
Functional and Molecular Characterisation of Heart Failure Progression in Mice and the Role of Myosin Regulatory Light Chains in the Recovery of Cardiac Muscle Function

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
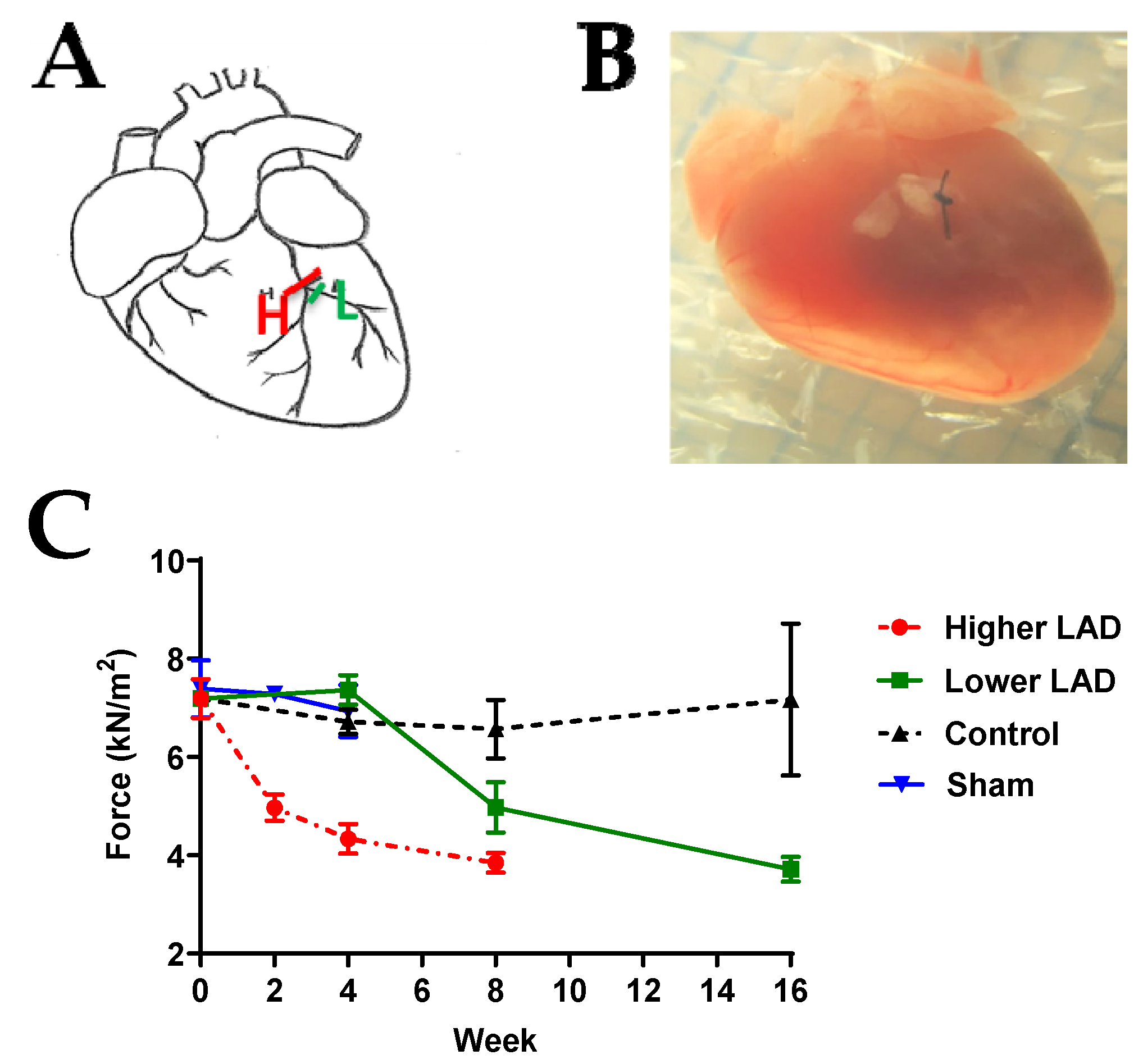
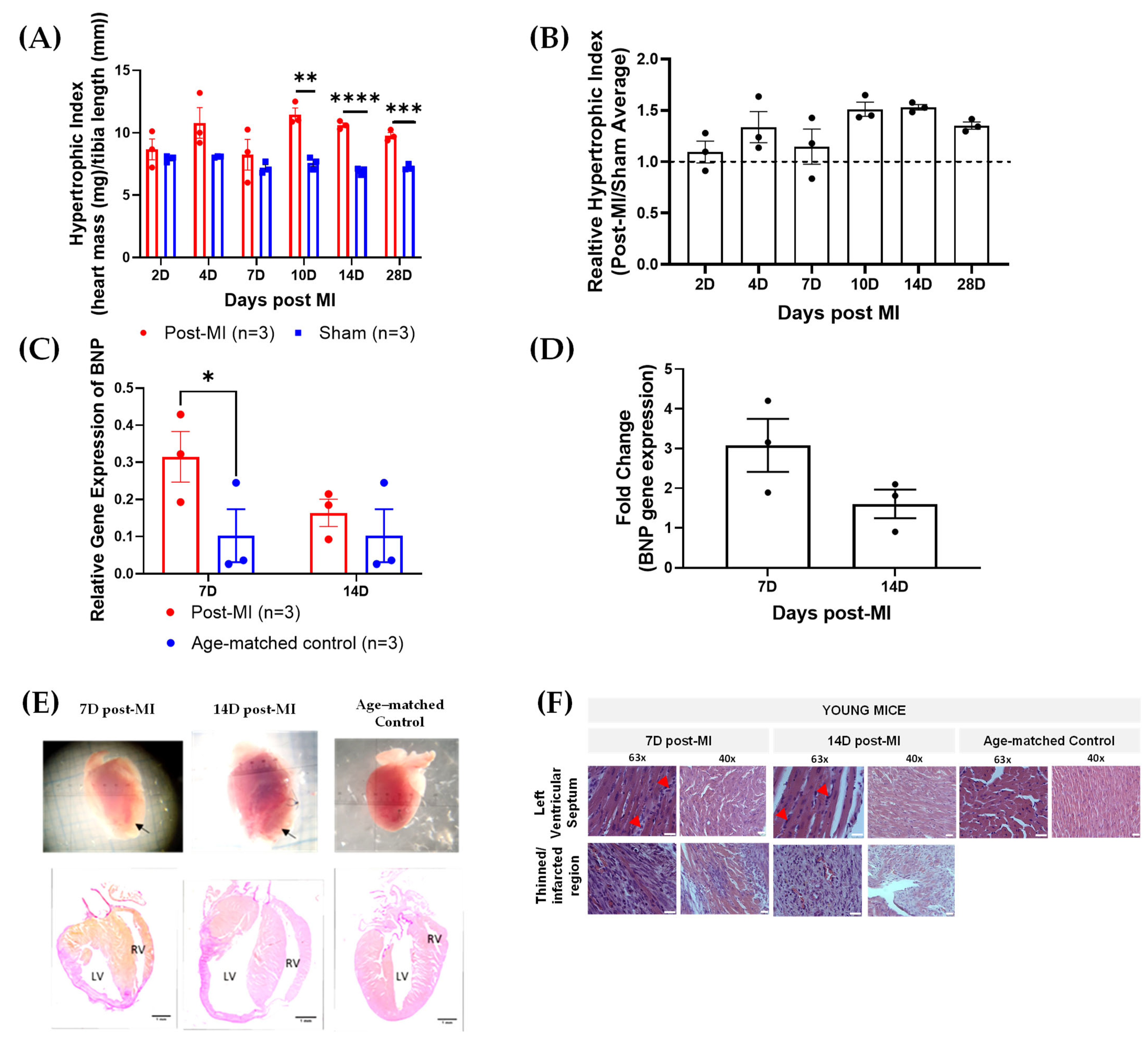
2.1. Myocardial Infarction (MI) Mouse Model Appropriate for HF Progression Study
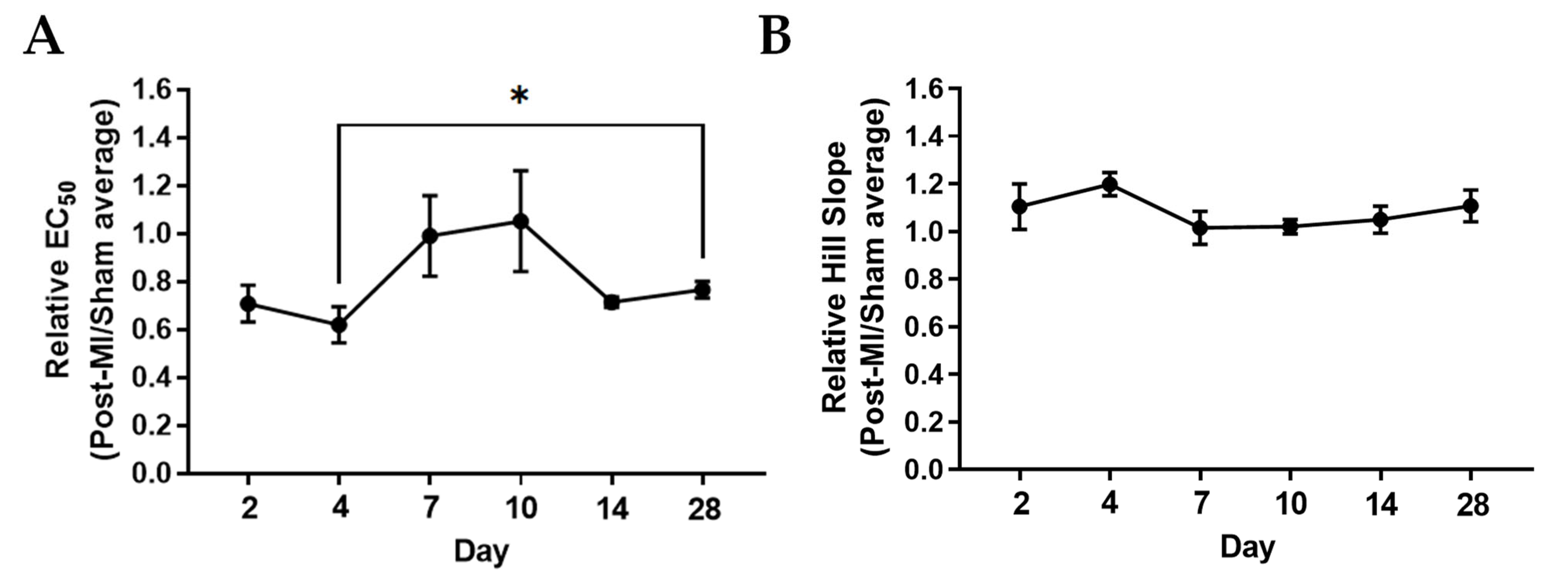
2.2. Ca2+ Sensitivity and Calcium-Induced Cooperativity May Not Be Key Regulators of Force Development during HF Progression
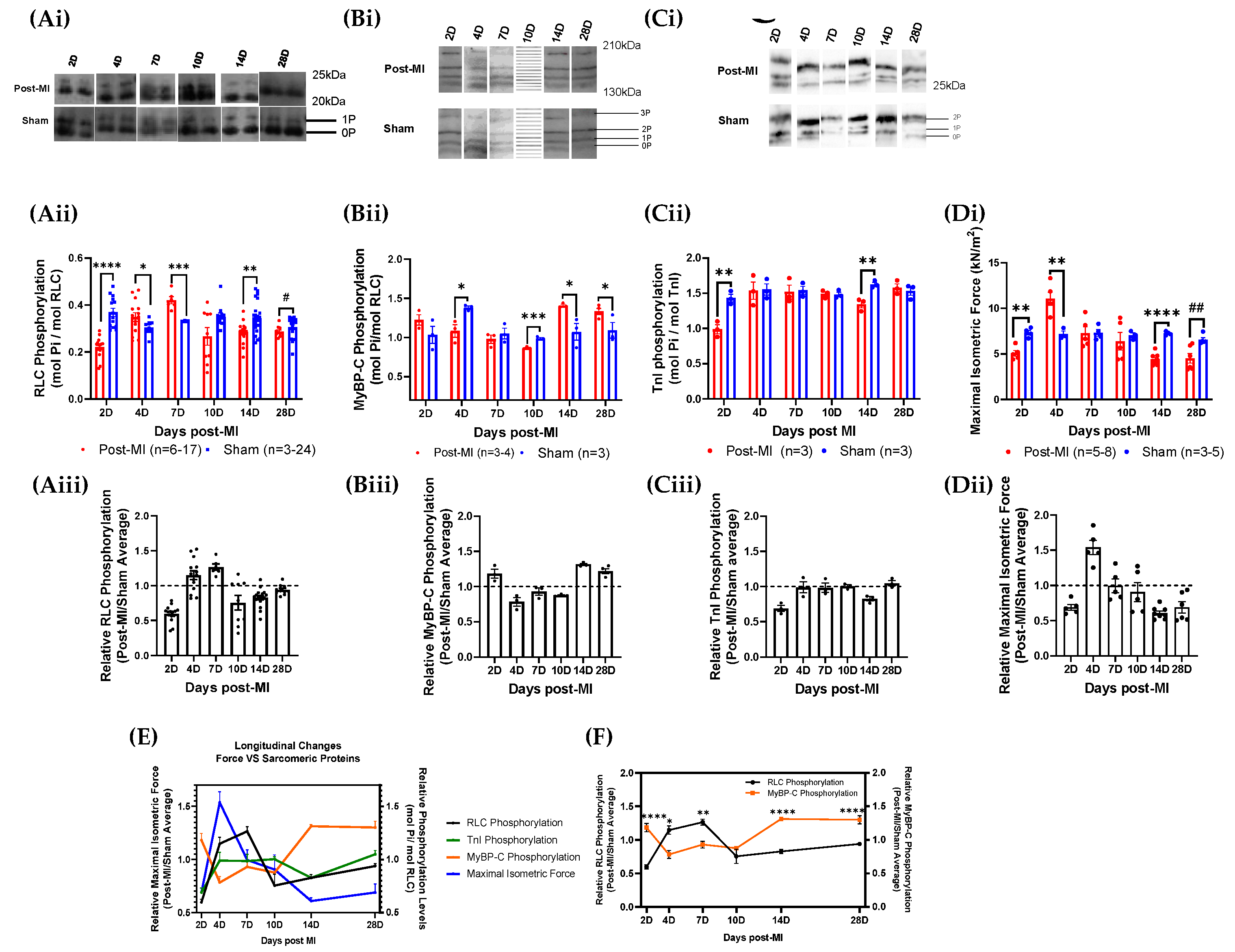
2.3. RLC Phosphorylation Transiently Changes to Sustain Cardiac Function during Compensatory Phase of Heart Failure
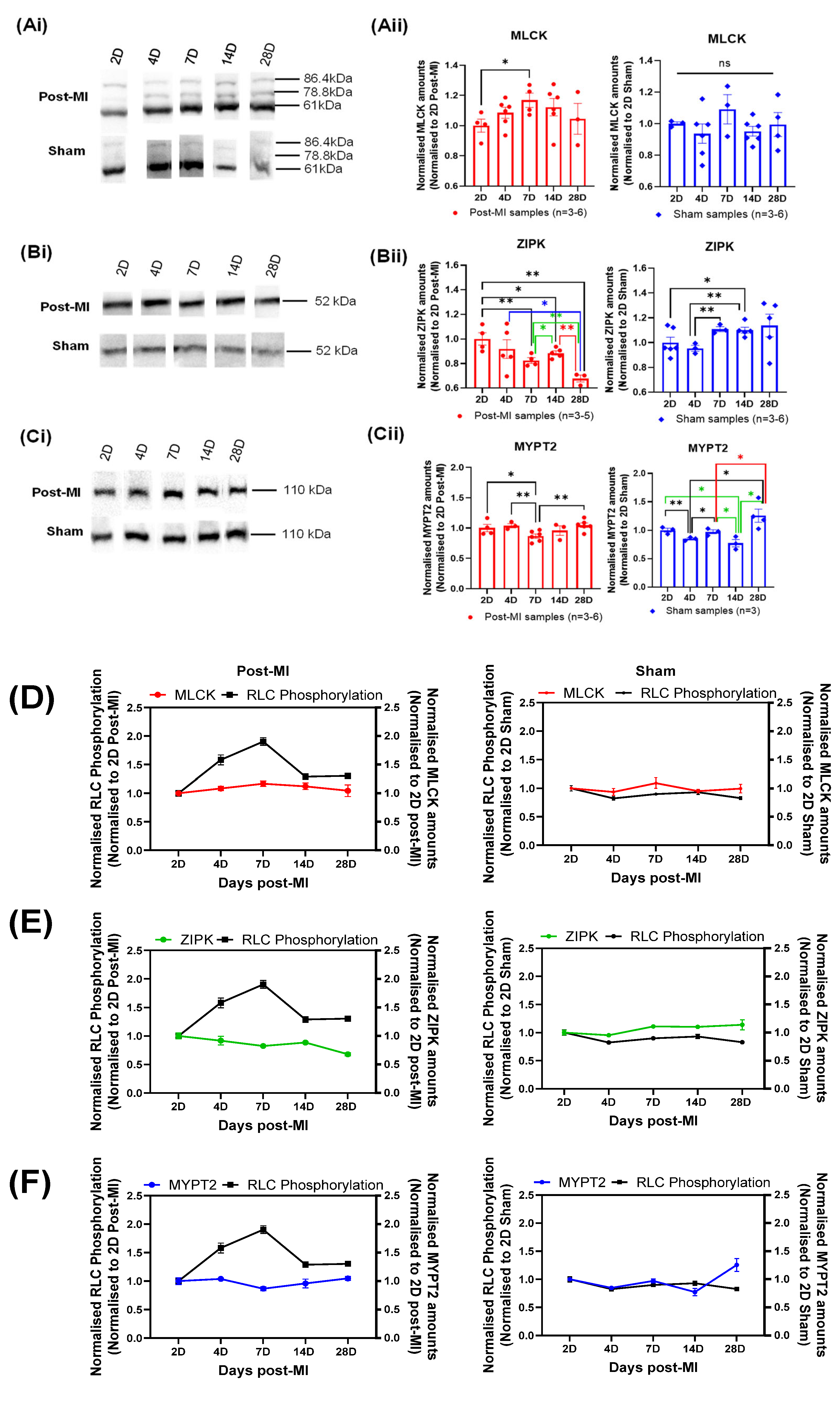
2.4. Post-Translational Modifications in Left Ventricular Tissue during HF Progression

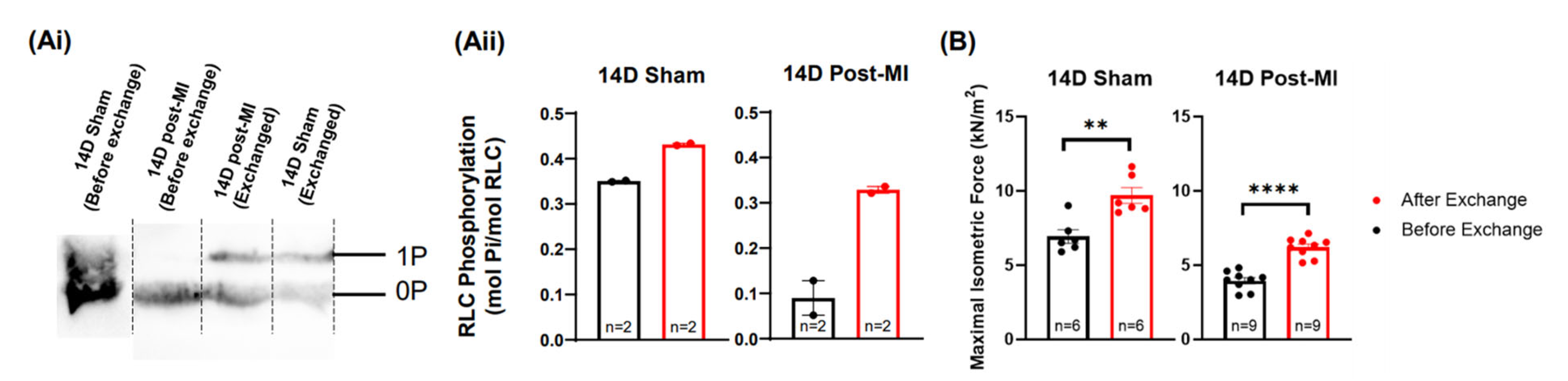
2.5. Modulation of the Phosphorylation Level of RLC Induces Recovery of the Contractile Performance of Papillary Muscle In Vitro
2.6. Enzyme Quantification of Left Ventricular Tissue during HF Progression
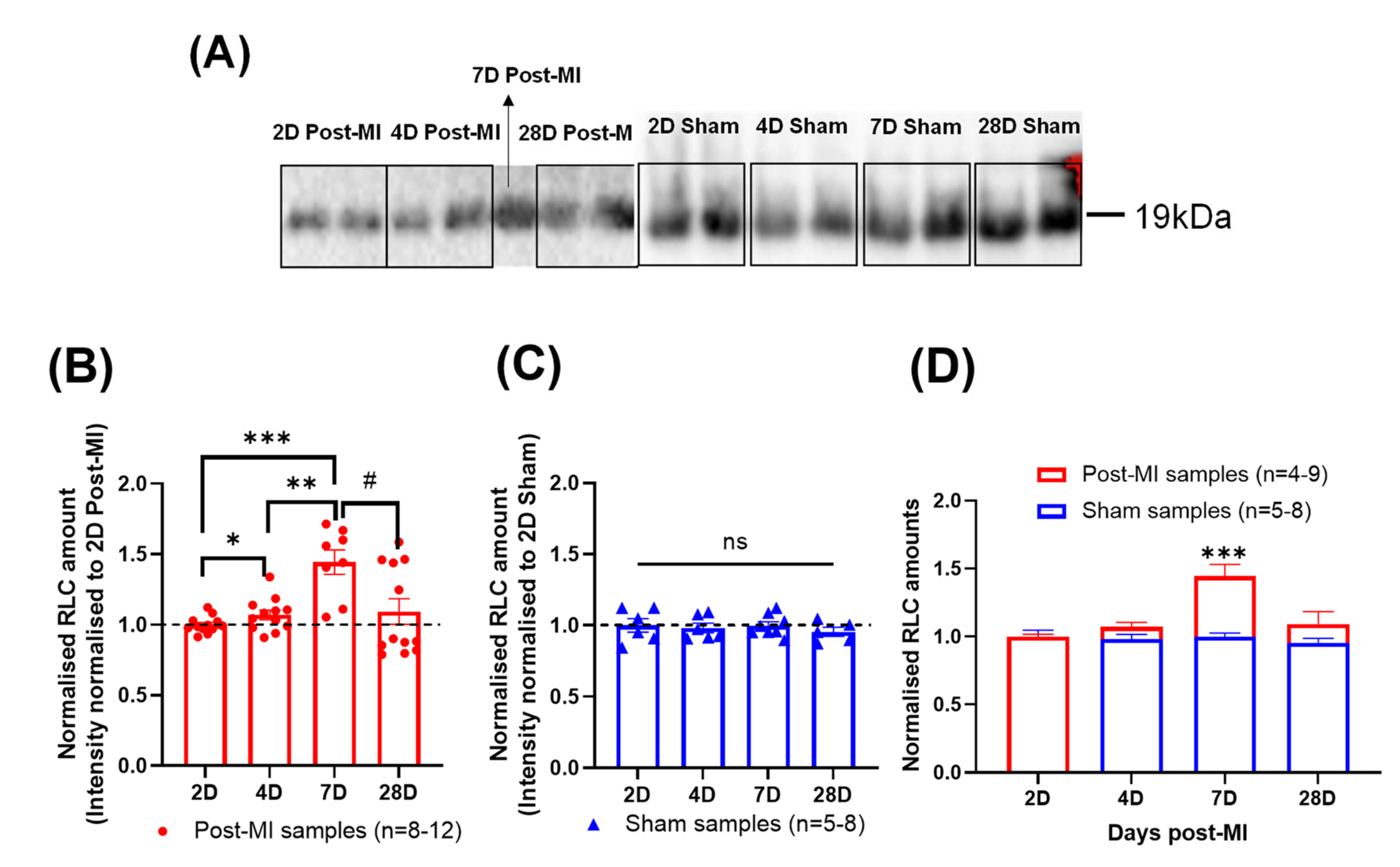
2.7. Differential RLC Expression in Left Ventricular Tissue during HF Progression
3. Discussion
4. Materials and Methods
4.1. Generation of Chronic Myocardial Infarction (MI) Mouse Model
4.2. Papillary Preparation for Maximal Isometric Force and Ca2+ Sensitivity Measurements
4.3. Measurement of Sarcomeric Protein Phosphorylation
4.4. Quantifications of Enzymes That Regulate RLC Phosphorylation
4.5. Expression and Purification of Recombinant RLC (rRLC)
4.6. In Vitro rRLC Phosphorylation
4.7. Phosphorylated rRLC Exchange
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Amy Groenewegen, F.H.R.; Mosterd, A.; Arno, W.H. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- Huang, W.; Lee, S.G.; How, C.H. Management of the heart failure patient in the primary care setting. Singap. Med. J. 2020, 61, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.S.; La Bonte, L.R.; Xu, Y.; Yang, Z.; French, B.A.; Guilford, W.H. Alterations to myofibrillar protein function in nonischemic regions of the heart early after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H654–H659. [Google Scholar] [CrossRef]
- Sutton, M.G.S.J.; Sharpe, N. Left Ventricular Remodeling After Myocardial Infarction. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Pfeffer, J.M.; Pfeffer, M.A.; Fletcher, P.J.; Braunwald, E. Progressive ventricular remodeling in rat with myocardial infarction. Am. J. Physiol. 1991, 260 Pt 2, H1406–H1414. [Google Scholar] [CrossRef]
- Usui, T.; Okada, M.; Yamawaki, H. Zipper interacting protein kinase (ZIPK): Function and signaling. Apoptosis 2014, 19, 387–391. [Google Scholar] [CrossRef]
- Daniels, M.C.G.; Naya, T.; Rundell, V.L.M.; Tombe, P.P.D. Development of contractile dysfunction in rat heart failure: Hierarchy of cellular events. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2007, 293, R284–R292. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C.; Caorsi, V.; Kampourakis, T.; Sikkel, M.B.; West, T.G.; Leung, M.-C. Myosin Regulatory Light Chain (RLC) Phosphorylation Change as a Modulator of Cardiac Muscle Contraction in Disease. J. Biol. Chem. 2013, 288, 13446–13454. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C.N.; Sikkel, M.B.; Caorsi, V.; Vydyanath, A.; Torre, I.; Copeland, O.N. A post-MI power struggle: Adaptations in cardiac power occur at the sarcomere level alongside MyBP-C and RLC phosphorylation. Am. J. Physiol. -Heart Circ. Physiol. 2016, 311, H465–H475. [Google Scholar] [CrossRef]
- Tardiff, J.C.; Carrier, L.; Bers, D.M.; Poggesi, C.; Ferrantini, C.; Coppini, R. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Scruggs, S.B.; Solaro, R.J. The significance of regulatory light chain phosphorylation in cardiac physiology. Arch. Biochem. Biophys. 2011, 510, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.N.; Battiprolu, P.K.; Cowley, P.M.; Chen, G.; Gerard, R.D.; Pinto, J.R. Constitutive phosphorylation of cardiac myosin regulatory light chain in vivo. J. Biol. Chem. 2015, 290, 10703–10716. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C.N.; West, T.G.; Ferenczi, M.A. Revisiting Frank-Starling: Regulatory light chain phosphorylation alters the rate of force redevelopment (ktr) in a length-dependent fashion. J. Physiol. 2016, 594, 5237–5254. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, C.; West, T.G.; Curtin, N.A.; Ferenczi, M.A. Stretch of Contracting Cardiac Muscle Abruptly Decreases the Rate of Phosphate Release at High and Low Calcium. J. Biol. Chem. 2012, 287, 25696–25705. [Google Scholar] [CrossRef] [Green Version]
- Karabina, A.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Myosin regulatory light chain phosphorylation enhances cardiac beta-myosin in vitro motility under load. Arch Biochem. Biophys. 2015, 580, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Markandran, K.; Poh, J.W.; Ferenczi, M.A.; Cheung, C. Regulatory Light Chains in Cardiac Development and Disease. Int. J. Mol. Sci. 2021, 22, 4351. [Google Scholar] [CrossRef]
- Huang, J.; Shelton, J.M.; Richardson, J.A.; Kamm, K.E.; Stull, J.T. Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J. Biol. Chem. 2008, 283, 19748–19756. [Google Scholar] [CrossRef] [Green Version]
- Granzier, H.L.; de Tombe, P.P. Myosin light chain phosphorylation to the rescue. Proc. Natl. Acad. Sci. USA 2015, 112, 9148–9149. [Google Scholar] [CrossRef] [Green Version]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic. Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hamm, C. Role of B-type natriuretic peptide (BNP) and NT-proBNP in clinical routine. Heart 2006, 92, 843–849. [Google Scholar] [CrossRef]
- George, J.; Struthers, A.D. Chapter 31—Natriuretic Peptides. In Comprehensive Hypertension; Lip, G.Y.H., Hall, J.E., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 349–362. [Google Scholar]
- Kalsmith, B.M. Role of the Brain Natriuretic Peptide in Heart Failure Management. Circ. Heart Fail. 2009, 2, 379. [Google Scholar] [CrossRef] [Green Version]
- Nishikimi, T.; Maeda, N.; Matsuoka, H. The role of natriuretic peptides in cardioprotection. Cardiovasc. Res. 2006, 69, 318–328. [Google Scholar]
- Sunil, K.; Nadar, M.M.S. Biomarkers in Routine Heart Failure Clinical Care. Card. Fail. Rev. 2019, 5, 50–56. [Google Scholar]
- Hanna, K.; Gaggin, J.L.J.J. Biomarkers and diagnostics in heart failure. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 2442–2450. [Google Scholar]
- Hirata, Y.M.A.; Aoyagi, T.; Yamaoki, K.; Komuro, I.; Suzuki, T.; Ashida, T.; Sugiyama, T.; Hada, Y.; Kuwajima, I.; Nishinaga, M.; et al. Measurement of plasma brain natriuretic peptide level as a guide for cardiac overload. Cardiovasc. Res. 2001, 51, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.-H.; Biesiadecki, B.J.; Ziolo, M.T.; Davis, J.P.; Janssen, P.M.L. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Front. Physiol. 2016, 7, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-B.; Irving, M. The molecular basis of the steep force-calcium relation in heart muscle. J. Mol. Cell. Cardiol. 2010, 48, 859–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, J.D. Congestive Heart Failure. Pediatrics 1956, 18, 491. [Google Scholar] [CrossRef]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Voors, A.A.; Girerd, N.; Billotte, M.; Anker, S.D.; Cleland, J.G. Heart failure etiologies and clinical factors precipitating for worsening heart failure: Findings from BIOSTAT-CHF. Eur. J. Intern. Med. 2020, 71, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Cowie, M.R.; Wood, D.A.; Coats, A.J.S.; Thompson, S.G.; Poole-Wilson, P.A.; Suresh, V. Incidence and aetiology of heart failure; a population-based study. Eur. J. Intern. Med. 1999, 20, 421–428. [Google Scholar] [CrossRef]
- McMurray, J.J.; Stewart, S. Epidemiology, aetiology, and prognosis of heart failure. Heart 2000, 83, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Tarnavski, O.; McMullen, J.R.; Schinke, M.; Nie, Q.; Kong, S.; Izumo, S. Mouse cardiac surgery: Comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol. Genomics 2004, 16, 349–360. [Google Scholar] [CrossRef]
- van den Borne, S.W.M.; van de Schans, V.A.M.; Strzelecka, A.E.; Vervoort-Peters, H.T.M.; Lijnen, P.M.; Cleutjens, J.P.M. Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovasc. Res. 2009, 84, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Morano, I. Tuning the human heart molecular motors by myosin light chains. J. Mol. Med. 1999, 77, 544–555. [Google Scholar] [CrossRef]
- van der Velden, J.; Merkus, D.; de Beer, V.; Hamdani, N.; Linke, W.A.; Boontje, N.M. Transmural heterogeneity of myofilament function and sarcomeric protein phosphorylation in remodeled myocardium of pigs with a recent myocardial infarction. Front. Physiol. 2011, 2, 83. [Google Scholar] [CrossRef] [Green Version]
- Vargas, L.A.; Pinilla, O.A.; Díaz, R.G.; Sepúlveda, D.E.; Swenson, E.R.; Pérez, N.G. Carbonic anhydrase inhibitors reduce cardiac dysfunction after sustained coronary artery ligation in rats. Cardiovasc. Pathol. 2016, 25, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2–9. [Google Scholar] [CrossRef]
- Baudet, S. Hypertrophy and dilation: A TOTally new story? Cardiovasc. Res. 2000, 46, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Bahit, M.C.; Kochar, A.; Granger, C.B. Post-Myocardial Infarction Heart Failure. JACC Heart Fail. 2018, 6, 179–186. [Google Scholar] [CrossRef]
- Willott, R.H.; Gomes, A.V.; Chang, A.N.; Parvatiyar, M.S.; Pinto, J.R.; Potter, J.D. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J. Mol. Cell Cardiol. 2010, 48, 882–892. [Google Scholar] [CrossRef]
- Marston, S.B.; de Tombe, P.P. Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: Increased or decreased? J. Mol. Cell. Cardiol. 2008, 45, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef]
- Yadav, S.; Szczesna-Cordary, D. Pseudophosphorylation of cardiac myosin regulatory light chain: A promising new tool for treatment of cardiomyopathy. Biophys. Rev. 2017, 9, 57–64. [Google Scholar]
- van der Velden, J.; Papp, Z.; Zaremba, R.; Boontje, N.M.; de Jong, J.W.; Owen, V.J. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef]
- van der Velden, J.; Papp, Z.; Boontje, N.M.; Zaremba, R.; de Jong, J.W.; Janssen, P.M.L. The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovasc. Res. 2003, 57, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, K.; Akopian, G.; Jinadasa, P.; Gluckman, T.L.; Terhakopian, A.; Massey, B. Myocardial infarction and regulatory myosin light chain. J. Mol. Cell. Cardiol. 1997, 29, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Roy, S. Papillary muscles of left ventricle-Morphological variations and it’s clinical relevance. Indian Heart J. 2018, 70, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Kristine, E.; Kamm, J.T.S. Signaling to Myosin Regulatory Light Chain in Sarcomeres. J. Biol. Chem. 2011, 286, 9941–9947. [Google Scholar]
- Chan, J.Y.; Takeda, M.; Briggs, L.E.; Graham, M.L.; Lu, J.T.; Horikoshi, N. Identification of cardiac-specific myosin light chain kinase. Circ. Res. 2008, 102, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chakravorty, S.; Song, W.; Ferenczi, M.A. Phosphorylation of the regulatory light chain of myosin in striated muscle: Methodological perspectives. Eur. Biophys. J. 2016, 45, 779–805. [Google Scholar] [CrossRef] [Green Version]
- Szczesna, D.; Ghosh, D.; Li, Q.; Gomes, A.V.; Guzman, G.; Arana, C. Familial Hypertrophic Cardiomyopathy Mutations in the Regulatory Light Chains of Myosin Affect Their Structure, Ca2+Binding, and Phosphorylation*. J. Biol. Chem. 2001, 276, 7086–7092. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, J.E.; Murray, M.T. Textbook of Natural Medicine-E-Book; Elsevier Health Sciences: St. Louis, MI, USA, 2012. [Google Scholar]
- Lewis, S.E.M.; Kelly, F.J.; Goldspink, D.F. Pre- and post-natal growth and protein turnover in smooth muscle, heart and slow- and fast-twitch skeletal muscles of the rat. Biochem. J. 1984, 217, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Margossian, S.S.; White, H.D.; Caulfield, J.B.; Norton, P.; Taylor, S.; Slayter, H.S. Light chain 2 profile and activity of human ventricular myosin during dilated cardiomyopathy. Identification of a causal agent for impaired myocardial function. Circulation 1992, 85, 1720–1733. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.; Buckingham, M. Manipulating Myosin Light Chain 2 Isoforms In Vivo. Circ. Res. 1997, 80, 751–753. [Google Scholar] [CrossRef]
- Palermo, J.; Gulick, J.; Ng, W.; Grupp, I.L.; Grupp, G.; Robbins, J. Remodeling the mammalian heart using transgenesis. Cell. Mol. Biol. Res. 1995, 41, 501–509. [Google Scholar]
- Chadin, A.V.; Belokurova, M.V.; Stepanova, O.V.; Ivanova, M.V.; Shirinskiĭ, V.P. [Content of myosin-activating protein kinases in myocardium of patients with dilated cardiomyopathy and in the animal heart]. Biofizika 2006, 51, 924–928. [Google Scholar]
- Olsson, M.C.; Patel, J.R.; Fitzsimons, D.P.; Walker, J.W.; Moss, R.L. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2712–H2718. [Google Scholar] [CrossRef] [Green Version]
- Osamu Seguchi, S.T.; Yamazaki, S.; Asakura, M.; Asano, Y.; Shintani, Y.; Wakeno, M.; Minamino, T.; Kondo, H.; Furukawa, H.; Nakamaru, K.; et al. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J. Clin. Investig. 2007, 117, 2812–2824. [Google Scholar] [CrossRef] [Green Version]
- Haystead, T.A. ZIP kinase, a key regulator of myosin protein phosphatase 1. Cell. Signal. 2005, 17, 1313–1322. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Zhang, H.; Zeng, W.; Li, S.; Chen, C. ZIPK mediates endothelial cell contraction through myosin light chain phosphorylation and is required for ischemic-reperfusion injury. FASEB J. 2019, 33, 9062–9074. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Surks, H.K.; Mochizuki, S.; Mochizuki, N.; Mendelsohn, M.E. Identification and Characterization of Zipper-interacting Protein Kinase as the Unique Vascular Smooth Muscle Myosin Phosphatase-associated Kinase. J. Biol. Chem. 2004, 279, 42055–42061. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.N.; Chen, G.; Gerard, R.D.; Kamm, K.E.; Stull, J.T. Cardiac myosin is a substrate for zipper-interacting protein kinase (ZIPK). J. Biol. Chem. 2010, 285, 5122–5126. [Google Scholar] [CrossRef] [Green Version]
- Ihara, E.; MacDonald, J.A. The regulation of smooth muscle contractility by zipper-interacting protein kinase. Can. J. Physiol. Pharmacol. 2007, 85, 79–87. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okamoto, R.; Ito, M.; Goto, I.; Fujita, S.; Konishi, K. New Isoform of Cardiac Myosin Light Chain Kinase and the Role of Cardiac Myosin Phosphorylation in α1-Adrenoceptor Mediated Inotropic Response. PLoS ONE 2015, 10, e0141130. [Google Scholar] [CrossRef]
- Arimura, T.; Muchir, A.; Kuwahara, M.; Morimoto, S.; Ishikawa, T.; Du, C.-K. Overexpression of heart-specific small subunit of myosin light chain phosphatase results in heart failure and conduction disturbance. Am. J. Physiol. -Heart Circ. Physiol. 2018, 314, H1192–H1202. [Google Scholar] [CrossRef]
- Fujioka, M.; Takahashi, N.; Odai, H.; Araki, S.; Ichikawa, K.; Feng, J. A new isoform of human myosin phosphatase targeting/regulatory subunit (MYPT2): cDNA cloning, tissue expression, and chromosomal mapping. Genomics 1998, 49, 59–68. [Google Scholar] [CrossRef]
- Moorhead, G.; Johnson, D.; Morrice, N.; Cohen, P. The major myosin phosphatase in skeletal muscle is a complex between the beta-isoform of protein phosphatase 1 and the MYPT2 gene product. FEBS Lett. 1998, 438, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Nishio, H.; Ichikawa, K.; Hartshorne, D.J. Evidence for Myosin-Binding Phosphatase in Heart Myofibrils. Biochem. Biophys. Res. Commun. 1997, 236, 570–575. [Google Scholar] [CrossRef]
- Chang, A.N.; Kamm, K.E.; Stull, J.T. Role of myosin light chain phosphatase in cardiac physiology and pathophysiology. J. Mol. Cell. Cardiol. 2016, 101, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.H.; Chang, A.N.; Huang, J.; He, W.; Sweeney, H.L.; Zhu, M. Constitutive phosphorylation of myosin phosphatase targeting subunit-1 in smooth muscle. J. Physiol. 2014, 592, 3031–3051. [Google Scholar] [CrossRef] [Green Version]
- Khromov, A.; Choudhury, N.; Stevenson, A.S.; Somlyo, A.V.; Eto, M. Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J. Biol. Chem. 2009, 284, 21569–21579. [Google Scholar] [CrossRef] [Green Version]
- Venema, R.C.; Raynor, R.L.; Noland, T.A., Jr.; Kuo, J.F. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochem. J. 1993, 294 Pt 2, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Eikemo, H.; Moltzau, L.R.; Hussain, R.I.; Nguyen, C.H.; Qvigstad, E.; Levy, F.O. CaMKII in addition to MLCK contributes to phosphorylation of regulatory light chain in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 471, 219–225. [Google Scholar] [CrossRef]
- Rajashree, R.; Blunt, B.C.; Hofmann, P.A. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1736–H1743. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Martin, L.A.; Hahn, H.S.; Dorn, G.W. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005, 97, 1156–1163. [Google Scholar] [CrossRef]
- Jacques, A.M.; Copeland, O.; Messer, A.E.; Gallon, C.E.; King, K.; McKenna, W.J. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J. Mol. Cell. Cardiol. 2008, 45, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Copeland, O.N.; Sadayappan, S.; Messer, A.E.; Steinen, G.J.M.; van der Velden, J.; Marston, S.B. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 2010, 49, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Kuster, D.W.; Bawazeer, A.C.; Zaremba, R.; Goebel, M.; Boontje, N.M.; van der Velden, J. Cardiac myosin binding protein C phosphorylation in cardiac disease. J. Muscle Res. Cell. Motil. 2012, 33, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, D.K.; Kobilka, B.K.G. Protein-Coupled Receptors: Functional and Mechanistic Insights through Altered Gene Expression. Physiol. Rev. 1998, 78, 35–52. [Google Scholar] [CrossRef]
- Boivin, B.; Lavoie, C.; Vaniotis, G.; Baragli, A.; Villeneuve, L.R.; Ethier, N. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc. Res. 2006, 71, 69–78. [Google Scholar] [CrossRef]
- Ratti, J.; Rostkova, E.; Gautel, M.; Pfuhl, M. Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 2011, 286, 12650–12658. [Google Scholar] [CrossRef] [Green Version]
- Messer, A.E.; Jacques, A.M.; Marston, S.B. Troponin phosphorylation and regulatory function in human heart muscle: Dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 2007, 42, 247–259. [Google Scholar] [CrossRef]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef] [Green Version]
- Layland, J.; Solaro, R.J.; Shah, A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Salhi, H.E.; Hassel, N.C.; Siddiqui, J.K.; Brundage, E.A.; Ziolo, M.T.; Janssen, P.M. Myofilament Calcium Sensitivity: Mechanistic Insight into TnI Ser-23/24 and Ser-150 Phosphorylation Integration. Front. Physiol. 2016, 7, 567. [Google Scholar] [CrossRef] [Green Version]
- Schechter, M.A.; Hsieh, M.K.H.; Njoroge, L.W.; Thompson, J.W.; Soderblom, E.J.; Feger, B.J. Phosphoproteomic profiling of human myocardial tissues distinguishes ischemic from non-ischemic end stage heart failure. PLoS ONE 2014, 9, e104157. [Google Scholar]
- Lam, M.P.Y.; Ping, P.; Murphy, E. Proteomics Research in Cardiovascular Medicine and Biomarker Discovery. J. Am. Coll. Cardiol. 2016, 68, 2819–2830. [Google Scholar]
- Awinda, P.O.; Watanabe, M.; Bishaw, Y.; Huckabee, A.M.; Agonias, K.B.; Kazmierczak, K. Mavacamten decreases maximal force and Ca2+ sensitivity in the N47K-myosin regulatory light chain mouse model of hypertrophic cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H881–H890. [Google Scholar] [CrossRef]
- Vitale, G.; Ferrantini, C.; Piroddi, N.; Scellini, B.; Pioner, J.M.; Colombini, B. The relation between sarcomere energetics and the rate of isometric tension relaxation in healthy and diseased cardiac muscle. J. Muscle Res. Cell Motility 2021, 42, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Li, C.; Hu, S.; Xue, F.; Kang, Y.J.; Zhang, W. An appropriate loading control for western blot analysis in animal models of myocardial ischemic infarction. Biochem. Biophys. Rep. 2017, 12, 108–113. [Google Scholar] [CrossRef]
- Tubbs, J.T.; Kissling, G.E.; Travlos, G.S.; Goulding, D.R.; Clark, J.A.; King-Herbert, A.P. Effects of buprenorphine, meloxicam, and flunixin meglumine as postoperative analgesia in mice. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 185–191. [Google Scholar]
- Tremoleda, J.L.; Kerton, A.; Gsell, W. Anaesthesia and physiological monitoring during in vivo imaging of laboratory rodents: Considerations on experimental outcomes and animal welfare. EJNMMI Res. 2012, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- McClellan, G.; Kulikovskaya, I.; Winegrad, S. Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys. J. 2001, 81, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Kampourakis, T.; Irving, M. Phosphorylation of myosin regulatory light chain controls myosin head conformation in cardiac muscle. J. Mol. Cell. Cardiol. 2015, 85, 199–206. [Google Scholar] [CrossRef] [Green Version]
- OpenWetWare. Electrophoresis. 2017. Available online: https://openwetware.org/mediawiki/index.php?title=Electrophoresis&oldid=980725 (accessed on 19 May 2020).
- Chemiluminescent Western Blotting. Available online: https://www.thermofisher.com/sg/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/chemiluminescent-western-blotting.html (accessed on 19 May 2020).
- Markandran, K.; Xuan, J.V.L.E.; Yu, H.; Shun, L.M.; Ferenczi, M.A. Mn2+-Phos-Tag Polyacrylamide for the Quantification of Protein Phosphorylation Levels. Curr. Protoc. 2021, 1, e221. [Google Scholar] [CrossRef]
- Yang, Z.; Sweeney, H.L. Restoration of Phosphorylation-dependent Regulation to the Skeletal Muscle Myosin Regulatory Light Chain. J. Biol. Chem. 1995, 270, 24646–24649. [Google Scholar] [CrossRef] [Green Version]
- Fusi, L.; Huang, Z.; Irving, M. The Conformation of Myosin Heads in Relaxed Skeletal Muscle: Implications for Myosin-Based Regulation. Biophys, J. 2015, 109, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Szczesna, D.; Zhao, J.; Jones, M.; Zhi, G.; Stull, J.; Potter, J.D. Phosphorylation of the regulatory light chains of myosin affects Ca2+ sensitivity of skeletal muscle contraction. J. Appl. Physiol. 2002, 92, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C. The Role of Myosin Regulatory LIGHT Chain Phosphorylation in Cardiac Health and Disease; Imperial College London: London, UK, 2015. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Post-MI Group | Sham Group | ||||||
|---|---|---|---|---|---|---|---|
| Time-Point | 2D | 4D | 7D | 10D | 14D | 28D | Average of All Time Points |
| (µM) | 1.02 ± 0.11 | 0.898 ± 0.11 | 1.43 ± 0.24 | 1.51 ± 0.3 | 1.03 ± 0.03 | 1.10 ± 0.05 | 1.34 ± 0.09 |
| Hill Slope | 1.48 ± 0.26 | 1.50 ± 0.11 | 1.2 ± 0.18 | 1.27 ± 0.19 | 1.28 ± 0.08 | 1.44 ± 0.19 | 1.25 ± 0.19 |
| Sham Samples | 4D | 7D | 14D | 28D |
|---|---|---|---|---|
| MLCK | ↓ | ↑ | ↓ | ↑ |
| ZIPK | ↓ | ↑(**) | - | ↑ |
| MYPT2 | ↓(**) | ↑(*) | ↓(*) | ↑(*) |
| RLC Phosphorylation | ↓(**) | ↑(*) | ↑ | ↓(*) |
| Post-MI samples | 4D | 7D | 14D | 28D |
| MLCK | ↑ | ↑ | ↓ | ↓ |
| ZIPK | ↓ | ↓ | ↑(*) | ↓(**) |
| MYPT2 | ↑ | ↓(**) | ↑ | ↑ |
| RLC Phosphorylation | ↑(****) | ↑(*) | ↓(****) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markandran, K.; Yu, H.; Song, W.; Lam, D.T.U.H.; Madathummal, M.C.; Ferenczi, M.A. Functional and Molecular Characterisation of Heart Failure Progression in Mice and the Role of Myosin Regulatory Light Chains in the Recovery of Cardiac Muscle Function. Int. J. Mol. Sci. 2022, 23, 88. https://doi.org/10.3390/ijms23010088
Markandran K, Yu H, Song W, Lam DTUH, Madathummal MC, Ferenczi MA. Functional and Molecular Characterisation of Heart Failure Progression in Mice and the Role of Myosin Regulatory Light Chains in the Recovery of Cardiac Muscle Function. International Journal of Molecular Sciences. 2022; 23(1):88. https://doi.org/10.3390/ijms23010088
Chicago/Turabian StyleMarkandran, Kasturi, Haiyang Yu, Weihua Song, Do Thuy Uyen Ha Lam, Mufeeda Changaramvally Madathummal, and Michael A. Ferenczi. 2022. "Functional and Molecular Characterisation of Heart Failure Progression in Mice and the Role of Myosin Regulatory Light Chains in the Recovery of Cardiac Muscle Function" International Journal of Molecular Sciences 23, no. 1: 88. https://doi.org/10.3390/ijms23010088