
Morphological Characterization and Integrated Transcriptome and Proteome Analysis of Organ Development Defective 1 (odd1) Mutant in Cucumis sativus L.

 and
and
Abstract
:1. Introduction
2. Results
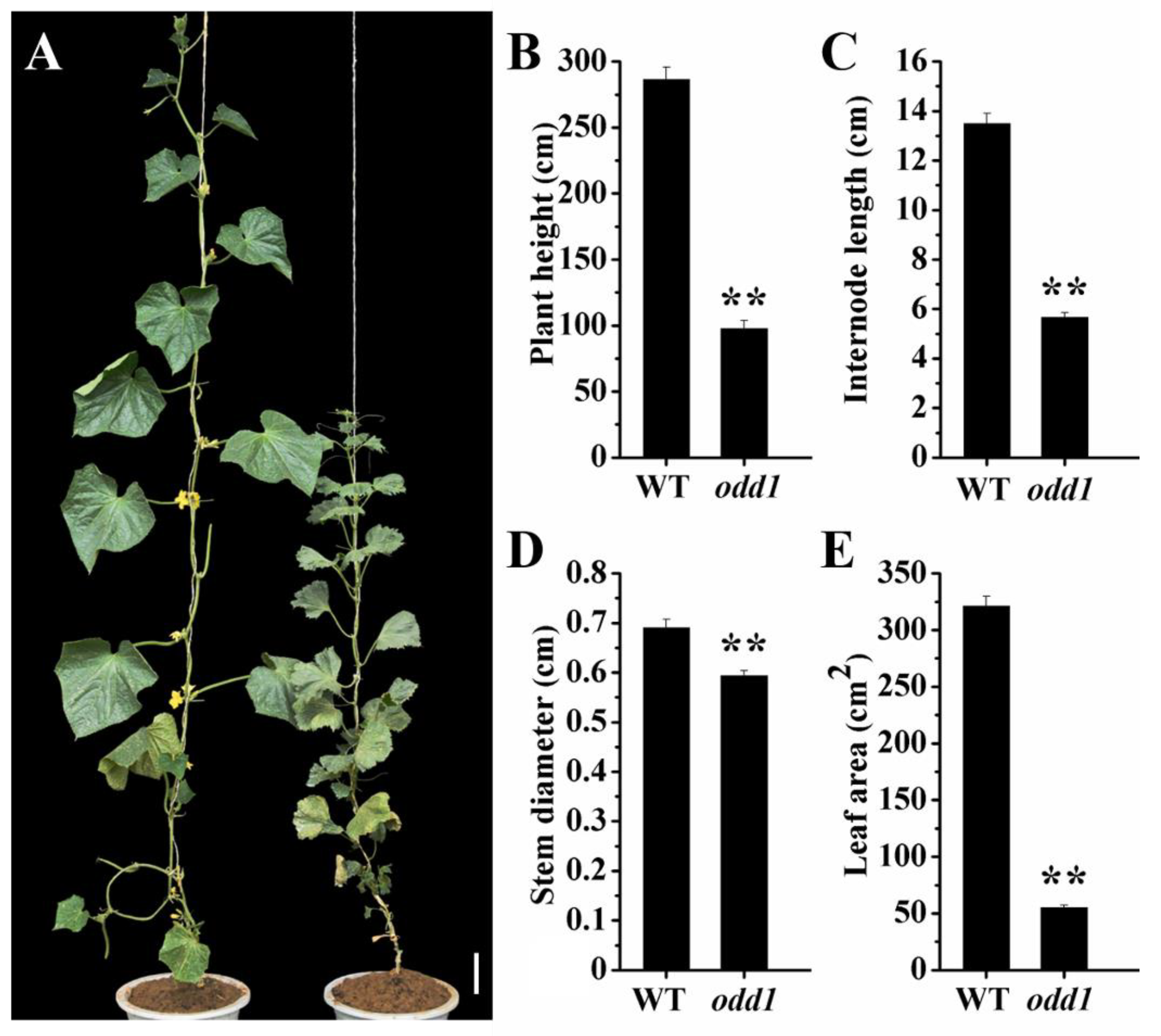
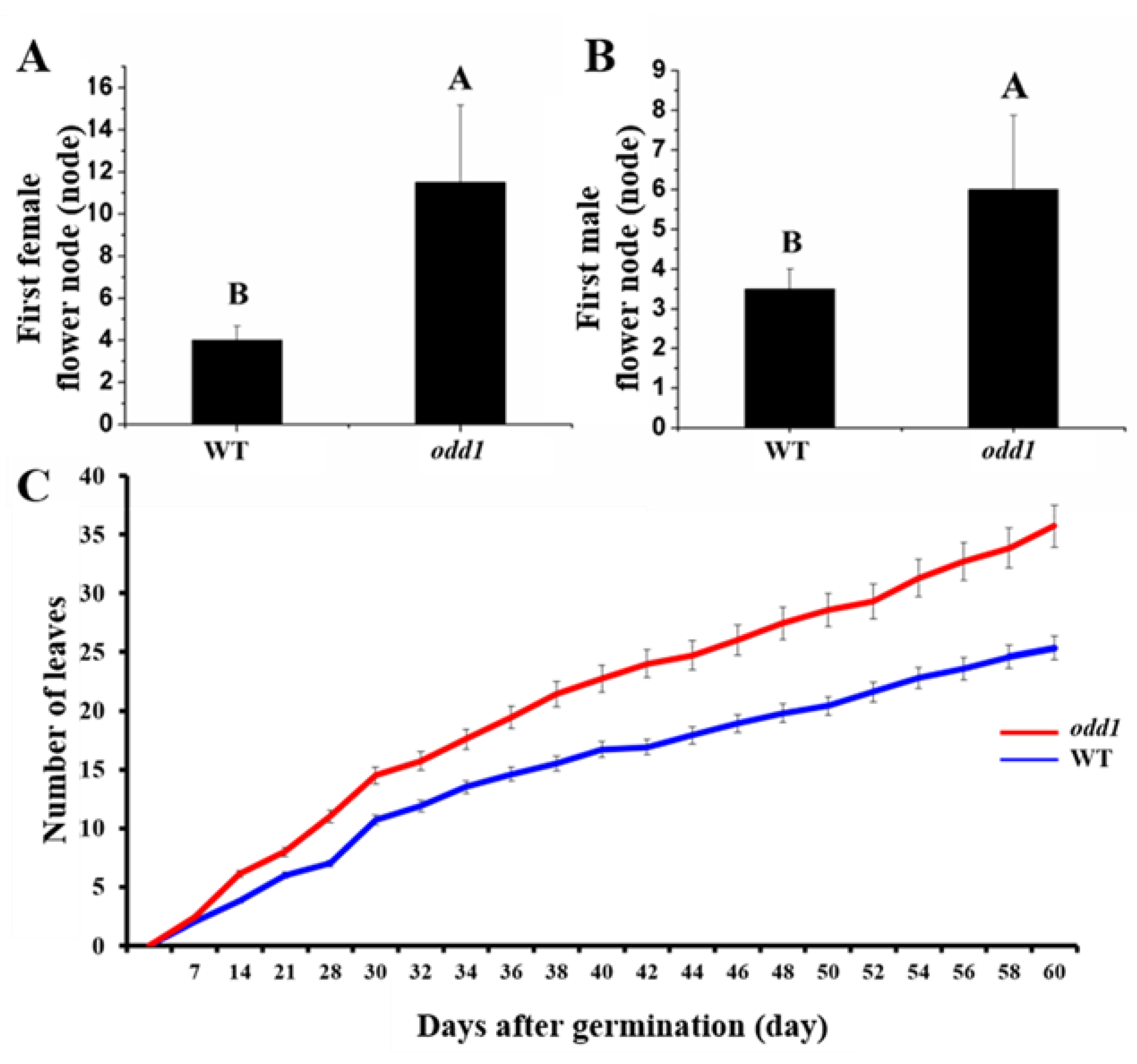
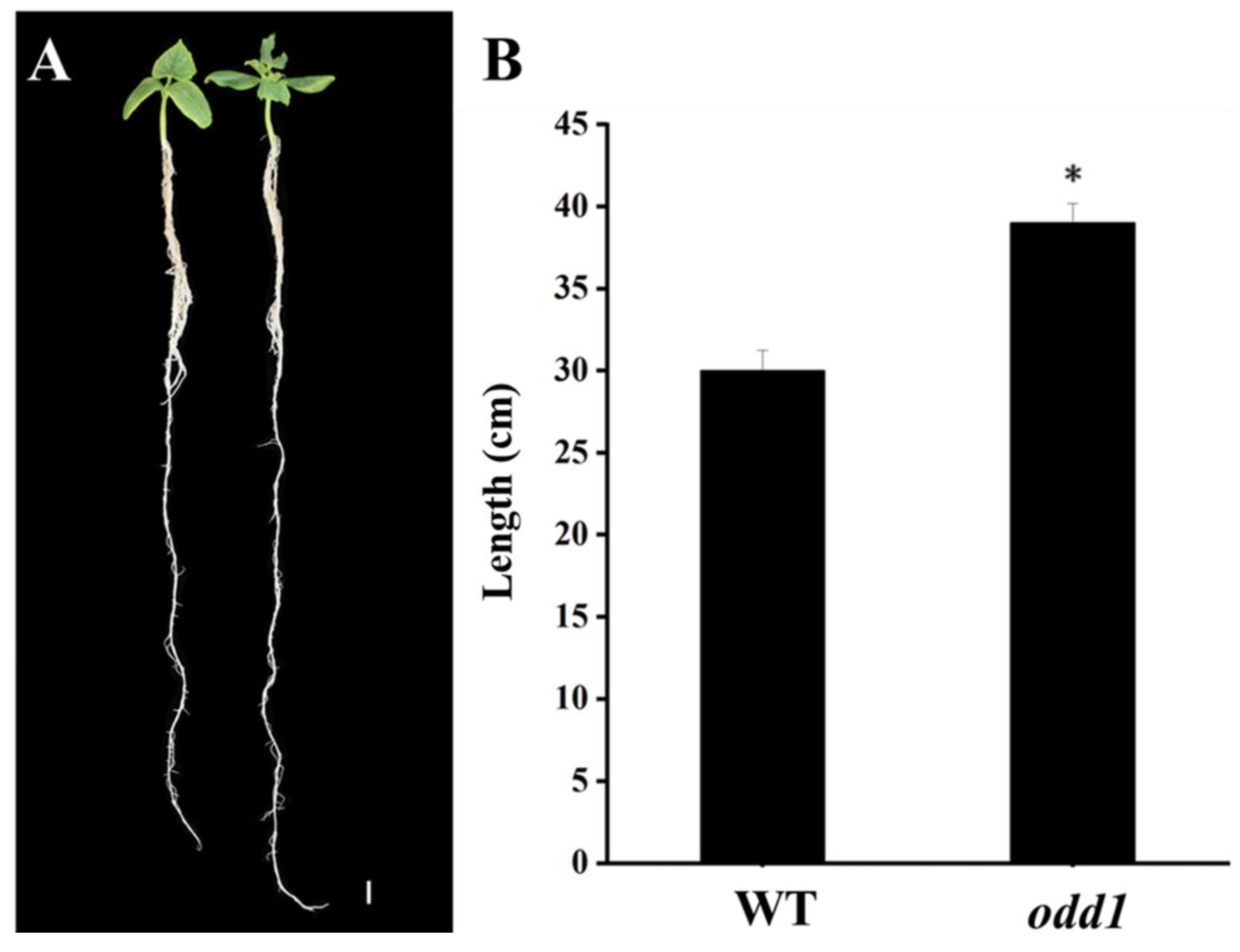
2.1. Phenotypic and Physiological Characterization of odd1 Mutant
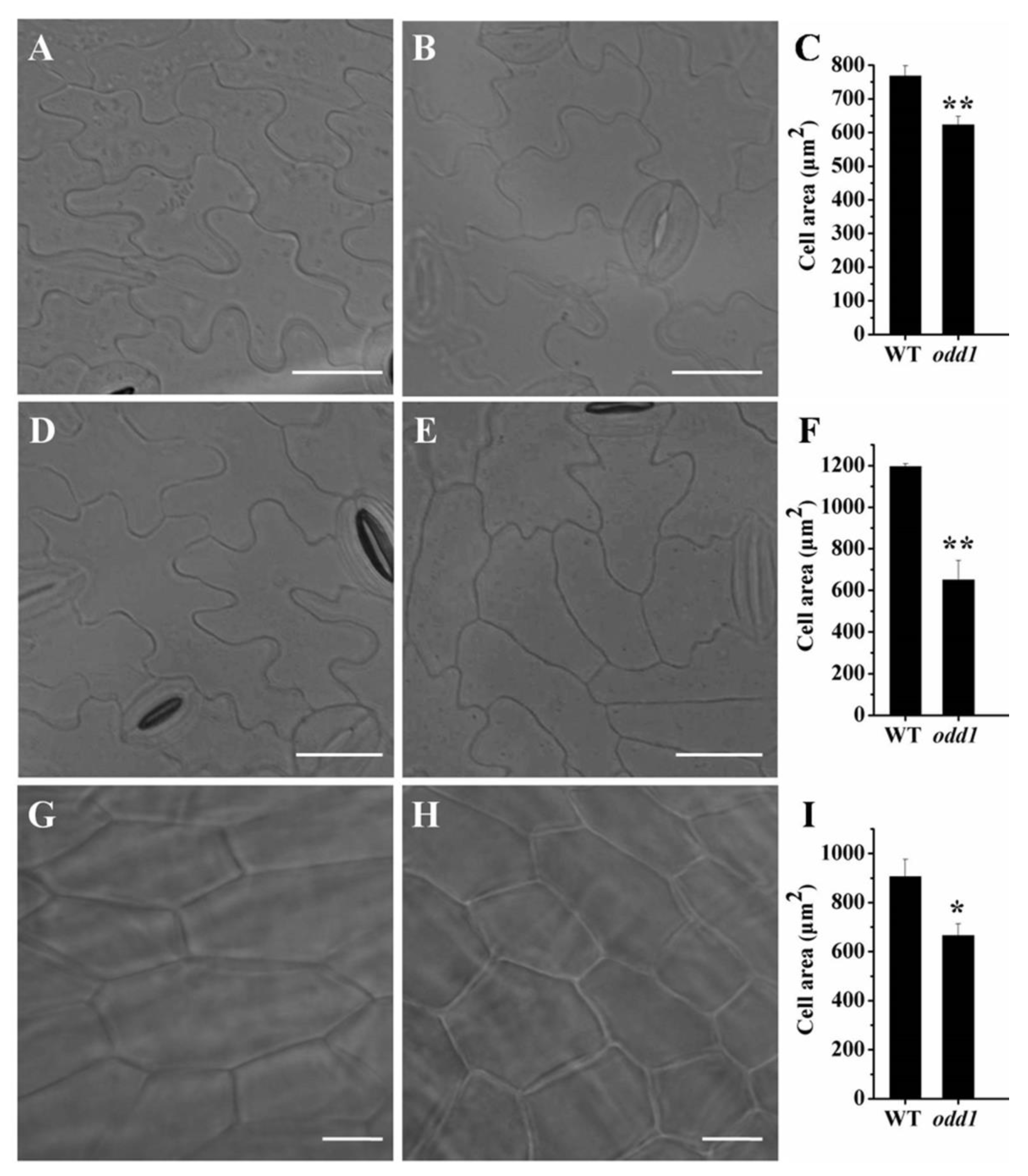
2.2. Histological and Anatomical Features of odd1 Mutant
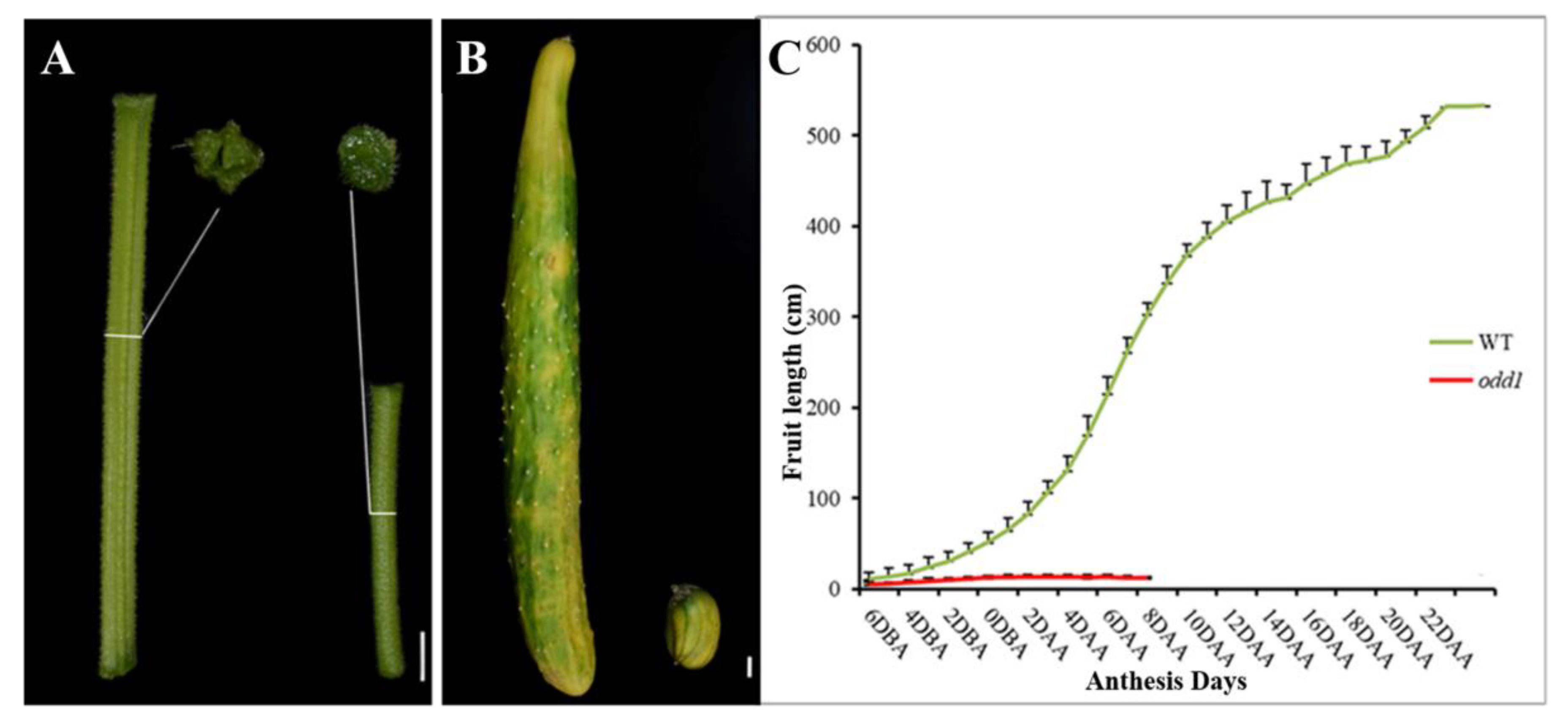
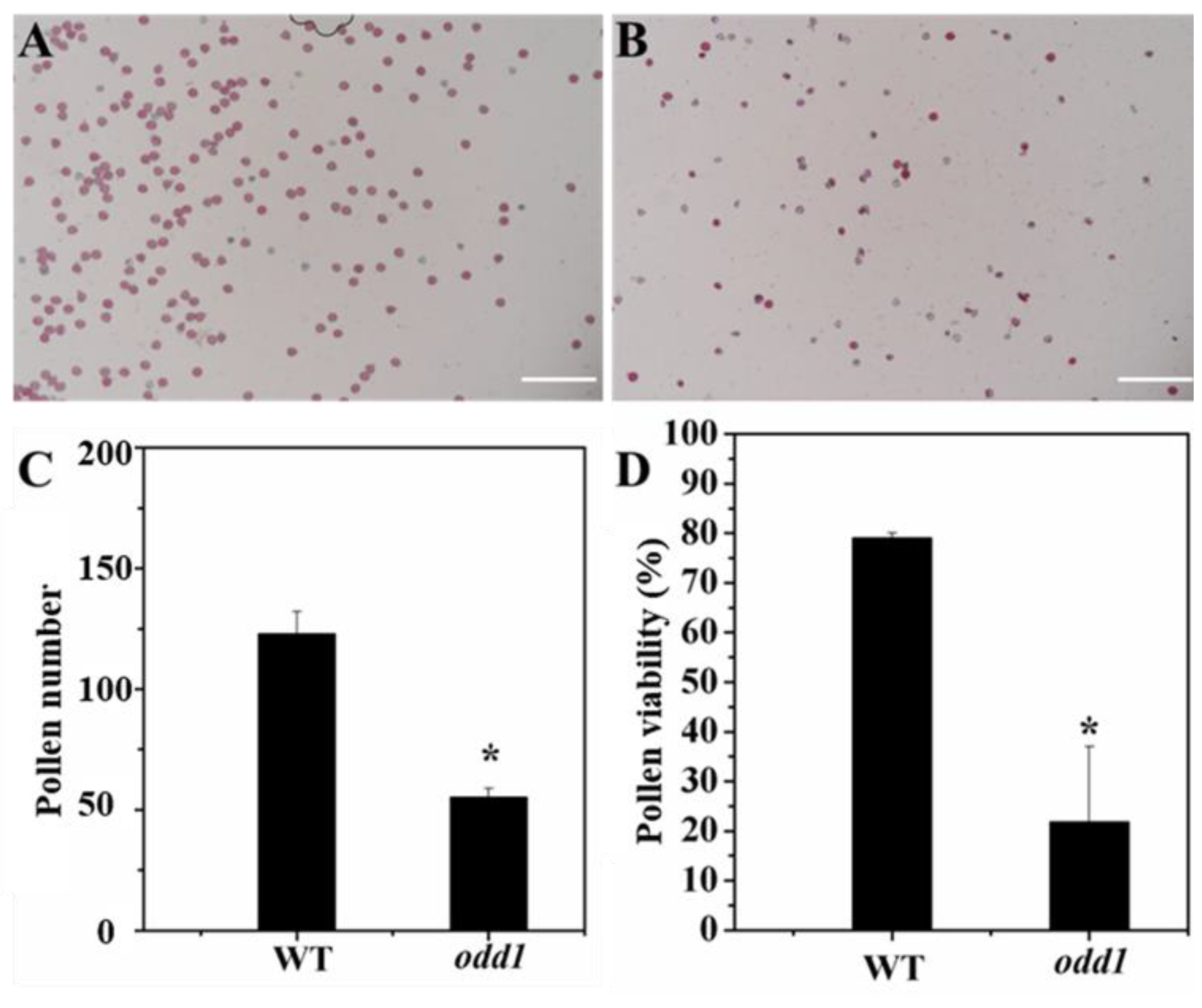
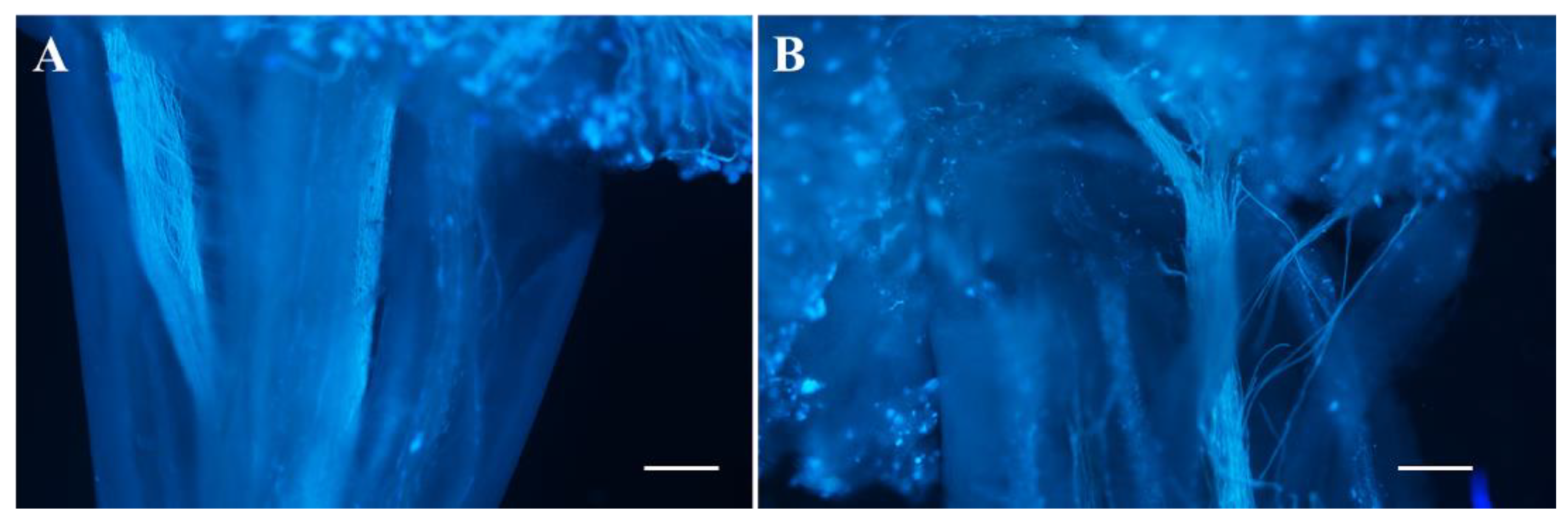
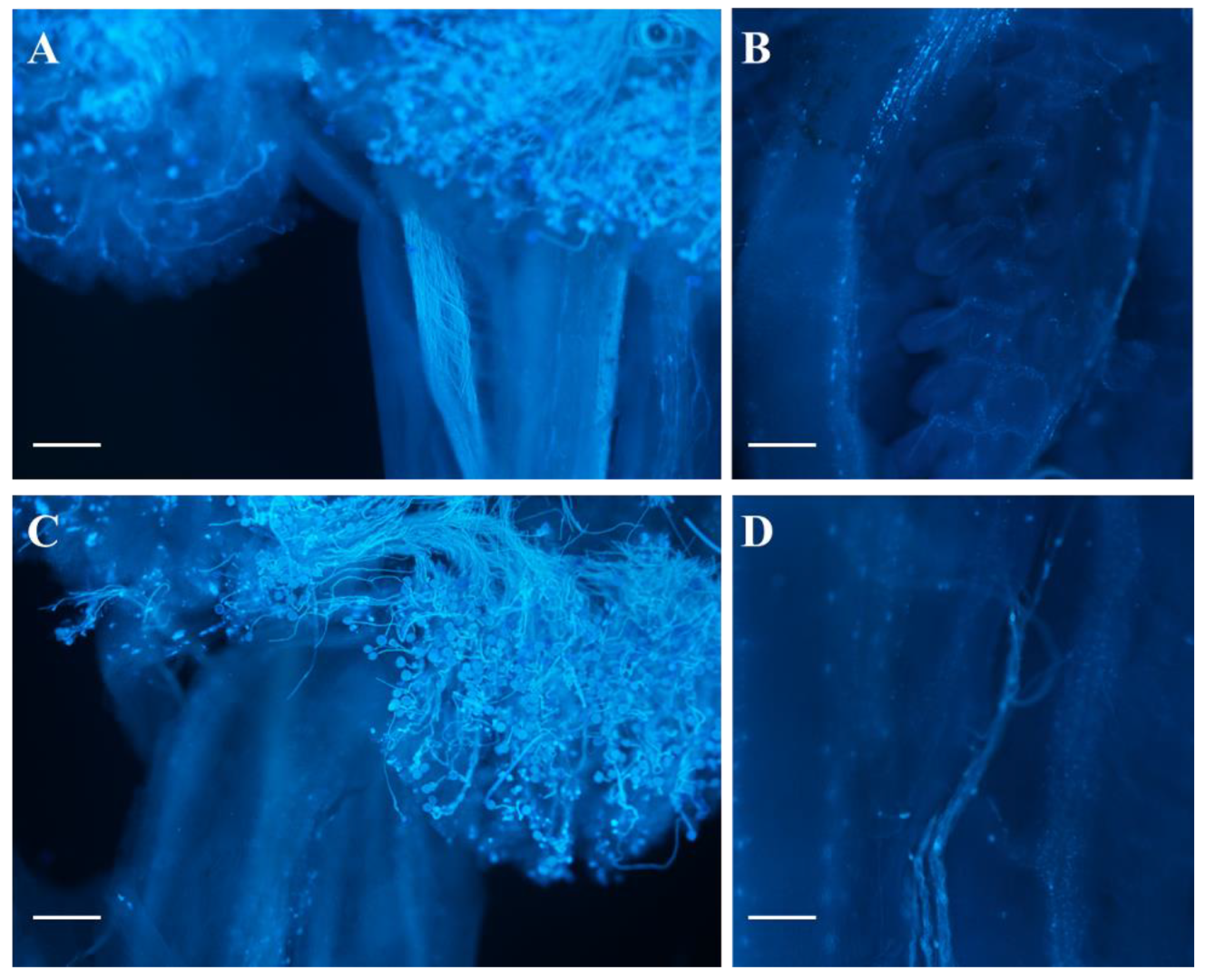
2.3. The Female Sterility of odd1
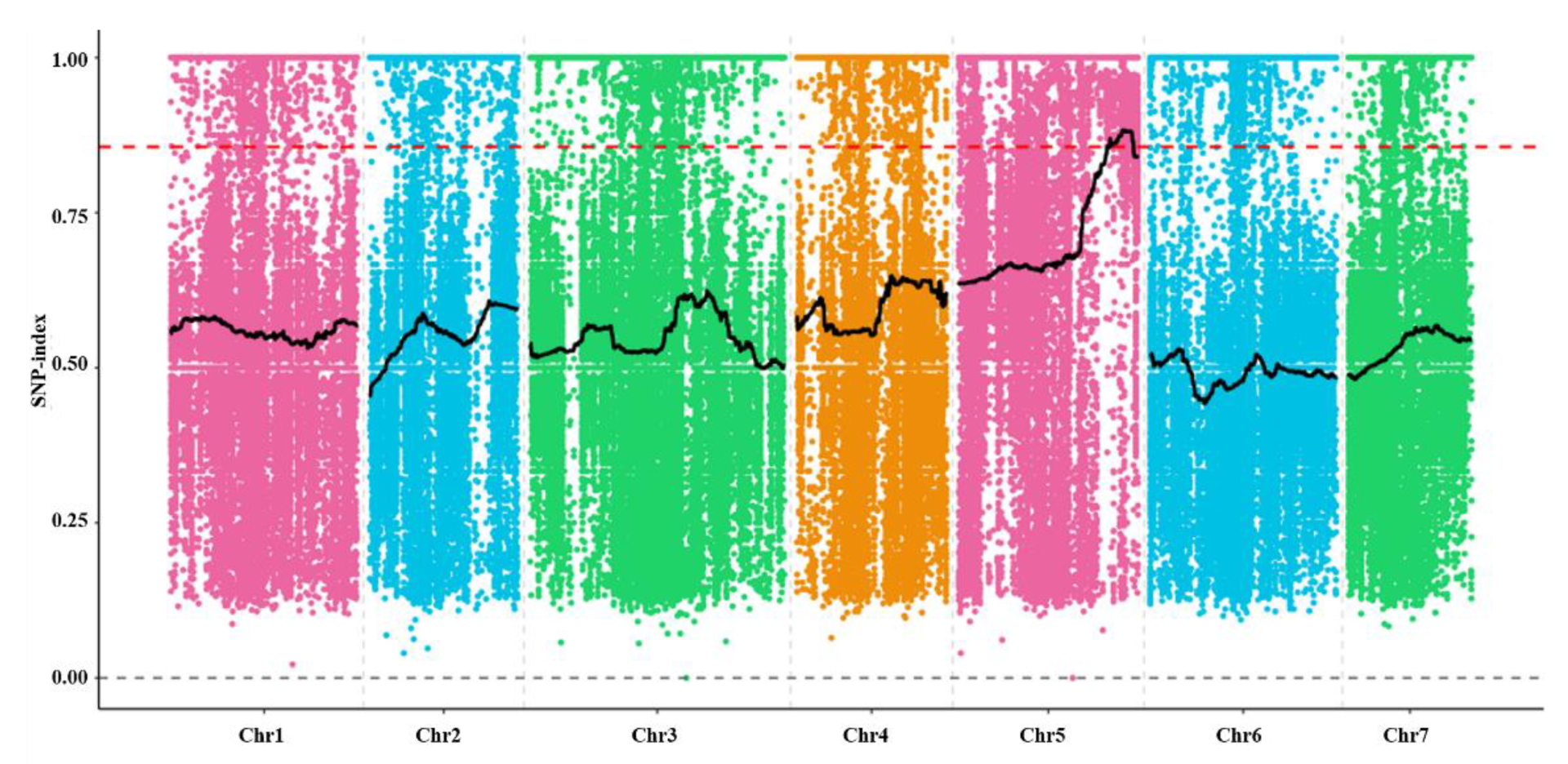
2.4. Genetic Characteristic and Mapping of odd1 Mutant
2.5. The Transcriptomic and Proteomic Profiles of odd1 Mutant
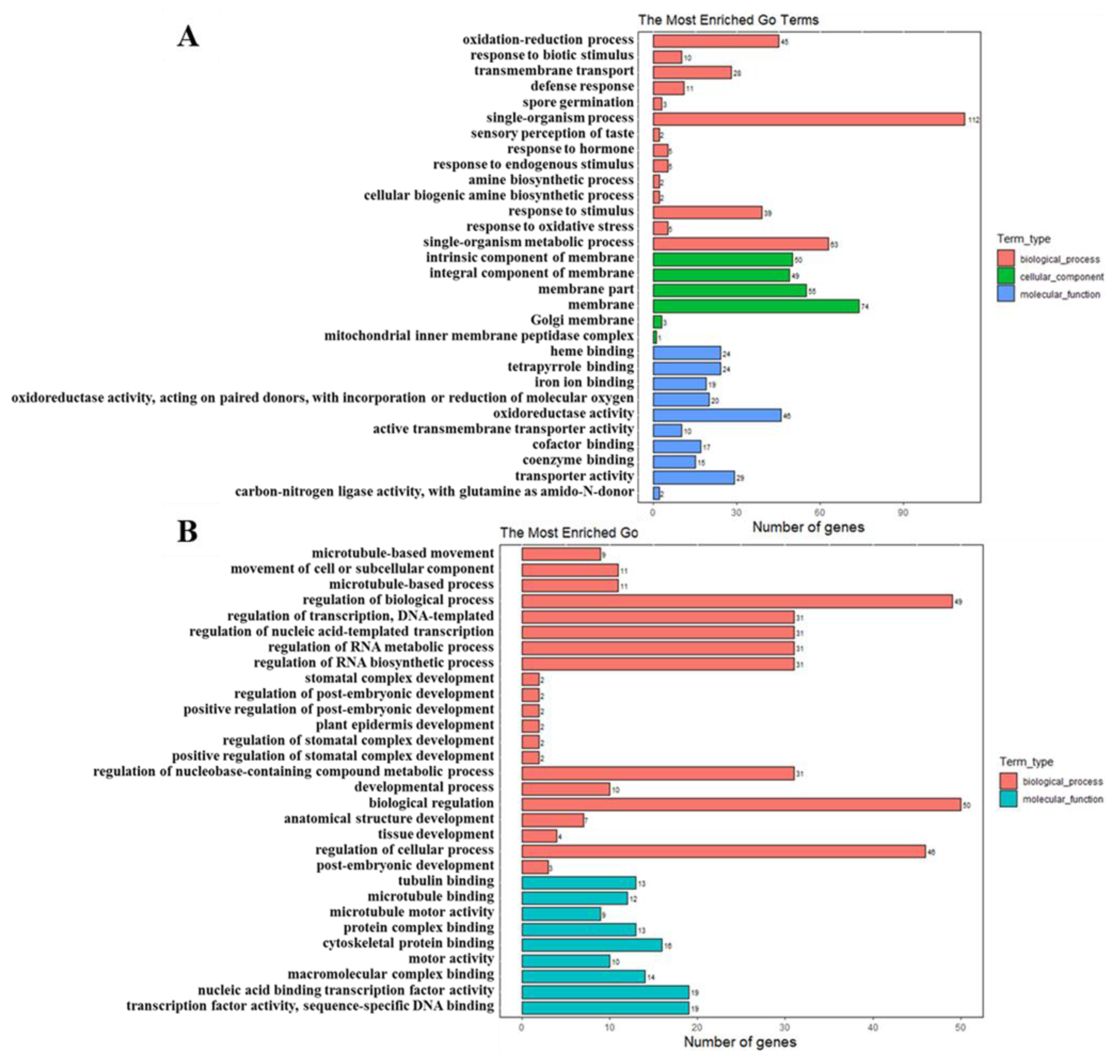
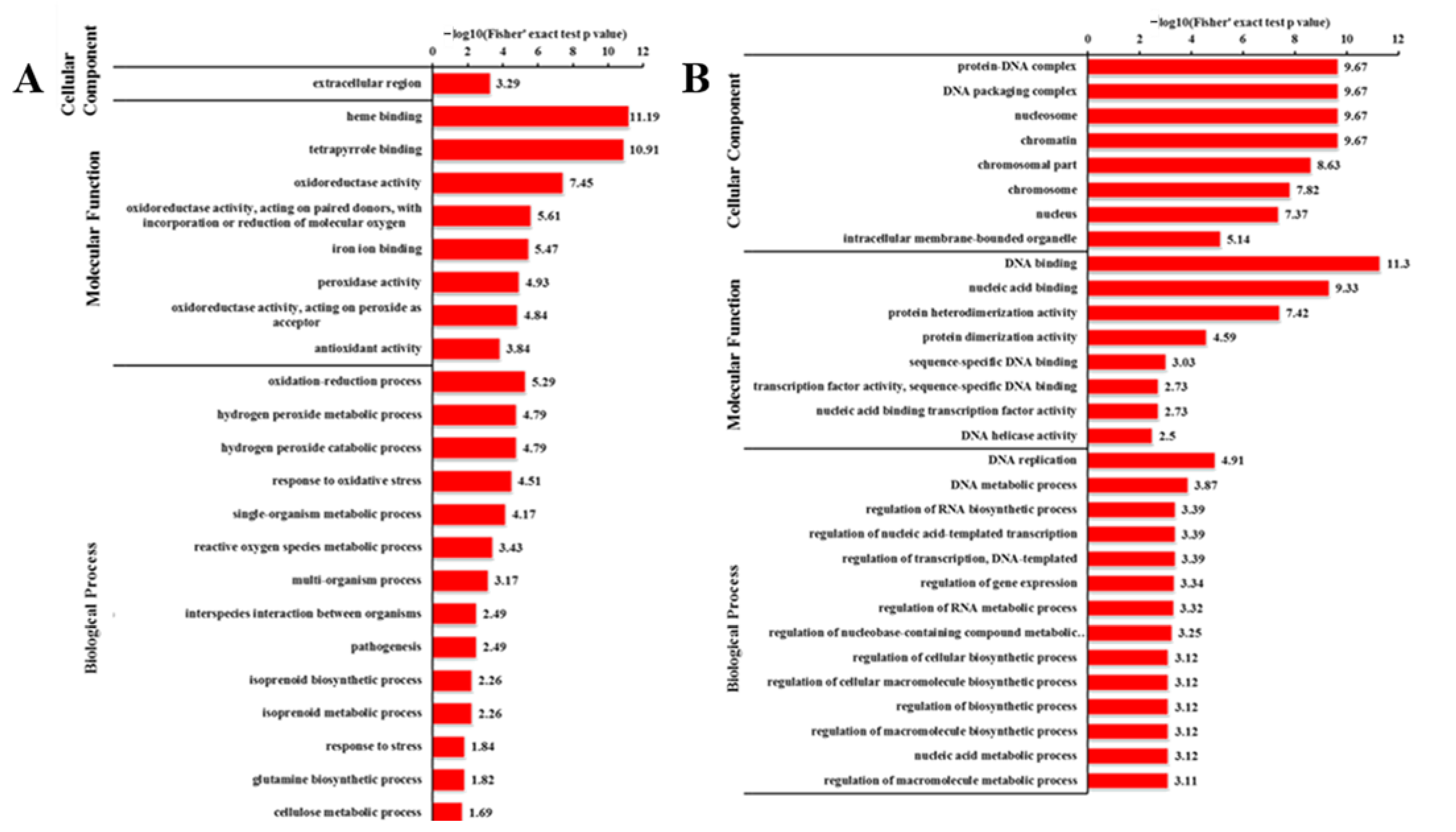
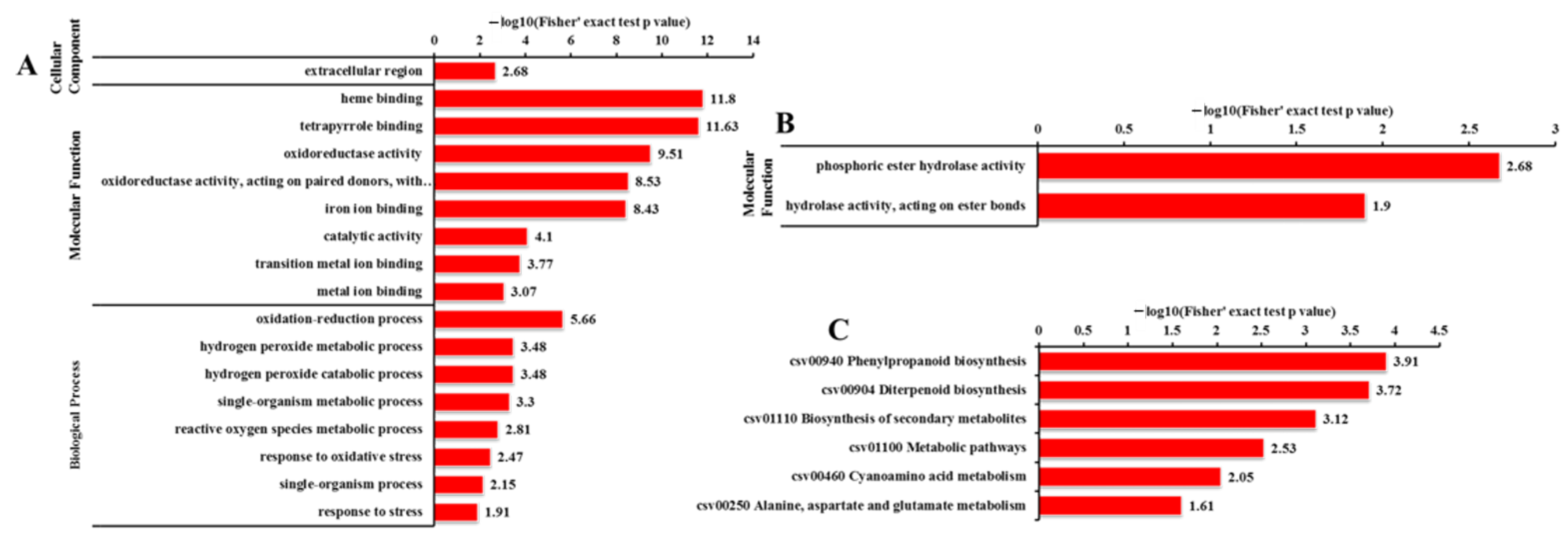
2.6. GO Analysis of DEGs and DEPs
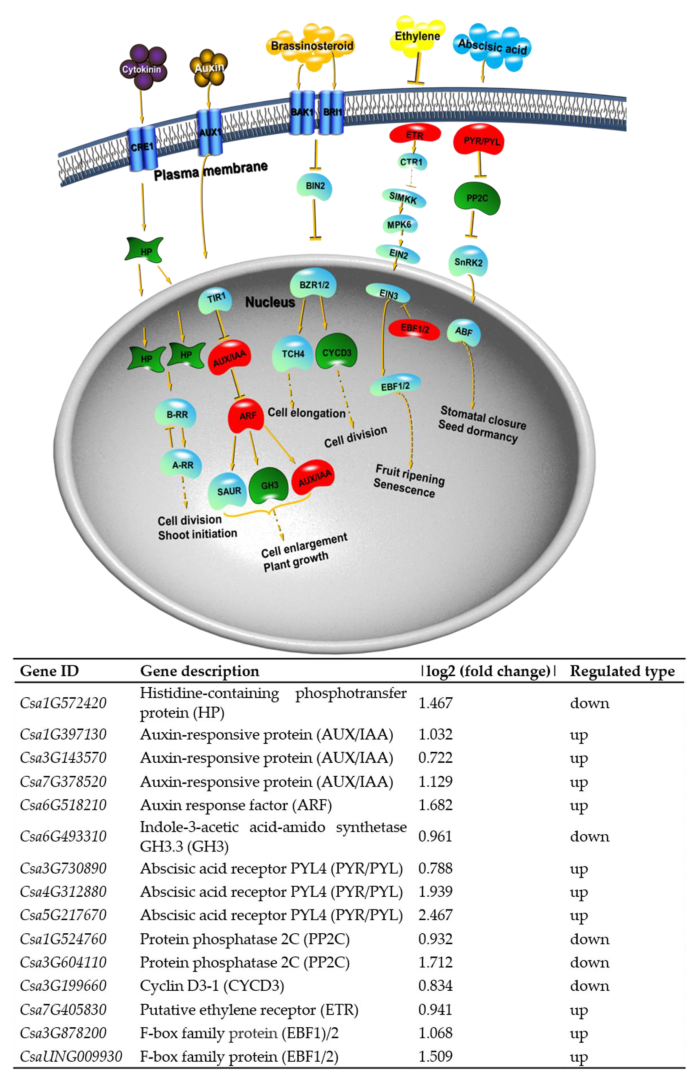
2.7. Transcription Factors Are Involved in Cucumber Growth and Development and Resistance Control
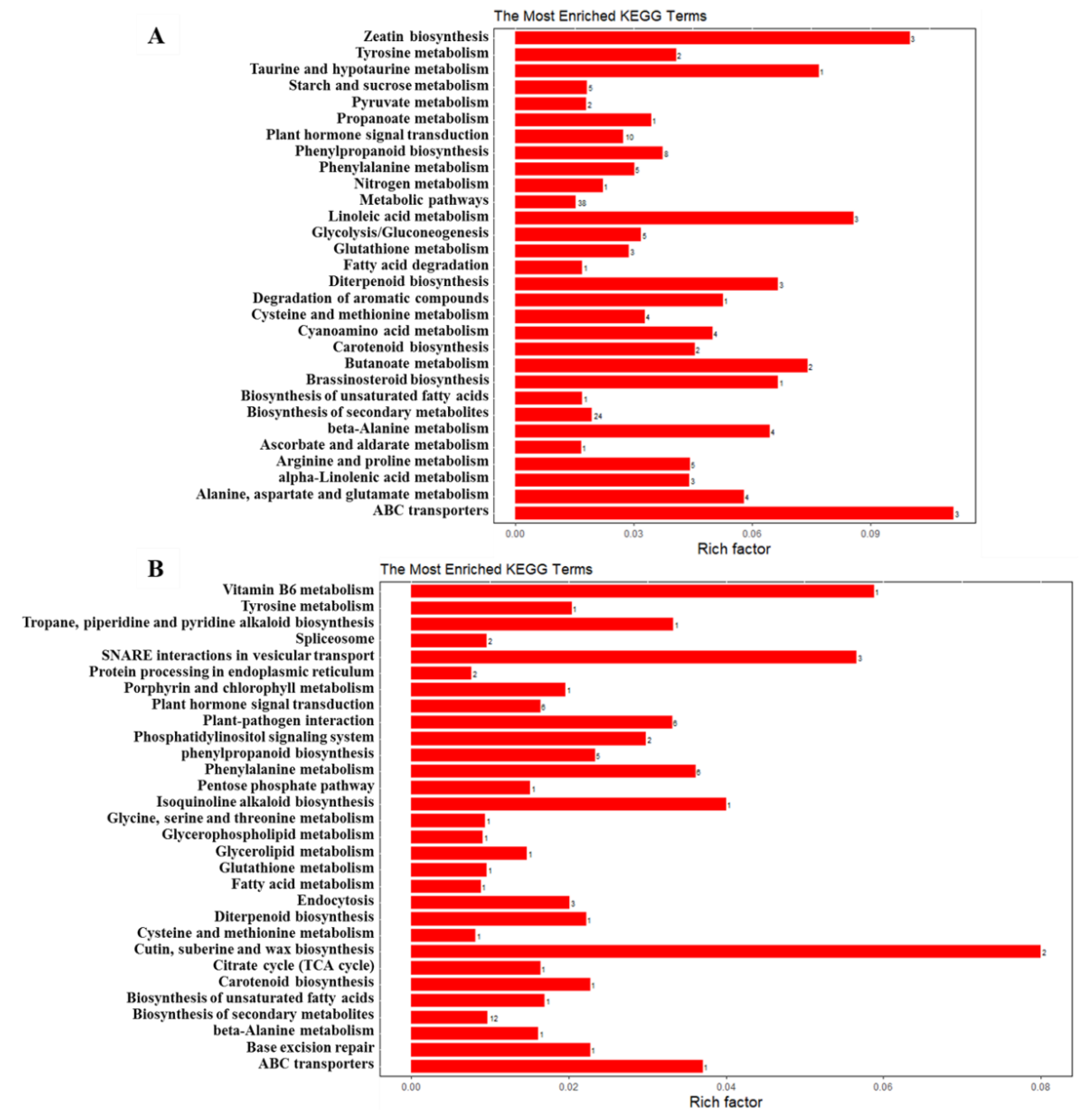
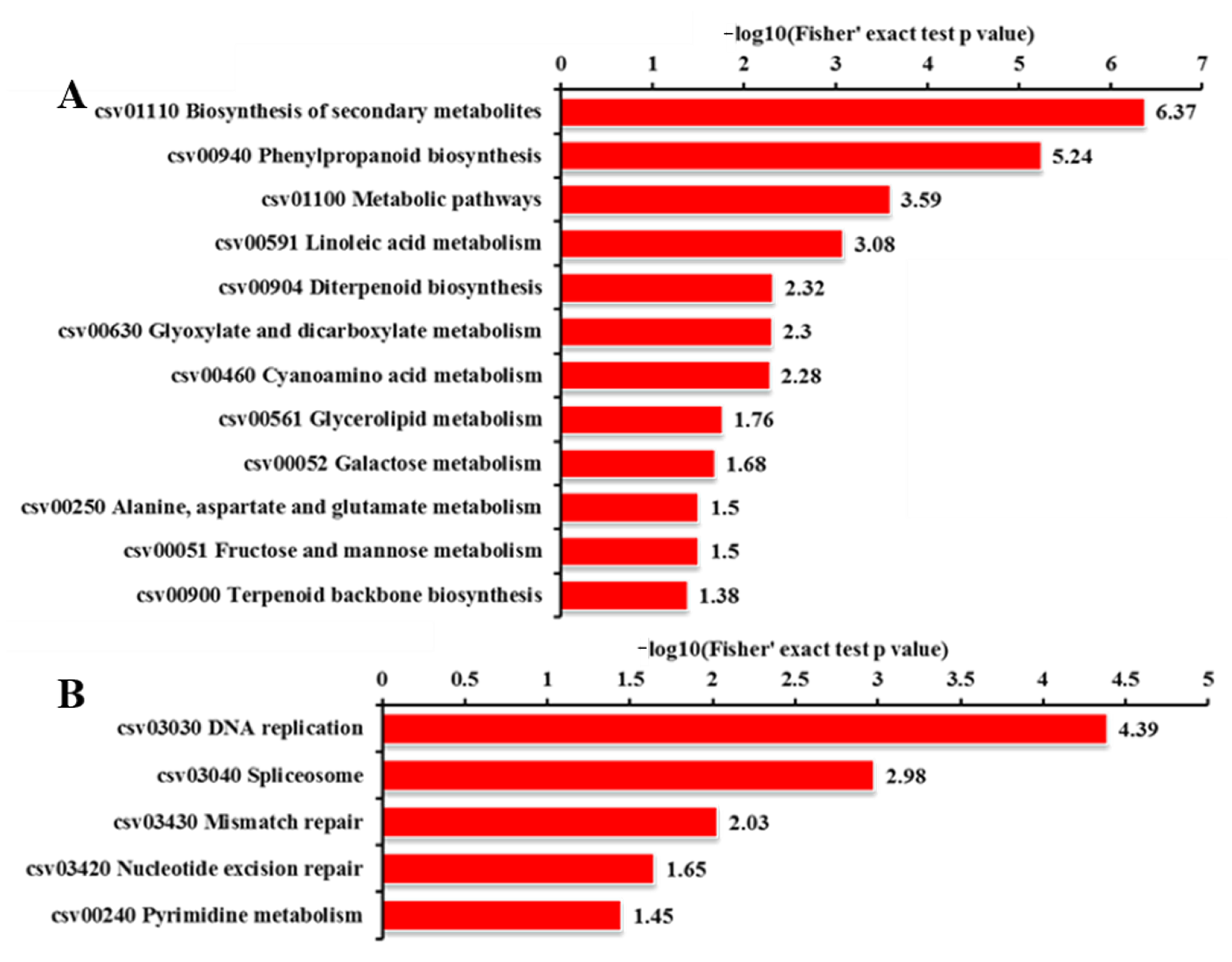
2.8. KEGG Pathway Analysis for DEGs and DEPs
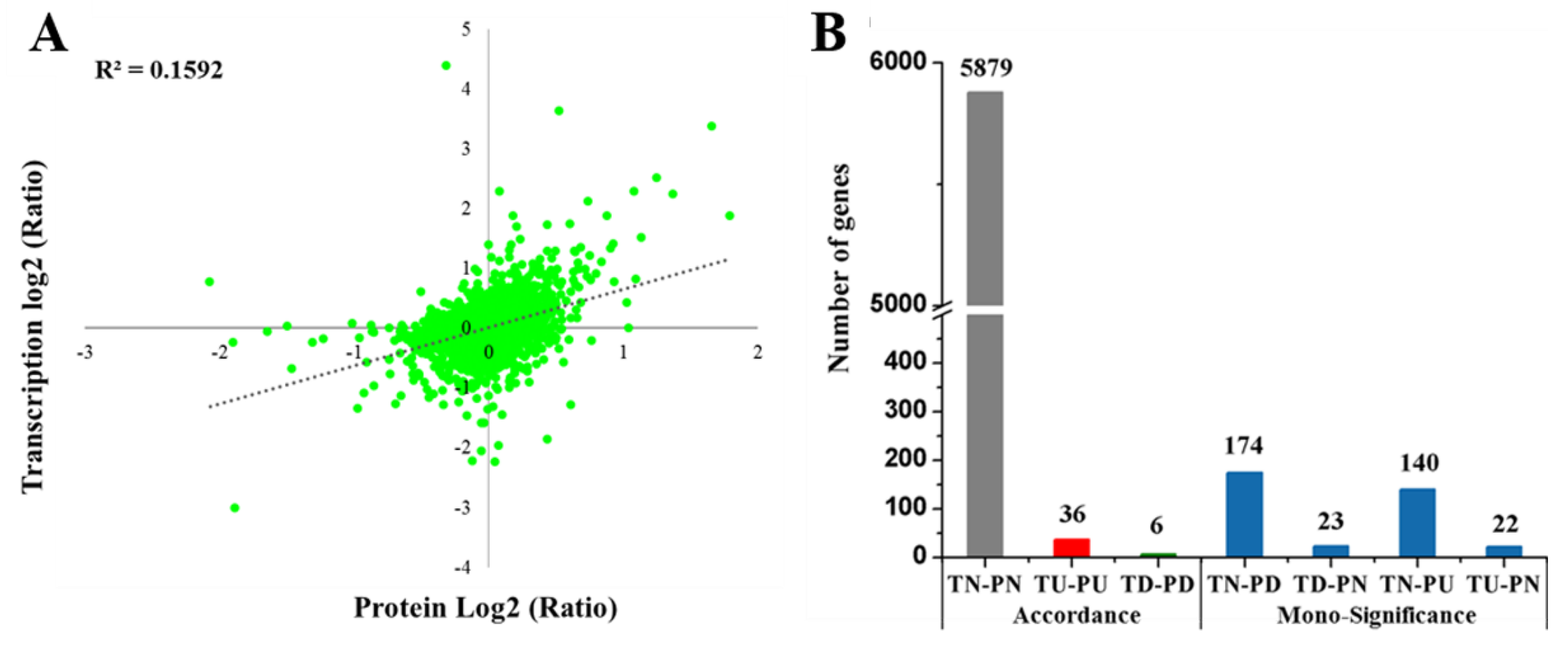
2.9. Comparison of Transcriptome and Proteome Data
2.10. Analysis of the 42 Cor-DEG-DEP Genes
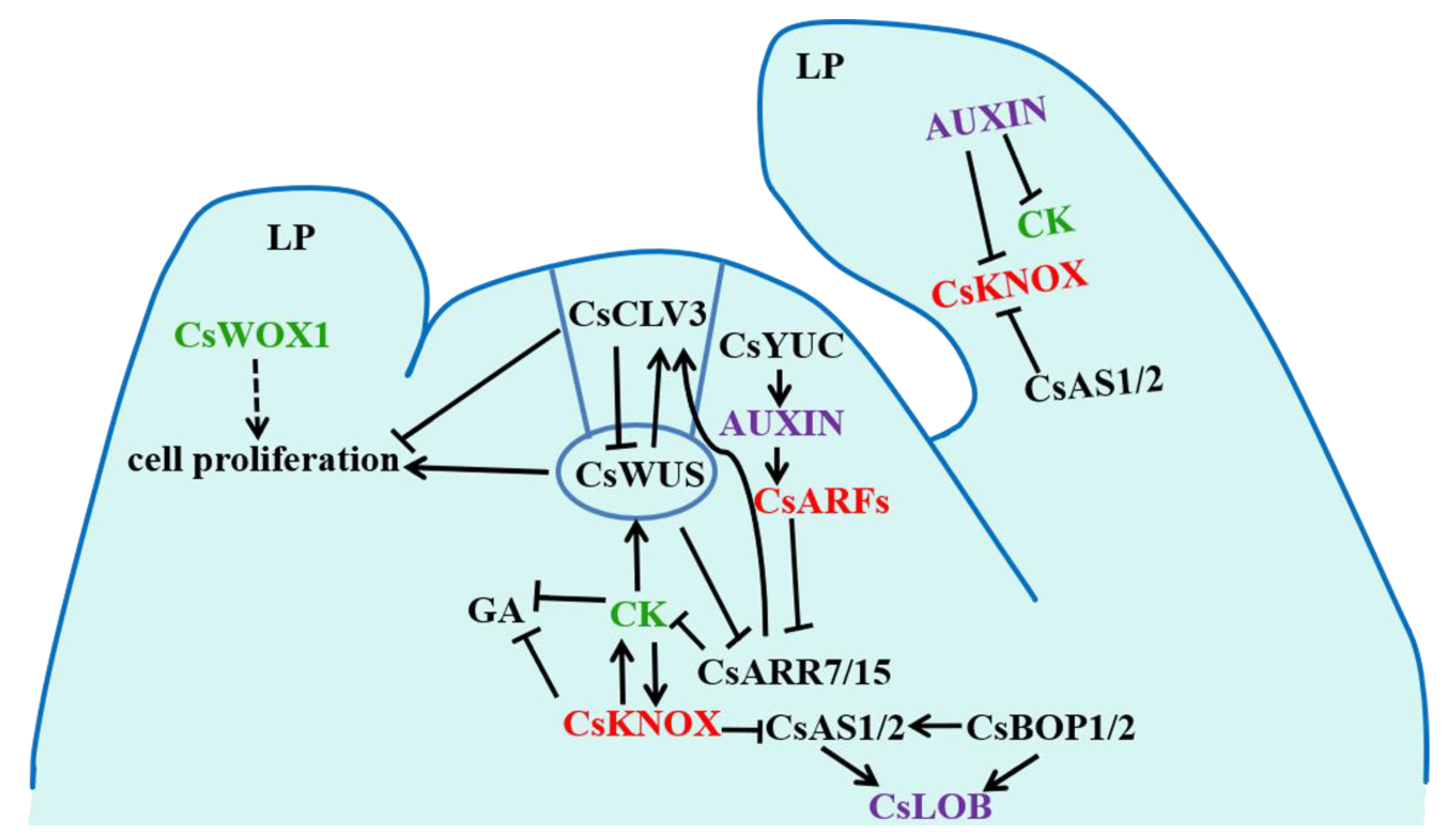
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Phenotypic Characterization and Photosynthesis-Related Parameters in the WT and odd1 Mutant
4.3. Cell Investigation of odd1 Mutant
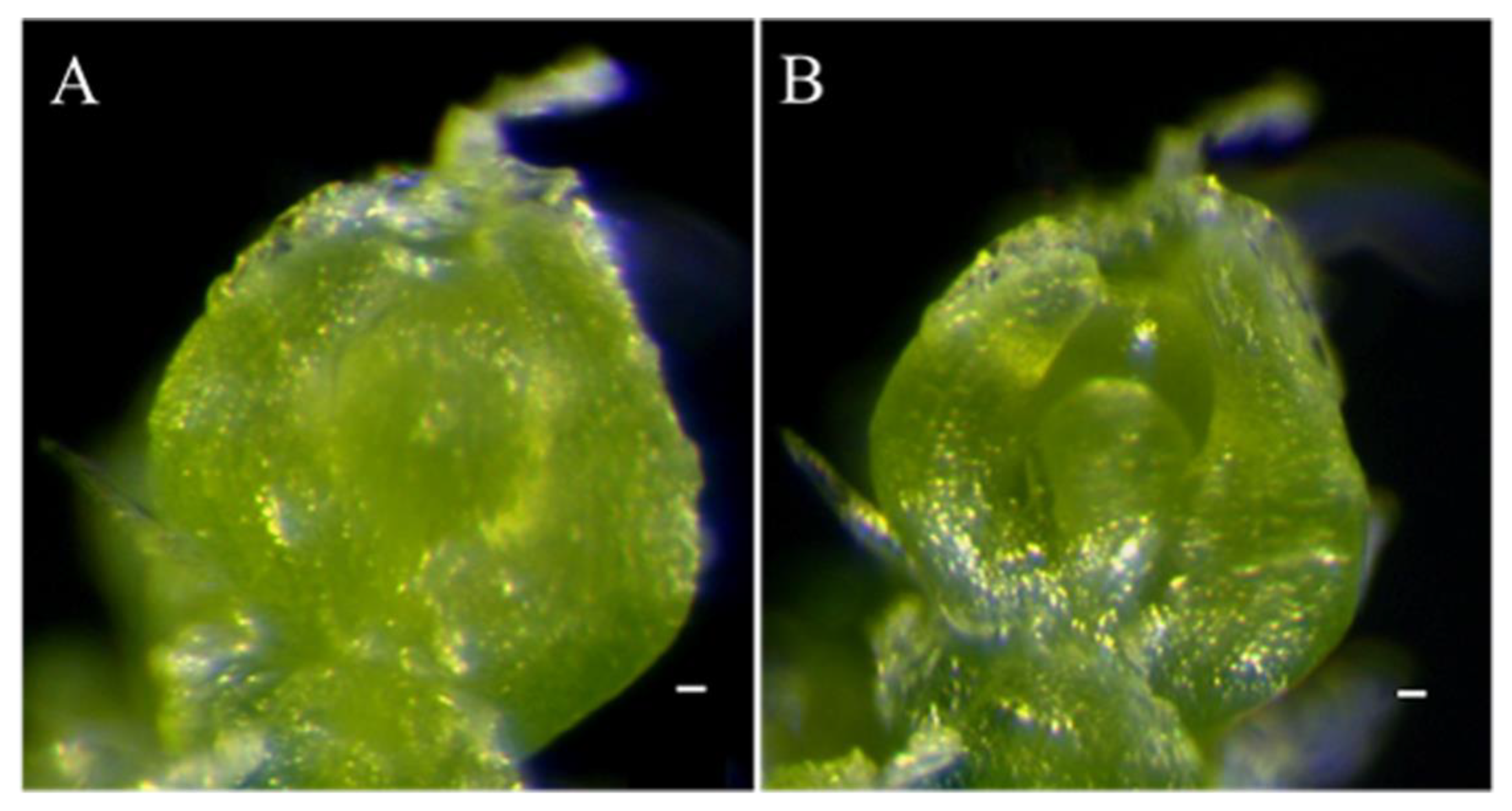
4.4. Observation of SAM
4.5. Pollen Quantity and Vigor Identification
4.6. Observation of Pollen Germination In Vivo
4.7. Gene Preliminary Mapping
4.8. RNA Extraction, Library Construction, and Sequencing
4.9. Bioinformatics Analysis of RNA-Seq Data
4.10. Quantitative Real-Time PCR
4.11. Protein Extraction
4.12. Trypsin Digestion
4.13. Tandem Mass Tag Labeling
4.14. HPLC Fractionation
4.15. LC-MS/MS Analysis
4.16. Database Search
4.17. Bioinformatic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, H.; Tian, Y.; Lu, X.; Liu, X. The inheritance of two novel subgynoecious genes in cucumber (Cucumis sativus L.). Sci. Hortic. 2011, 127, 464–467. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, J.; Zhang, X. Genetic regulation of shoot architecture in cucumber. Hortic. Res. 2021, 8, 143. [Google Scholar] [CrossRef]
- Gou, C.; Zhu, P.; Meng, Y.; Yang, F.; Xu, Y.; Xia, P.; Chen, J.; Li, J. Evaluation and Genetic Analysis of Parthenocarpic Germplasms in Cucumber. Genes 2022, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X.; et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 2013, 45, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.R. Inter-Relations of vegetative and reproductive growth, with special reference to indeterminate plants. Bot. Rev. 1962, 28, 353–410. [Google Scholar] [CrossRef]
- Weng, Y.; Johnson, S.; Staub, J.E.; Huang, S. An Extended Intervarietal Microsatellite Linkage Map of Cucumber, Cucumis sativus L. HortScience 2010, 45, 882–886. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.S.; Chen, Z.J.; Liu, X.F.; Che, G.; Gu, R.; Zhao, J.Y.; Wang, Z.Y.; Hou, Y.; Zhang, X.L. CsLFY is required for shoot meristem maintenance via interaction with WUSCHEL in cucumber (Cucumis sativus). New Phytol. 2018, 218, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, L.; Pathak, M.; Li, D.; He, X.; Weng, Y. Fine genetic mapping of cp: A recessive gene for compact (dwarf) plant architecture in cucumber, Cucumis sativus L. Theor. Appl. Genet. 2011, 123, 973–983. [Google Scholar] [CrossRef]
- Kubicki, B.; Soltysiak, U.; Korzeniewska, A. Induced mutation in cucumber (Cucumis sativus L.) V. Compact type of growth. Genet. Pol. 1986, 27, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, W.; Qin, Y.; Pan, Y.; Wang, X.; Weng, Y.; Chen, P.; Li, Y. The Cytochrome P450 Gene CsCYP85A1 Is a Putative Candidate for Super Compact-1 (Scp-1) Plant Architecture Mutation in Cucumber (Cucumis sativus L.). Front. Plant Sci. 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, S.; Niu, H.; Tao, Q.; Wang, S.; Gong, Z.; Li, S.; Weng, Y.; Li, Z. A mutant in the CsDET2 gene leads to a systemic brassinosteriod deficiency and super compact phenotype in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2017, 130, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, F.; Zhao, Z.; Hu, L.; Liu, H.; Cheng, Z.; Weng, Y.; Chen, P.; Li, Y. Mutations in CsPID encoding a Ser/Thr protein kinase are responsible for round leaf shape in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2018, 131, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, H.; Zhao, J.; Pan, Y.; Cheng, S.; Lietzow, C.D.; Wen, C.; Zhang, X.; Weng, Y. Littleleaf (LL) encodes a WD40 repeat domain-containing protein associated with organ size variation in cucumber. Plant J. 2018, 95, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Niu, H.; Li, C.; Shen, G.; Liu, X.; Weng, Y.; Wu, T.; Li, Z. WUSCHEL-related homeobox1 (WOX1) regulates vein patterning and leaf size in Cucumis sativus. Hortic. Res. 2020, 7, 182. [Google Scholar] [CrossRef]
- Wen, C.; Zhao, W.; Liu, W.; Yang, L.; Wang, Y.; Liu, X.; Xu, Y.; Ren, H.; Guo, Y.; Li, C.; et al. CsTFL1 inhibits determinate growth and terminal flower formation through interaction with CsNOT2a in cucumber (Cucumis sativus L.). Development 2019, 146, dev180166. [Google Scholar] [CrossRef] [Green Version]
- Xin, T.; Zhang, Z.; Li, S.; Zhang, S.; Li, Q.; Zhang, Z.-H.; Huang, S.; Yang, X. Genetic Regulation of Ethylene Dosage for Cucumber Fruit Elongation. Plant Cell 2019, 31, 1063–1076. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, B.; Wang, S.; Lin, T.; Yang, L.; Zhao, Z.; Zhang, Z.; Huang, S.; Yang, X. Genome-wide Target Mapping Shows Histone Deacetylase Complex1 Regulates Cell Proliferation in Cucumber Fruit. Plant Physiol. 2019, 182, 167–184. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cao, C.; Zheng, S.; Zhang, H.; Liu, P.; Ge, Q.; Li, J.; Ren, Z. Transcriptomic analysis of short-fruit 1 (sf1) reveals new insights into the variation of fruit-related traits in Cucumis sativus. Sci. Rep. 2017, 7, 2950. [Google Scholar] [CrossRef]
- Chen, F.; Fu, B.; Pan, Y.; Zhang, C.; Wen, H.; Weng, Y.; Chen, P.; Li, Y. Fine mapping identifies CsGCN5 encoding a histone acetyltransferase as putative candidate gene for tendril-less1 mutation (td-1) in cucumber. Theor. Appl. Genet. 2017, 130, 1549–1558. [Google Scholar] [CrossRef]
- Rong, F.; Chen, F.; Huang, L.; Zhang, J.; Zhang, C.; Hou, D.; Cheng, Z.; Weng, Y.; Chen, P.; Li, Y. A mutation in class III homeodomain-leucine zipper (HD-ZIP III) transcription factor results in curly leaf (cul) in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2019, 132, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.J.; Cheng, Z.J.; Sang, Y.L.; Zhang, M.M.; Rong, X.F.; Wang, Z.W.; Tang, Y.Y.; Zhang, X.S. Type-B ARABIDOPSIS RESPONSE REGULATORs Specify the Shoot Stem Cell Niche by Dual Regulation of Wuschel. Plant Cell 2017, 29, 1357–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, K.F.X.; Schoof, H.; Haecker, A.; Lenhard, M.; Jürgens, G.; Laux, T. Role of WUSCHEL in Regulating Stem Cell Fate in the Arabidopsis Shoot Meristem. Cell 1998, 95, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef]
- Yadav, R.K.; Perales, M.; Gruel, J.; Girke, T.; Jönsson, H.; Reddy, G.V. WUSCHEL protein movement mediates stem cell homeostasis in the Arabidopsis shoot apex. Genes Dev. 2011, 25, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Brand, U.; Fletcher, J.C.; Hobe, M.; Meyerowitz, E.M.; Simon, R. Dependence of Stem Cell Fate in Arabidopsis on a Feedback Loop Regulated by CLV3 Activity. Science 2000, 289, 617–619. [Google Scholar] [CrossRef]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.; Jürgens, G.; Laux, T. The Stem Cell Population of Arabidopsis Shoot Meristems Is Maintained by a Regulatory Loop between the CLAVATA and WUSCHEL Genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Perales, M.; Reddy, G.V. Stem cell maintenance in shoot apical meristems. Curr. Opin. Plant Biol. 2011, 15, 10–16. [Google Scholar] [CrossRef]
- Somssich, M.; Je, B.I.; Simon, R.; Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 2016, 143, 3238–3248. [Google Scholar] [CrossRef] [Green Version]
- Pautler, M.; Tanaka, W.; Hirano, H.-Y.; Jackson, D. Grass Meristems I: Shoot Apical Meristem Maintenance, Axillary Meristem Determinacy and the Floral Transition. Plant Cell Physiol. 2013, 54, 302–312. [Google Scholar] [CrossRef]
- van der Graaff, E.; Laux, T.; Rensing, S.A. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, Y.; Tanaka, W.; Kojima, M.; Sakakibara, H.; Hirano, H.-Y. WUSCHEL-RELATED HOMEOBOX4 Is Involved in Meristem Maintenance and Is Negatively Regulated by the CLE Gene FCP1 in Rice. Plant Cell 2013, 25, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, L.; Aluru, M.; Aluru, S.; Yin, Y. Mechanisms and networks for brassinosteroid regulated gene expression. Curr. Opin. Plant Biol. 2013, 16, 545–553. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Bai, M.-Y.; Oh, E.; Zhu, J.-Y. Brassinosteroid Signaling Network and Regulation of Photomorphogenesis. Annu. Rev. Genet. 2012, 46, 701–724. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.-Y.; Fan, M.; Oh, E.; Wang, Z.-Y. A Triple Helix-Loop-Helix/Basic Helix-Loop-Helix Cascade Controls Cell Elongation Downstream of Multiple Hormonal and Environmental Signaling Pathways in Arabidopsis. Plant Cell 2012, 24, 4917–4929. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Fujiwara, S.; Mitsuda, N.; Ohme-Takagi, M. A Triantagonistic Basic Helix-Loop-Helix System Regulates Cell Elongation in Arabidopsis. Plant Cell 2012, 24, 4483–4497. [Google Scholar] [CrossRef] [Green Version]
- Dinneny, J.R.; Yadegari, R.; Fischer, R.L.; Yanofsky, M.F.; Weigel, D. The role of JAGGED in shaping lateral organs. Development 2004, 131, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Ohno, C.K.; Reddy, G.V.; Heisler, M.G.B.; Meyerowitz, E.M. The Arabidopsis JAGGED gene encodes a zinc finger protein that promotes leaf tissue development. Development 2004, 131, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Dinneny, J.R.; Weigel, D.; Yanofsky, M.F. NUBBIN and JAGGED define stamen and carpel shape in Arabidopsis. Development 2006, 133, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteom. 2018, 179, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, L.; Gao, Z.; Jiang, M.; Liu, Y.; Zhou, L.; He, Y.; Chen, H. Combined transcriptomic and proteomic analysis constructs a new model for light-induced anthocyanin biosynthesis in eggplant (Solanum melongena L.). Plant Cell Environ. 2017, 40, 3069–3087. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, J.; Wei, Q.; Wang, R.; Yang, W.; Ma, Y.; Chen, G.; Yu, Y. Proteomes and Ubiquitylomes Analysis Reveals the Involvement of Ubiquitination in Protein Degradation in Petunias. Plant Physiol. 2017, 173, 668–687. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jiang, C.; Zhang, W.; Wang, H.; Li, K.; Liu, X.; Liu, Z.; Wu, Y.; Huang, C.; Hu, X. Morphological Characterization and Transcriptome Analysis of New Dwarf and Narrow-Leaf (dnl2) Mutant in Maize. Int. J. Mol. Sci. 2022, 23, 795. [Google Scholar] [CrossRef]
- Haecker, A.; Groß-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Vandenbussche, M.; Horstman, A.; Zethof, J.; Koes, R.; Rijpkema, A.S.; Gerats, T. Differential recruitment of WOX transcription factors for lateral development and organ fusion in Petunia and Arabidopsis. Plant Cell 2009, 21, 2269–2283. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Niu, L.; McHale, N.A.; Ohme-Takagi, M.; Mysore, K.S.; Tadege, M. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc. Natl. Acad. Sci. USA 2012, 110, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Liu, X.; Tong, C.; Wang, H.; Li, S.; Lu, L.; Pan, Y.; Zhang, X.; Weng, Y.; Li, Z. The WUSCHEL-related homeobox1 gene of cucumber regulates reproductive organ development. J. Exp. Bot. 2018, 69, 5373–5387. [Google Scholar] [CrossRef]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef]
- Agarwal, P.; Arora, R.; Ray, S.; Singh, A.K.; Singh, V.P.; Takatsuji, H.; Kapoor, S.; Tyagi, A.K. Genome-wide identification of C2H2 zinc-finger gene family in rice and their phylogeny and expression analysis. Plant Mol. Biol. 2007, 65, 467–485. [Google Scholar] [CrossRef]
- Gaiser, J.C.; Robinson-Beers, K.; Gasser, C.S. The Arabidopsis SUPERMAN Gene Mediates Asymmetric Growth of the Outer Integument of Ovules. Plant Cell 1995, 7, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.B.; Hagen, G.; Guilfoyle, T. The Roles of Auxin Response Factor Domains in Auxin-Responsive Transcription. Plant Cell 2003, 15, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, A.W.; Bartel, B. A receptor for auxin. Plant Cell 2005, 17, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Quint, M.; Gray, W.M. Auxin signaling. Curr. Opin. Plant Biol. 2006, 9, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Z.; Dixon, R.A. On–Off Switches for Secondary Cell Wall Biosynthesis. Mol. Plant 2012, 5, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Balazadeh, S.; Siddiqui, H.; Allu, A.D.; Matallana-Ramirez, L.P.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Kohler, B.; Mueller-Roeber, B. A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 2010, 62, 250–264. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.-H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef] [Green Version]
- He, X.J.; Mu, R.L.; Cao, W.H.; Zhang, Z.G.; Zhang, J.S.; Chen, S.Y. AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J. 2010, 44, 903–916. [Google Scholar] [CrossRef]
- Martín-Trillo, M.; Cubas, P. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2010, 15, 31–39. [Google Scholar] [CrossRef]
- Lee, D.-K.; Geisler, M.; Springer, P.S. Lateral Organ Fusion1 and Lateral Organ Fusion2 function in lateral organ separation and axillary meristem formation in Arabidopsis. Development 2009, 136, 2423–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Sun, W.; Singh, R.; Zheng, Y.; Cao, Z.; Li, M.; Lunde, C.; Hake, S.; Zhang, Z. GRF-interacting factor1 Regulates Shoot Architecture and Meristem Determinacy in Maize. Plant Cell 2018, 30, 360–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Kiełbowicz-Matuk, A. Involvement of plant C2H2-type zinc finger transcription factors in stress responses. Plant Sci. 2012, 185-186, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Geng, J.; Du, Y.; Zhao, Q.; Zhang, W.; Fang, Q.; Yin, Z.; Li, J.; Yuan, X.; Fan, Y.; et al. Heat shock transcription factor (Hsf) gene family in common bean (Phaseolus vulgaris): Genome-wide identification, phylogeny, evolutionary expansion and expression analyses at the sprout stage under abiotic stress. BMC Plant Biol. 2022, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmuller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef] [Green Version]
- Castilhos, G.; Lazzarotto, F.; Spagnolo-Fonini, L.; Bodanese-Zanettini, M.H.; Margis-Pinheiro, M. Possible roles of basic helix-loop-helix transcription factors in adaptation to drought. Plant Sci. 2014, 223, 1–7. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Alves, M.S.; Dadalto, S.P.; Gonçalves, A.B.; De Souza, G.B.; Barros, V.A.; Fietto, L.G. Plant bZIP Transcription Factors Responsive to Pathogens: A Review. Int. J. Mol. Sci. 2013, 14, 7815–7828. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 97–103. [Google Scholar] [CrossRef]
- Liu, W.; Tai, H.; Li, S.; Gao, W.; Zhao, M.; Xie, C.; Li, W.X. bHLH122 is important for drought and osmotic stress resistance in Arabidopsis and in the repression of ABA catabolism. New Phytol. 2014, 201, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.S.; Chen, M.; Li, L.C.; Ma, Y.Z. Functions and Application of the AP2/ERF Transcription Factor Family in Crop Improvement. J. Integr. Plant Biol. 2011, 53, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-G.; Zhang, W.-K.; He, S.-J.; Zhang, J.-S.; Liu, Q.; Chen, S.-Y. An EREBP/AP2-type protein in Triticum aestivum was a DRE-binding transcription factor induced by cold, dehydration and ABA stress. Theor. Appl. Genet. 2003, 106, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.K.; Palmer, R.G. Molecular mapping of four ovule lethal mutants in soybean. Theor. Appl. Genet. 2004, 108, 577–585. [Google Scholar] [CrossRef]
- Palmer, R.G.; Sandhu, D.; Curran, K.; Bhattacharyya, M.K. Molecular mapping of 36 soybean male-sterile, female-sterile mutants. Theor. Appl. Genet. 2008, 117, 711–719. [Google Scholar] [CrossRef]
- Raval, J.; Baumbach, J.; Ollhoff, A.R.; Pudake, R.N.; Palmer, R.G.; Bhattacharyya, M.K.; Sandhu, D. A candidate male-fertility female-fertility gene tagged by the soybean endogenous transposon, Tgm9. Funct. Integr. Genom. 2013, 13, 67–73. [Google Scholar] [CrossRef]
- Baumbach, J.; Rogers, J.P.; Slattery, R.A.; Narayanan, N.N.; Xu, M.; Palmer, R.G.; Bhattacharyya, M.K.; Sandhu, D. Segregation distortion in a region containing a male-sterility, female-sterility locus in soybean. Plant Sci. 2012, 195, 151–156. [Google Scholar] [CrossRef]
- Teng, C.; Du, D.; Xiao, L.; Yu, Q.; Shang, G.; Zhao, Z. Mapping and Identifying a Candidate Gene (Bnmfs) for Female-Male Sterility through Whole-Genome Resequencing and RNA-Seq in Rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 2086. [Google Scholar] [CrossRef]
- Ling, D.H.; Ma, Z.R.; Cheng, M.F. Female sterility in indica rice produced by somatic culture. Acta Genet. Sin. 1991, 18, 446–451. [Google Scholar]
- Awasthi, A.; Paul, P.; Kumar, S.; Verma, S.K.; Prasad, R.; Dhaliwal, H. Abnormal endosperm development causes female sterility in rice insertional mutant OsAPC6. Plant Sci. 2012, 183, 167–174. [Google Scholar] [CrossRef]
- Nishiyama, R.; Watanabe, Y.; Leyva-Gonzalez, M.A.; Van Ha, C.; Fujita, Y.; Tanaka, M.; Seki, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Herrera-Estrella, L.; et al. Arabidopsis AHP2, AHP3, and AHP5 histidine phosphotransfer proteins function as redundant negative regulators of drought stress response. Proc. Natl. Acad. Sci. USA 2013, 110, 4840–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C.E.; Li, J.; Argueso, C.; Gonzalez, M.; Lee, E.; Lewis, M.W.; Maxwell, B.B.; Perdue, T.D.; Schaller, G.E.; Alonso, J.M.; et al. The Arabidopsis Histidine Phosphotransfer Proteins Are Redundant Positive Regulators of Cytokinin Signaling. Plant Cell 2006, 18, 3073–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, T. Recent advances in brassinosteroid molecular genetics. Curr. Opin. Plant Biol. 1998, 1, 378–383. [Google Scholar] [CrossRef]
- Clouse, S.D.; Sasse, J.M. BRASSINOSTEROIDS: Essential Regulators of Plant Growth and Development. Annu. Rev. Plant Biol. 1998, 49, 427–451. [Google Scholar] [CrossRef] [Green Version]
- Davière, J.-M.; Wild, M.; Regnault, T.; Baumberger, N.; Eisler, H.; Genschik, P.; Achard, P. Class I TCP-DELLA Interactions in Inflorescence Shoot Apex Determine Plant Height. Curr. Biol. 2014, 24, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.-L.; Yao, J.; Mei, C.-S.; Tong, X.-H.; Zeng, L.-J.; Li, Q.; Xiao, L.-T.; Sun, T.-P.; Li, J.; Deng, X.-W.; et al. Plant hormone jasmonate prioritizes defense over growth by interfering with gibberellin signaling cascade. Proc. Natl. Acad. Sci. USA 2012, 109, E1192–E1200. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A Natural Allele of a Transcription Factor in Rice Confers Broad-Spectrum Blast Resistance. Cell 2017, 170, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Huang, J.F.; Wang, R.C. Change Law of Hyperspectral Data in Related with Chlorophyll and Carotenoid in Rice at Different Developmental Stages. Rice Sci. 2004, 11, 274–282. [Google Scholar] [CrossRef]
- Han, X.; Hu, Y.; Zhang, G.; Jiang, Y.; Chen, X.; Yu, D. Jasmonate Negatively Regulates Stomatal Development in Arabidopsis Cotyledons. Plant Physiol. 2018, 176, 2871–2885. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Chlorophyll a (mg/g FW) | Chlorophyll b (mg/g FW) | Carotenoid (mg/g FW) | Pn (μmol·m−2·s−1) | Gs (mmol·m−2·s−1) | Ci (μmol·mol−1) | Tr (mmol·m−2·s−1) |
|---|---|---|---|---|---|---|---|
| WT | 9.34 ± 0.39 a | 2.95 ± 0.15 a | 1.94 ± 0.09 a | 26.51 ± 1.25 a | 817.24 ± 109.69 a | 242.60 ± 6.27 a | 6.11 ± 0.46 a |
| odd1 | 6.99 ± 0.63 b | 2.51 ± 0.23 b | 1.52 ± 0.18 b | 21.30 ± 1.81 b | 465.6 ± 94.81 b | 221.00 ± 13.77 b | 5.51 ± 0.46 a |
| Types | Seeds |
|---|---|
| odd1 (selfing) | no |
| WT (selfing) | yes |
| odd1♀ × WT♂ | no |
| WT♀ × odd1♂ | yes |
| Populations | # of Plants Observed | # WT | # odd1 | Expected WT to odd1 Ratio | χ2 Value | p Value |
|---|---|---|---|---|---|---|
| (Chinese long 9930×odd1) F1 | 16 | 16 | 0 | 1:0 | / | / |
| (F1 selfing) F2 | 200 | 143 | 57 | 3:1 | 1.127 | 0.289 |
| (F1×odd1) Ft | 200 | 97 | 103 | 1:1 | 0.125 | 0.724 |
| Category | Proteins | mRNAs |
|---|---|---|
| Unique protein/gene detected | 6283 | 24,118 |
| Significantly changed proteins/genes | 356 | 565 |
| Up-regulated | 176 | 314 |
| Down-regulated | 180 | 251 |
| Gene ID | Gene Description | Fold Change (odd1/WT) | p Value | Regulated Type |
|---|---|---|---|---|
| Csa2G238880 | Non-symbiotic hemoglobin 1 | 0.271 | 0.000162 | Down |
| Csa3G872170 | Gibberellin-regulated protein | 2.382 | 0.000364 | Up |
| Csa6G085120 | Hfr-2-like protein | 3.154 | 0.000364 | Up |
| Csa1G187170 | Unknown protein | 2.141 | 0.000456 | Up |
| Csa2G055560 | Choline dehydrogenase | 2.113 | 0.000776 | Up |
| Csa6G085110 | Hfr-2-like protein | 3.462 | 0.000864 | Up |
| Csa6G109750 | UDP-glucosyltransferase, putative | 1.423 | 0.000962 | Up |
| Csa2G023940 | Lipoxygenase | 2.198 | 0.000978 | Up |
| Csa7G073410 | Leucine-rich repeat receptor-likeserine/threonine-protein kinase | 1.588 | 0.000979 | Up |
| Csa5G410730 | Glutamine synthetase | 1.875 | 0.00112 | Up |
| Csa1G188680 | Xyloglucan endotransglucosylase/hydrolase | 1.56 | 0.00136 | Up |
| Csa3G903550 | Putative cytochrome P450superfamily protein | 1.742 | 0.00164 | Up |
| Csa2G009470 | Betaine aldehyde dehydrogenase | 0.778 | 0.00184 | Down |
| Csa1G151000 | Stress responsive A/B barrel domainfamily protein | 1.386 | 0.00268 | Up |
| Csa6G088160 | Cytochrome P450, putative | 1.686 | 0.00274 | Up |
| Csa6G522760 | Ycf23 protein | 0.701 | 0.00274 | Down |
| Csa3G698490 | Cytochrome P450 | 1.346 | 0.00338 | Up |
| Csa7G414480 | Short-chain dehydrogenase/reductasefamily protein | 1.362 | 0.00356 | Up |
| Csa5G224130 | Cytochrome P450 | 1.307 | 0.0037 | Up |
| Csa1G541390 | Phosphatidylinositol transfer protein sfh5 | 0.72 | 0.00418 | Down |
| Csa7G447020 | Probable peptide/nitrate transporter | 1.458 | 0.00504 | Up |
| Csa3G903540 | Putative cytochrome P450superfamily protein | 1.795 | 0.00516 | Up |
| Csa6G490110 | Methylesterase | 1.585 | 0.0062 | Up |
| Csa3G778270 | Short-chain dehydrogenase/reductase 2 | 1.308 | 0.00648 | Up |
| Csa3G778280 | Short-chain dehydrogenase/reductase 2 | 1.44 | 0.00694 | Up |
| Csa6G088710 | Cytochrome P450 | 1.547 | 0.00728 | Up |
| Csa6G088700 | Anthranilate N-benzoyltransferaseprotein, putative | 1.647 | 0.00966 | Up |
| Csa7G451920 | Putative phosphatase | 0.526 | 0.00994 | Down |
| Csa1G044890 | Cytochrome P450 | 1.907 | 0.014 | Up |
| Csa4G285730 | Peroxidase | 1.668 | 0.0154 | Up |
| Csa6G183190 | Asparagine synthetase | 1.52 | 0.0162 | Up |
| Csa1G611290 | Beta-glucosidase D7 | 1.566 | 0.0171 | Up |
| Csa4G285740 | Peroxidase | 1.663 | 0.0172 | Up |
| Csa3G556210 | Glycerophosphodiesterphosphodiesterase | 0.51 | 0.0204 | Down |
| Csa4G288610 | Lipoxygenase | 1.425 | 0.0216 | Up |
| Csa1G044880 | Short-chain dehydrogenase/reductase 1 | 1.363 | 0.0232 | Up |
| Csa4G285760 | Peroxidase | 1.352 | 0.0237 | Up |
| Csa6G088170 | Cytochrome P450 | 1.304 | 0.0253 | Up |
| Csa5G576590 | Auxin efflux carrier | 1.279 | 0.0255 | Up |
| Csa5G636450 | Lipid A export ATP-binding/permeaseprotein MsbA | 1.269 | 0.0269 | Up |
| Csa3G172370 | MLP | 1.333 | 0.035 | Up |
| Csa3G435030 | Profilin | 1.401 | 0.0496 | Up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Ma, Z.; Chen, L.; Wang, Z.; Wang, C.; Wang, L.; Chen, C.; Ren, Z.; Cao, C. Morphological Characterization and Integrated Transcriptome and Proteome Analysis of Organ Development Defective 1 (odd1) Mutant in Cucumis sativus L. Int. J. Mol. Sci. 2022, 23, 5843. https://doi.org/10.3390/ijms23105843
Han J, Ma Z, Chen L, Wang Z, Wang C, Wang L, Chen C, Ren Z, Cao C. Morphological Characterization and Integrated Transcriptome and Proteome Analysis of Organ Development Defective 1 (odd1) Mutant in Cucumis sativus L. International Journal of Molecular Sciences. 2022; 23(10):5843. https://doi.org/10.3390/ijms23105843
Chicago/Turabian StyleHan, Jing, Zengguang Ma, Linjie Chen, Zaizhan Wang, Can Wang, Lina Wang, Chunhua Chen, Zhonghai Ren, and Chenxing Cao. 2022. "Morphological Characterization and Integrated Transcriptome and Proteome Analysis of Organ Development Defective 1 (odd1) Mutant in Cucumis sativus L." International Journal of Molecular Sciences 23, no. 10: 5843. https://doi.org/10.3390/ijms23105843