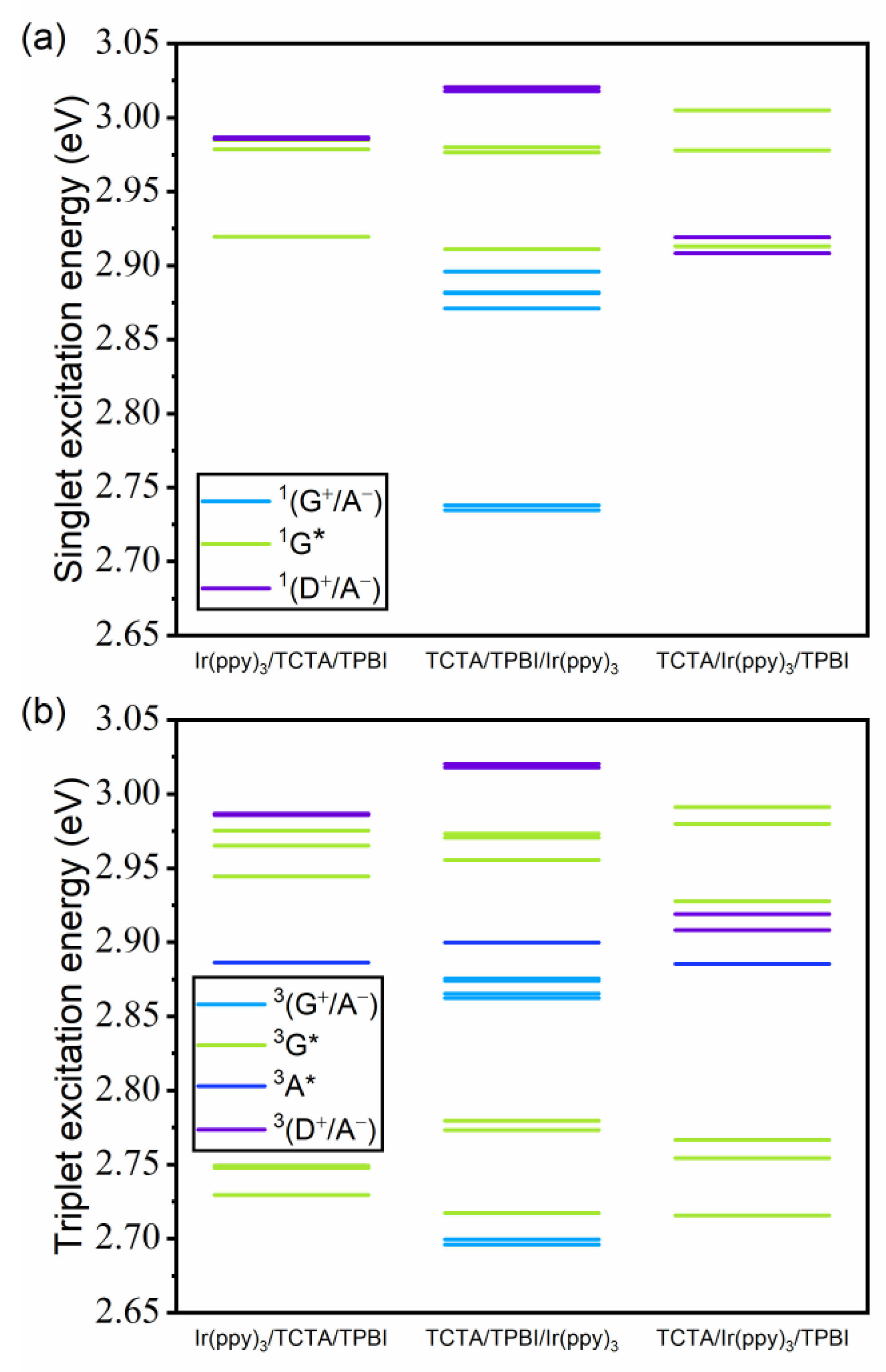
3.2.1. Energies of Singlet and Triplet Excited States
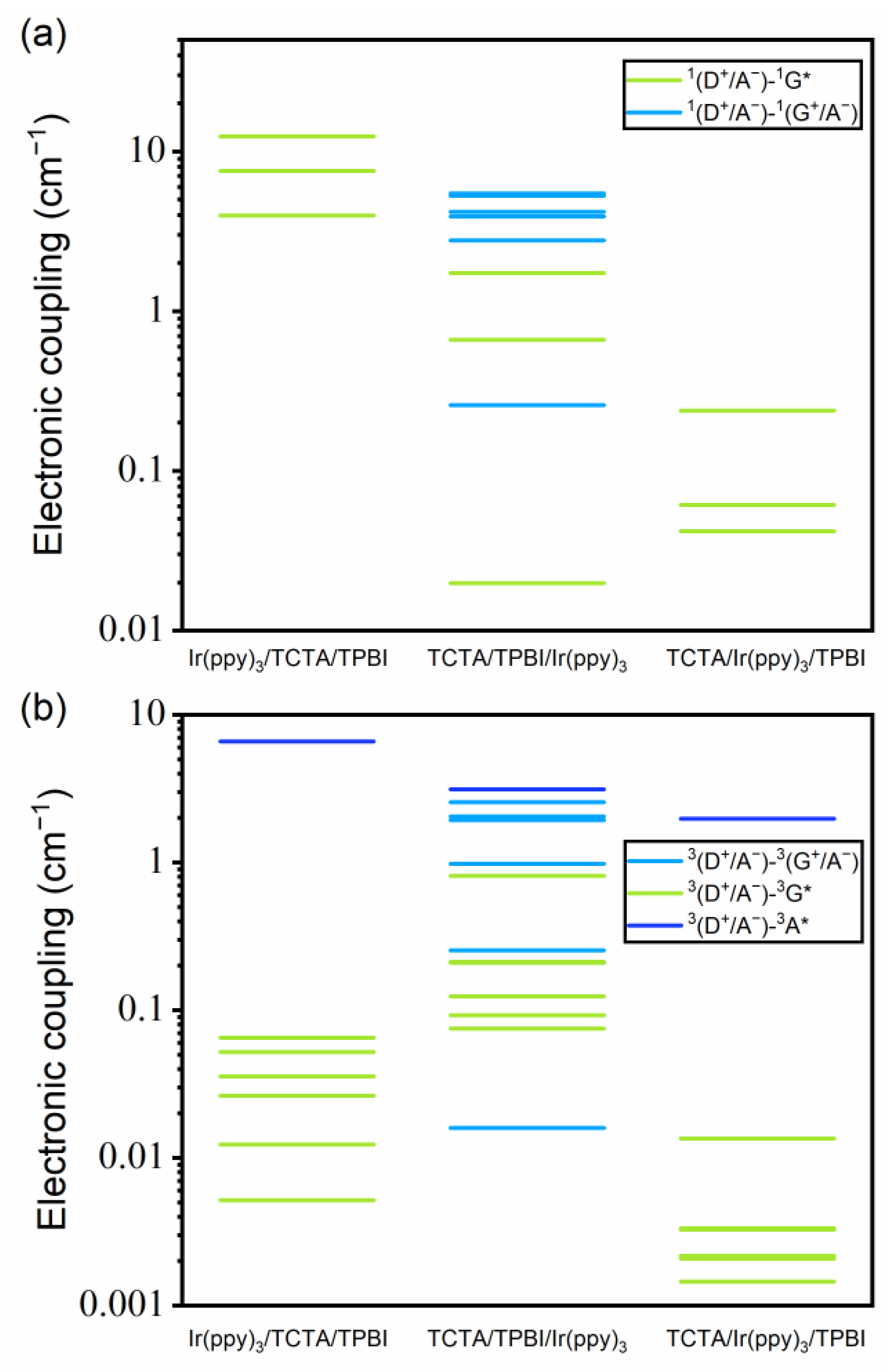
3.2.2. Electronic Couplings between the Excited States
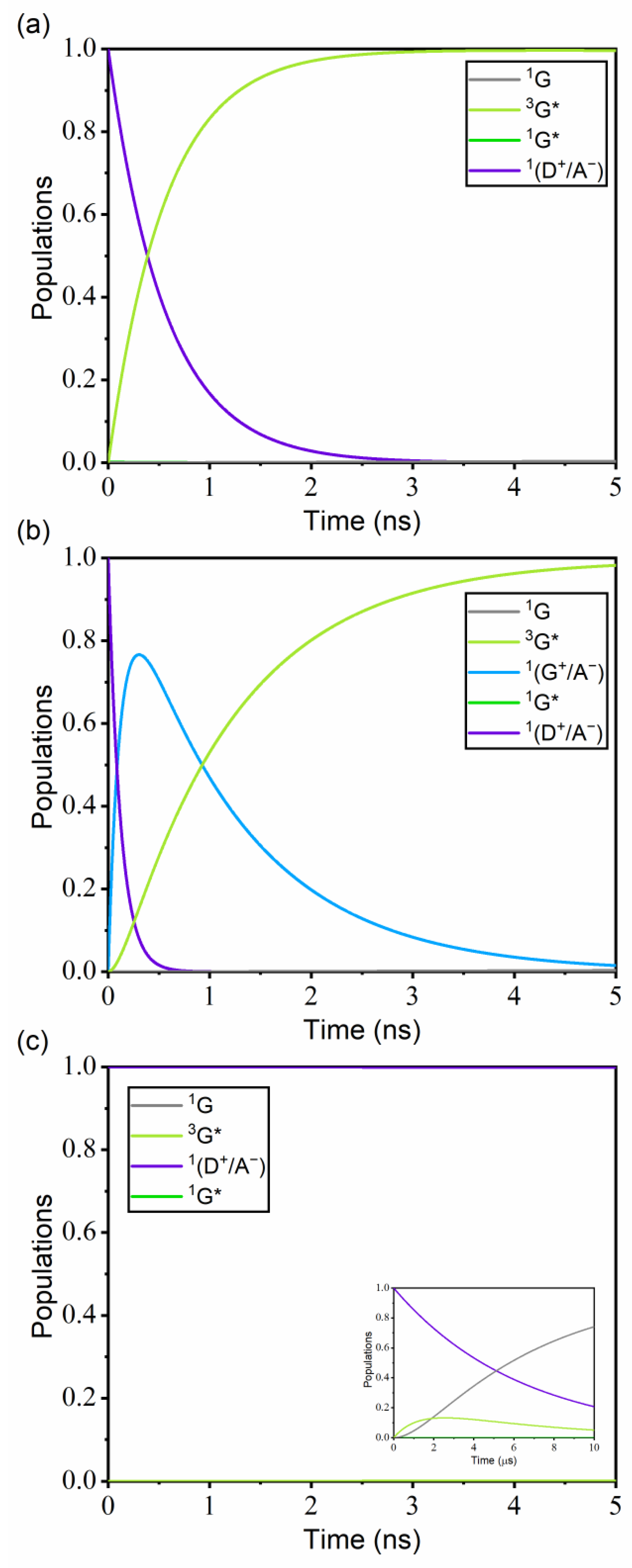
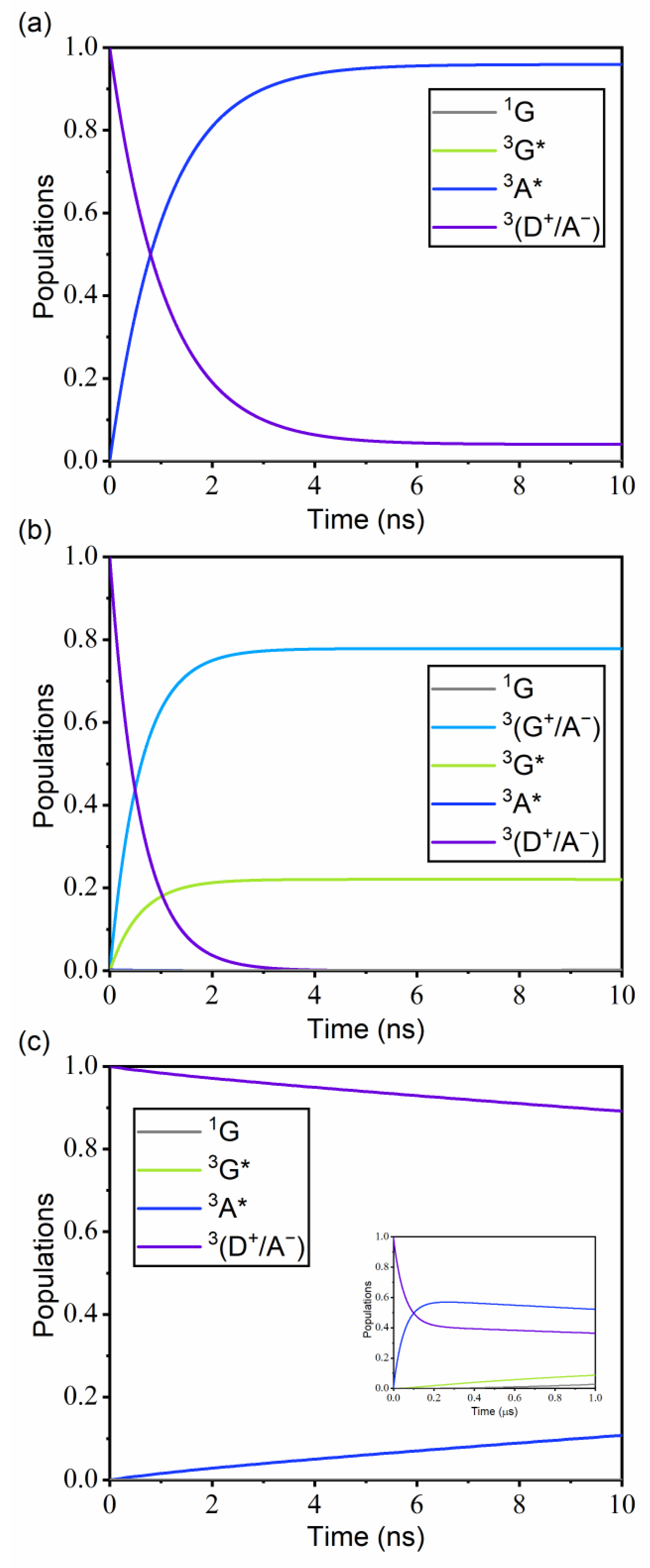
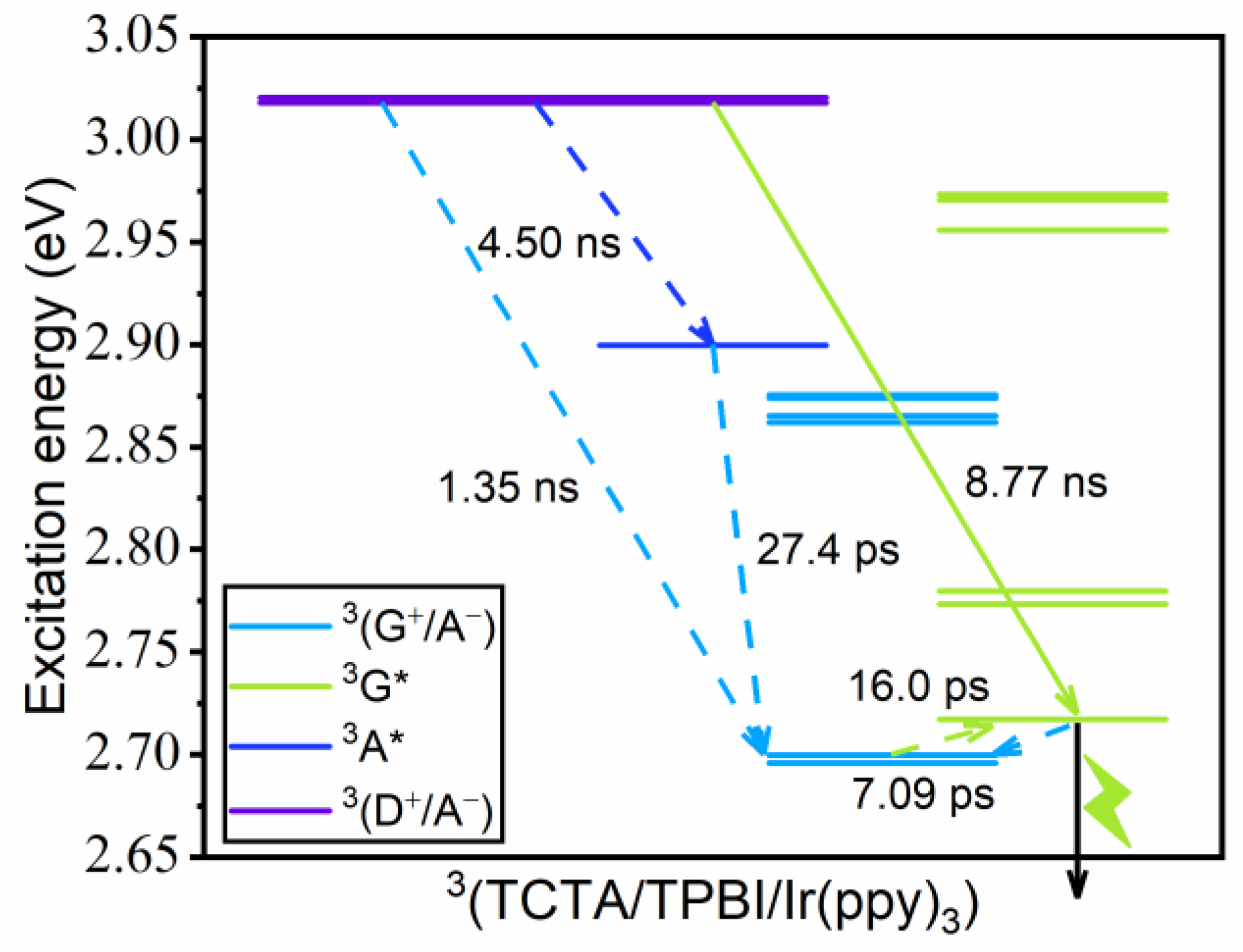
3.3. Kinetics of Guest Excitation Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luo, D.; Liao, C.-W.; Chang, C.-H.; Tsai, C.-C.; Lu, C.-W.; Chuang, T.C.; Chang, H.-H. Approach to fast screen the formation of an exciplex. J. Phys. Chem. C 2020, 124, 10175–10184. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Jeong, W.-I.; Mayr, C.; Park, Y.-S.; Kim, K.-H.; Lee, J.-H.; Moon, C.-K.; Brütting, W.; Kim, J.-J. Organic Light-Emitting diodes with 30% external quantum efficiency based on a horizontally oriented emitter. Adv. Funct. Mater. 2013, 23, 3896–3900. [Google Scholar] [CrossRef]
- Park, Y.-S.; Lee, S.; Kim, K.-H.; Kim, S.-Y.; Lee, J.-H.; Kim, J.-J. Exciplex-forming co-host for organic light-emitting diodes with ultimate efficiency. Adv. Funct. Mater. 2013, 23, 4914–4920. [Google Scholar] [CrossRef]
- Lee, J.Y. Mixed-host-emitting layer for high-efficiency organic light-emitting diodes. J. Inf. Disp. 2014, 15, 139–144. [Google Scholar] [CrossRef]
- Lee, S.; Kim, K.-H.; Limbach, D.; Park, Y.-S.; Kim, J.-J. Low roll-off and high efficiency orange organic light emitting diodes with controlled co-doping of green and red phosphorescent dopants in an exciplex forming co-host. Adv. Funct. Mater. 2013, 23, 4105–4110. [Google Scholar] [CrossRef]
- Song, W.; Lee, J.Y. Design strategy of exciplex host for extended operational lifetime. Org. Electron. 2017, 48, 285–290. [Google Scholar] [CrossRef]
- Jung, M.; Lee, J.Y. Exciplex hosts for blue phosphorescent organic light-emitting diodes. J. Inf. Disp. 2020, 21, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Shin, H.; Kim, K.-H.; Yoo, S.-J.; Huh, J.-S.; Kim, J.-J. An exciplex host for deep-blue phosphorescent organic light-emitting diodes. ACS Appl. Mater. Interfaces 2017, 9, 37883–37887. [Google Scholar] [CrossRef]
- Lee, J.-H.; Shin, H.; Kim, J.-M.; Kim, K.-H.; Kim, J.-J. Exciplex-forming co-host-based red phosphorescent organic light-emitting diodes with long operational stability and high efficiency. ACS Appl. Mater. Interfaces 2017, 9, 3277–3281. [Google Scholar] [CrossRef]
- Shin, H.; Lee, S.; Kim, K.-H.; Moon, C.-K.; Yoo, S.-J.; Lee, J.-H.; Kim, J.-J. Blue phosphorescent organic light-emitting diodes using an exciplex forming co-host with the external quantum efficiency of theoretical limit. Adv. Mater. 2014, 26, 4730–4734. [Google Scholar] [CrossRef]
- Liu, X.; Yao, B.; Zhang, Z.; Zhao, X.; Zhang, B.; Wong, W.-Y.; Cheng, Y.; Xie, Z. Power-efficient solution-processed red organic light-emitting diodes based on an exciplex host and a novel phosphorescent iridium complex. J. Mater. Chem. C 2016, 4, 5787–5794. [Google Scholar] [CrossRef]
- Liu, B.; Hu, S.; Zhang, L.; Xiao, P.; Huang, L.; Liu, C. Blue molecular emitter-free and doping-free white organic light-emitting diodes with high color rendering. IEEE Electron Device Lett. 2021, 42, 387–390. [Google Scholar] [CrossRef]
- Luo, D.; Xiao, Y.; Hao, M.; Zhao, Y.; Yang, Y.; Gao, Y.; Liu, B. Doping-free white organic light-emitting diodes without blue molecular emitter: An unexplored approach to achieve high performance via exciplex emission. Appl. Phys. Lett. 2017, 110, 061105. [Google Scholar] [CrossRef]
- Luo, D.; Li, X.-L.; Zhao, Y.; Gao, Y.; Liu, B. High-performance blue molecular emitter-free and doping-free hybrid white organic light-emitting diodes: An alternative concept to manipulate charges and excitons based on exciplex and electroplex emission. ACS Photonics 2017, 4, 1566–1575. [Google Scholar] [CrossRef]
- Wang, L.; Kou, Z.; Wang, B.; Zhou, J.; Lu, Z.; Li, L. Realizing high efficiency/CRI/color stability in the hybrid white organic light emitting diode by manipulating exciton energy transfer. Opt. Mater. 2021, 115, 111059. [Google Scholar] [CrossRef]
- Xiao, P.; Huang, J.; Yu, Y.; Yuan, J.; Luo, D.; Liu, B.; Liang, D. Recent advances of exciplex-based white organic light-emitting diodes. Appl. Sci. 2018, 8, 1449. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Yang, C.; Qin, J. Organic host materials for phosphorescent organic light-emitting diodes. Chem. Soc. Rev. 2011, 40, 2943–2970. [Google Scholar] [CrossRef]
- Chaskar, A.; Chen, H.-F.; Wong, K.-T. Bipolar host materials: A chemical approach for highly efficient electrophosphorescent devices. Adv. Mater. 2011, 23, 3876–3895. [Google Scholar] [CrossRef]
- Yeh, S.-J.; Wu, M.-F.; Chen, C.-T.; Song, Y.-H.; Chi, Y.; Ho, M.-H.; Hsu, S.-F.; Chen, C.H. New dopant and host materials for Blue-Light-Emitting phosphorescent organic electroluminescent devices. Adv. Mater. 2005, 17, 285–289. [Google Scholar] [CrossRef]
- Tokito, S.; Iijima, T.; Suzuri, Y.; Kita, H.; Tsuzuki, T.; Sato, F. Confinement of triplet energy on phosphorescent molecules for highly-efficient organic blue-light-emitting devices. Appl. Phys. Lett. 2003, 83, 569–571. [Google Scholar] [CrossRef]
- Nakanotani, H.; Masui, K.; Nishide, J.; Shibata, T.; Adachi, C. Promising operational stability of high-efficiency organic light-emitting diodes based on thermally activated delayed fluorescence. Sci. Rep. 2013, 3, 2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldo, M.A.; Lamansky, S.; Burrows, P.E.; Thompson, M.E.; Forrest, S.R. Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Appl. Phys. Lett. 1999, 75, 4–6. [Google Scholar] [CrossRef]
- Zhou, X.; Qin, D.S.; Pfeiffer, M.; Blochwitz-Nimoth, J.; Werner, A.; Drechsel, J.; Maennig, B.; Leo, K.; Bold, M.; Erk, P.; et al. High-efficiency electrophosphorescent organic light-emitting diodes with double light-emitting layers. Appl. Phys. Lett. 2002, 81, 4070–4072. [Google Scholar] [CrossRef]
- Adachi, C.; Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.C.; Karg, S.; Carter, S.A. Bipolar charge and current distributions in organic light-emitting diodes. J. Appl. Phys. 1997, 82, 1454–1460. [Google Scholar] [CrossRef]
- Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Phosphorescent materials for application to organic light emitting devices. Pure Appl. Chem. 1999, 71, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Adachi, C.; Baldo, M.A.; Forrest, S.R.; Thompson, M.E. High-efficiency organic electrophosphorescent devices with tris (2-phenylpyridine) iridium doped into electron-transporting materials. Appl. Phys. Lett. 2000, 77, 904–906. [Google Scholar] [CrossRef]
- Zhang, D.; Cai, M.; Zhang, Y.; Bin, Z.; Zhang, D.; Duan, L. Simultaneous enhancement of efficiency and stability of phosphorescent OLEDs based on efficient Förster energy transfer from interface exciplex. ACS Appl. Mater. Interfaces 2016, 8, 3825–3832. [Google Scholar] [CrossRef]
- Ban, X.; Sun, K.; Sun, Y.; Huang, B.; Jiang, W. Enhanced electron affinity and exciton confinement in exciplex-type host: Power efficient solution-processed blue phosphorescent OLEDs with low turn-on voltage. ACS Appl. Mater. Interfaces 2016, 8, 2010–2016. [Google Scholar] [CrossRef]
- Hsiao, C.-H.; Chen, Y.-H.; Lin, T.-C.; Hsiao, C.-C.; Lee, J.-H. Recombination zone in mixed-host organic light-emitting devices. Appl. Phys. Lett. 2006, 89, 163511. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.-I.; Lee, J.Y.; Chu, H.Y. Enhanced efficiency and reduced roll-off in blue and white phosphorescent organic light-emitting diodes with a mixed host structure. Appl. Phys. Lett. 2009, 94, 193305. [Google Scholar] [CrossRef]
- Lane, P.A.; Palilis, L.C.; O’Brien, D.F.; Giebeler, C.; Cadby, A.J.; Lidzey, D.G.; Campbell, A.J.; Blau, W.; Bradley, D.D.C. Origin of electrophosphorescence from a doped polymer light emitting diode. Phys. Rev. B 2001, 63, 235206. [Google Scholar] [CrossRef] [Green Version]
- Wetzelaer, G.A.H.; Kuik, M.; Nicolai, H.T.; Blom, P.W.M. Trap-assisted and Langevin-type recombination in organic light-emitting diodes. Phys. Rev. B 2011, 83, 165204. [Google Scholar] [CrossRef]
- Peng, Q.; Gao, N.; Li, W.; Chen, P.; Li, F.; Ma, Y. Investigation of energy transfer and charge trapping in dye-doped organic light-emitting diodes by magneto-electroluminescence measurement. Appl. Phys. Lett. 2013, 102, 193304. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, S.; Yoo, S.-J.; Kim, K.-H.; Kim, J.-J. Langevin and trap-assisted recombination in phosphorescent organic light emitting diodes. Adv. Funct. Mater. 2014, 24, 4681–4688. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, J.Y.; Park, T.J.; Jeon, W.S.; Kennedy, G.P.; Kwon, J.H. Small molecule host system for solution-processed red phosphorescent OLEDs. Synth. Met. 2010, 160, 631–635. [Google Scholar] [CrossRef]
- Park, Y.-S.; Jeong, W.-I.; Kim, J.-J. Energy transfer from exciplexes to dopants and its effect on efficiency of organic light-emitting diodes. J. Appl. Phys. 2011, 110, 124519. [Google Scholar] [CrossRef]
- Seo, S.; Shitagaki, S.; Ohsawa, N.; Inoue, H.; Suzuki, K.; Nowatari, H.; Yamazaki, S. Exciplex-triplet energy transfer: A new method to achieve extremely efficient organic light-emitting diode with external quantum efficiency over 30% and drive voltage below 3 V. Jpn. J. Appl. Phys. 2014, 53, 042102. [Google Scholar] [CrossRef]
- Song, W.; Lee, J.Y. Light emission mechanism of mixed host organic light-emitting diodes. Appl. Phys. Lett. 2015, 106, 123306. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Scholes, G.D. Long-range resonance energy transfer in molecular systems. Annu. Rev. Phys. Chem. 2003, 54, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.M.; Dattani, N.S. Why quantum coherence is not important in the Fenna–Matthews–Olsen complex. J. Chem. Theory Comput. 2015, 11, 3411–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaki, A.; Fleming, G.R. On the adequacy of the Redfield equation and related approaches to the study of quantum dynamics in electronic energy transfer. J. Chem. Phys. 2009, 130, 234110. [Google Scholar] [CrossRef] [PubMed]
- Renger, T. Theory of excitation energy transfer: From structure to function. Photosynth. Res. 2009, 102, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Fleming, G.R. Influence of phonons on exciton transfer dynamics: Comparison of the Redfield, Förster, and modified Redfield equations. Chem. Phys. 2002, 282, 163–180. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Garza, A.J.; Osman, O.I.; Wazzan, N.A.; Khan, S.B.; Asiri, A.M.; Scuseria, G.E. A computational study of the nonlinear optical properties of carbazole derivatives: Theory refines experiment. Theor. Chem. Acc. 2014, 133, 1458. [Google Scholar] [CrossRef]
- Pandey, L.; Doiron, C.; Sears, J.S.; Brédas, J.-L. Lowest excited states and optical absorption spectra of donor–acceptor copolymers for organic photovoltaics: A new picture emerging from tuned long-range corrected density functionals. Phys. Chem. Chem. Phys. 2012, 14, 14243–14248. [Google Scholar] [CrossRef] [Green Version]
- Karolewski, A.; Stein, T.; Baer, R.; Kümmel, S. Communication: Tailoring the optical gap in light-harvesting molecules. J. Chem. Phys. 2011, 134, 151101. [Google Scholar] [CrossRef]
- Stein, T.; Kronik, L.; Baer, R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Subotnik, J.E.; Vura-Weis, J.; Sodt, A.J.; Ratner, M.A. Predicting accurate electronic excitation transfer rates via Marcus theory with Boys or Edmiston− Ruedenberg localized diabatization. J. Phys. Chem. A 2010, 114, 8665–8675. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Moon, C.-K.; Huh, J.-S.; Kim, J.-M.; Kim, J.-J. Electronic structure and emission process of excited charge transfer states in solids. Chem. Mater. 2018, 30, 5648–5654. [Google Scholar] [CrossRef]
- Hung, W.-Y.; Chiang, P.-Y.; Lin, S.-W.; Tang, W.-C.; Chen, Y.-T.; Liu, S.-H.; Chou, P.-T.; Hung, Y.-T.; Wong, K.-T. Balance the carrier mobility to achieve high performance exciplex OLED using a triazine-based acceptor. ACS Appl. Mater. Interfaces 2016, 8, 4811–4818. [Google Scholar] [CrossRef]
- Angioni, E.; Chapran, M.; Ivaniuk, K.; Kostiv, N.; Cherpak, V.; Stakhira, P.; Lazauskas, A.; Tamulevičius, S.; Volyniuk, D.; Findlay, N.J.; et al. A single emitting layer white OLED based on exciplex interface emission. J. Mater. Chem. C 2016, 4, 3851–3856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-R.; Sears, J.S.; Yang, B.; Aziz, S.G.; Coropceanu, V.; Brédas, J.-L. Theoretical study of the local and charge-transfer excitations in model complexes of pentacene-C60 using tuned range-separated hybrid functionals. J. Chem. Theory Comput. 2014, 10, 2379–2388. [Google Scholar] [CrossRef]
- Lee, W.H.; Kim, D.H.; Jesuraj, P.J.; Hafeez, H.; Lee, J.C.; Choi, D.K.; Bae, T.-S.; Yu, S.M.; Song, M.; Kim, C.S.; et al. Improvement of charge balance, recombination zone confinement, and low efficiency roll-off in green phosphorescent OLEDs by altering electron transport layer thickness. Mater. Res. Express 2018, 5, 076201. [Google Scholar] [CrossRef]
- Hedley, G.J.; Ruseckas, A.; Samuel, I.D.W. Ultrafast luminescence in Ir(ppy)3. Chem. Phys. Lett. 2008, 450, 292–296. [Google Scholar] [CrossRef]
- Gertsen, A.S.; Koerstz, M.; Mikkelsen, K.V. Benchmarking triplet–triplet annihilation photon upconversion schemes. Phys. Chem. Chem. Phys. 2018, 20, 12182–12192. [Google Scholar] [CrossRef]
- Kleinschmidt, M.; Wüllen, C.V.; Marian, C.M. Intersystem-crossing and phosphorescence rates in fac-IrIII(ppy)3: A theoretical study involving multi-reference configuration interaction wavefunctions. J. Chem. Phys. 2015, 142, 094301. [Google Scholar] [CrossRef]
- de Vries, X.; Friederich, P.; Wenzel, W.; Coehoorn, R.; Bobbert, P.A. Triplet exciton diffusion in metalorganic phosphorescent host-guest systems from first principles. Phys. Rev. B 2019, 99, 205201. [Google Scholar] [CrossRef] [Green Version]
- Peach, M.J.G.; Williamson, M.J.; Tozer, D.J. Influence of triplet instabilities in TDDFT. J. Chem. Theory Comput. 2011, 7, 3578–3585. [Google Scholar] [CrossRef]
- de Moraes, I.R.; Scholz, S.; Lüssem, B.; Leo, K. Role of oxygen-bonds in the degradation process of phosphorescent organic light emitting diodes. Appl. Phys. Lett. 2011, 99, 053302. [Google Scholar] [CrossRef]
- Liu, B.; Wang, L.; Tao, H.; Xu, M.; Zou, J.; Ning, H.; Peng, J.; Cao, Y. Doping-free tandem white organic light-emitting diodes. Sci. Bull. 2017, 62, 1193–1200. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.W.; Cho, K.H.; Rhee, Y.M. Mechanism of Ir(ppy)3 Guest Exciton Formation with the Exciplex-Forming TCTA:TPBI Cohost within a Phosphorescent Organic Light-Emitting Diode Environment. Int. J. Mol. Sci. 2022, 23, 5940. https://doi.org/10.3390/ijms23115940
Park JW, Cho KH, Rhee YM. Mechanism of Ir(ppy)3 Guest Exciton Formation with the Exciplex-Forming TCTA:TPBI Cohost within a Phosphorescent Organic Light-Emitting Diode Environment. International Journal of Molecular Sciences. 2022; 23(11):5940. https://doi.org/10.3390/ijms23115940
Chicago/Turabian StylePark, Jae Whee, Kwang Hyun Cho, and Young Min Rhee. 2022. "Mechanism of Ir(ppy)3 Guest Exciton Formation with the Exciplex-Forming TCTA:TPBI Cohost within a Phosphorescent Organic Light-Emitting Diode Environment" International Journal of Molecular Sciences 23, no. 11: 5940. https://doi.org/10.3390/ijms23115940