
Targeting the ERβ/HER Oncogenic Network in KRAS Mutant Lung Cancer Modulates the Tumor Microenvironment and Is Synergistic with Sequential Immunotherapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
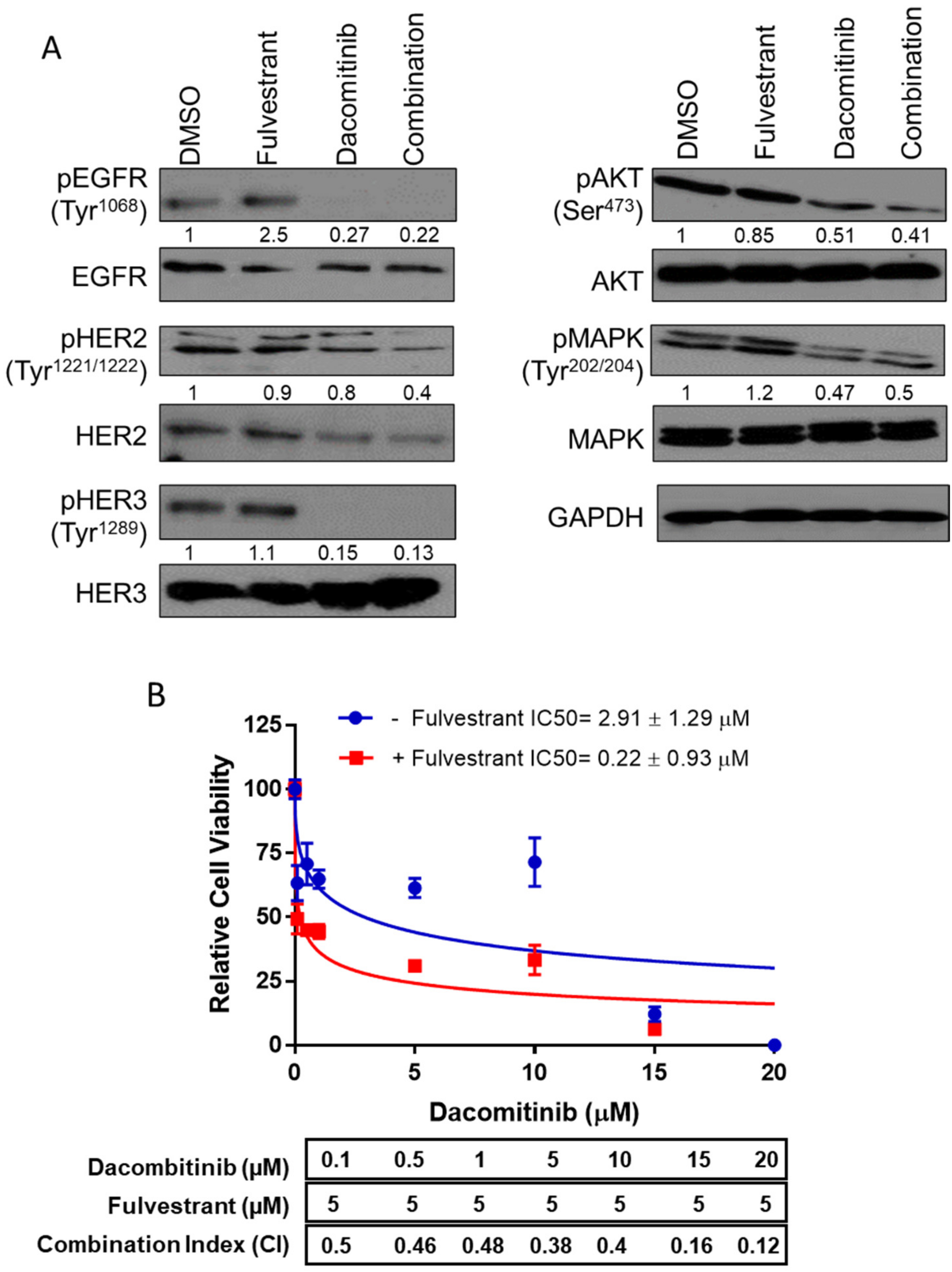
2. Results
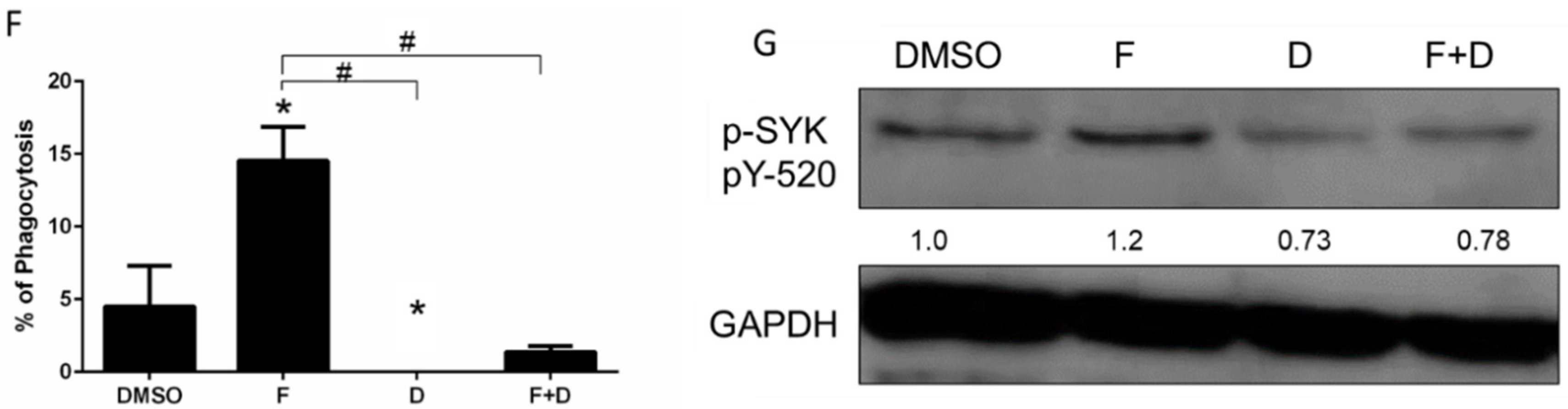
2.1. While Potently Inhibiting KRAS+ Lung Tumor Cell Growth, F+D Induced PD-1 Expression in Bone Marrow Derived Macrophages
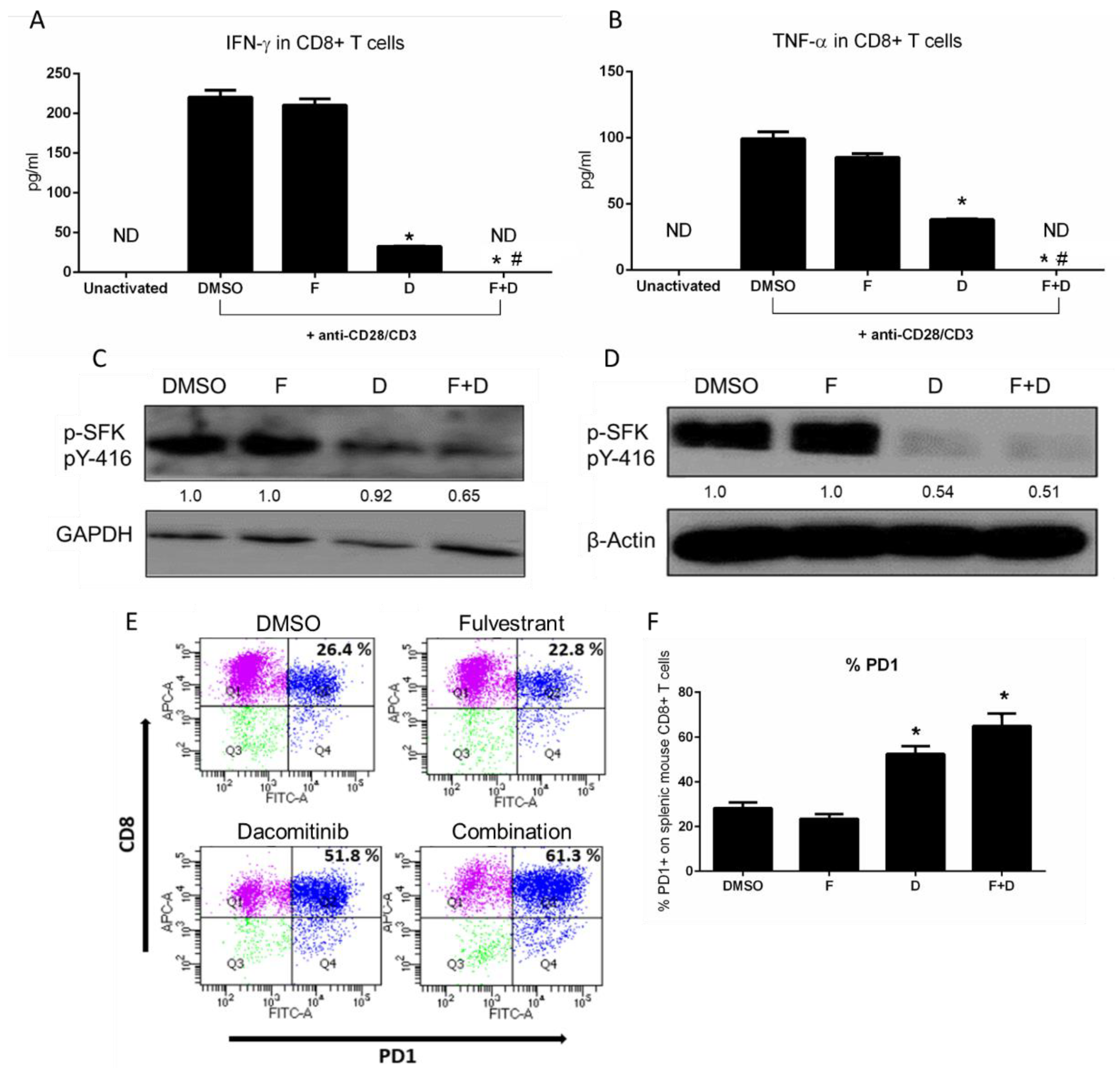
2.2. F+D Inhibited CD8+ T Cell Activity and Induced PD-1
2.3. Triple Therapy Increases CD8+ T Lymphocytes and Produces an Inflamed TME
2.4. The Triple Therapy Given Sequentially Significantly Improved the Antitumor Effect
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents
4.2. Cell Viability Assay
4.3. Isolation and Culture of Murine BMDMs
4.4. Harvesting and Activation of Mouse CD8+ T Cells
4.5. Immunoblotting
4.6. ELISA Assays
4.7. Macrophage Phagocytic Activity
4.8. Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.9. Flow Cytometry Analysis
4.10. In Vivo Animal Studies
4.11. Immunohistochemistry (IHC)
4.12. Cytometric Bead Array
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yang, X.; Huang, Y.; Zhao, M.; Li, M.; Ma, K.; Yin, J.; Zhan, C.; Wang, Q. Trends in the incidence, treatment, and survival of patients with lung cancer in the last four decades. Cancer Manag. Res. 2019, 11, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Panni, R.Z.; Linehan, D.C.; DeNardo, D.G. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy 2013, 5, 1075–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csöszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Almotlak, A.A.; Farooqui, M.; Siegfried, J.M. Inhibiting Pathways Predicted from a Steroid Hormone Gene Signature Yields Synergistic Antitumor Effects in NSCLC. J. Thorac. Oncol. 2020, 15, 62–79. [Google Scholar] [CrossRef] [Green Version]
- Kumai, T.; Ohkuri, T.; Nagato, T.; Matsuda, Y.; Oikawa, K.; Aoki, N.; Kimura, S.; Celis, E.; Harabuchi, Y.; Kobayashi, H. Targeting HER-3 to elicit antitumor helper T cells against head and neck squamous cell carcinoma. Sci. Rep. 2015, 5, 16280. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations that Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, D.H.; Griner, L.M.; Keller, J.M.; Hu, X.; Southall, N.; Marugan, J.; David, J.; Ferrer, M.; Palena, C. Targeting Estrogen Receptor Signaling with Fulvestrant Enhances Immune and Chemotherapy-Mediated Cytotoxicity of Human Lung Cancer. Clin. Cancer Res. 2016, 22, 6204–6216. [Google Scholar] [CrossRef] [Green Version]
- Stabile, L.P.; Kumar, V.; Gaither-Davis, A.; Huang, E.H.; Vendetti, F.P.; Devadassan, P.; Dacic, S.; Bao, R.; Steinman, R.A.; Burns, T.F.; et al. Syngeneic tobacco carcinogen–induced mouse lung adenocarcinoma model exhibits PD-L1 expression and high tumor mutational burden. JCI Insight 2021, 6, e145307. [Google Scholar] [CrossRef]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib restrains K-RAS–driven lung tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruspig, B.; Monteverde, T.; Neidler, S.; Hock, A.; Kerr, E.; Nixon, C.; Clark, W.; Hedley, A.; Laing, S.; Coffelt, S.B.; et al. The ERBB network facilitates KRAS–driven lung tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2565. [Google Scholar] [CrossRef] [Green Version]
- Siegfried, J.M.; Lin, Y.; Diergaarde, B.; Lin, H.M.; Dacic, S.; Pennathur, A.; Weissfeld, J.L.; Romkes, M.; Nukui, T.; Stabile, L.P. Expression of PAM50 Genes in Lung Cancer: Evidence that Interactions between Hormone Receptors and HER2/HER3 Contribute to Poor Outcome. Neoplasia 2015, 17, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Perrotta, F.; Barra, G.; Malapelle, U.; Rocco, D.; De Palma, R. Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 4931. [Google Scholar] [CrossRef] [Green Version]
- Bruno, D.; Dowlati, A. Immunotherapy in EGFR mutant non-small cell lung cancer: When, who and how? Transl. Lung Cancer Res. 2019, 8, 710–714. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bally, A.P.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-κB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.-K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.-S.; Son, Y.-J.; Ryou, C.; Sung, G.-H.; Kim, J.-H.; Cho, J.Y. Functional Roles of Syk in Macrophage-Mediated Inflammatory Responses. Mediat. Inflamm. 2014, 2014, 270302. [Google Scholar] [CrossRef] [Green Version]
- Deronic, A.; Tahvili, S.; Leanderson, T.; Ivars, F. The anti-tumor effect of the quinoline-3-carboxamide tasquinimod: Blockade of recruitment of CD11b(+) Ly6C(hi) cells to tumor tissue reduces tumor growth. BMC Cancer 2016, 16, 440. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Geahlen, R.L. Getting Syk: Spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 2014, 35, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [Green Version]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib with Gefitinib in Patients with Advanced Non–Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhang, J.-T.; Liu, S.-Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.-Y.; Xu, C.-R.; Si-Yang, L.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. OncoImmunology 2017, 6, e1356145. [Google Scholar] [CrossRef] [Green Version]
- Isomoto, K.; Haratani, K.; Hayashi, H.; Shimizu, S.; Tomida, S.; Niwa, T.; Yokoyama, T.; Fukuda, Y.; Chiba, Y.; Kato, R.; et al. Impact of EGFR-TKI Treatment on the Tumor Immune Microenvironment in EGFR Mutation–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Ayeni, D.; Miller, B.; Kuhlmann, A.; Ho, P.-C.; Robles-Oteiza, C.; Gaefele, M.; Levy, S.; de Miguel, F.J.; Perry, C.; Guan, T.; et al. Tumor regression mediated by oncogene withdrawal or erlotinib stimulates infiltration of inflammatory immune cells in EGFR mutant lung tumors. J. Immunother. Cancer 2019, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Huijberts, S.C.F.A.; Van Geel, R.M.J.M.; Van Brummelen, E.M.J.; Opdam, F.L.; Marchetti, S.; Steeghs, N.; Pulleman, S.; Thijssen, B.; Rosing, H.; Monkhorst, K.; et al. Phase I study of lapatinib plus trametinib in patients with KRAS-mutant colorectal, non-small cell lung, and pancreatic cancer. Cancer Chemother. Pharmacol. 2020, 85, 917–930. [Google Scholar] [CrossRef]
- van Brummelen, E.M.J.; Huijberts, S.; van Herpen, C.; Desar, I.; Opdam, F.; van Geel, R.; Marchetti, S.; Steeghs, N.; Monkhorst, K.; Thijssen, B.; et al. Phase I Study of Afatinib and Selumetinib in Patients with KRAS-Mutated Colorectal, Non-Small Cell Lung, and Pancreatic Cancer. Oncologist 2021, 26, 290-e545. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.M.J.M.; van Brummelen, E.M.J.; Eskens, F.A.L.M.; Huijberts, S.C.F.A.; de Vos, F.Y.F.L.; Lolkema, M.P.J.K.; Devriese, L.A.; Opdam, F.L.; Marchetti, S.; Steeghs, N.; et al. Phase 1 study of the pan-HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD-0325901 in patients with KRAS-mutation-positive colorectal, non-small-cell lung and pancreatic cancer. Br. J. Cancer 2020, 122, 1166–1174. [Google Scholar] [CrossRef]
- Moya-Horno, I.; Viteri, S.; Karachaliou, N.; Rosell, R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC). Ther. Adv. Med. Oncol. 2018, 10, 1758834017745012. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Yang, J.C.-H.; Yu, H.; Kim, S.-W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, A.W.; Patel, S.; Boucher, K.; Cannon, L.; Esplin, M.; Luckart, J.; Graves, N.; Van Duren, T.; Akerley, W. Phase I Trial of Targeted EGFR or ALK Therapy with Ipilimumab in Metastatic NSCLC with Long-Term Follow-Up. Target. Oncol. 2019, 14, 417–421. [Google Scholar] [CrossRef]
- Creelan, B.C.; Yeh, T.C.; Kim, S.-W.; Nogami, N.; Kim, D.-W.; Chow, L.Q.M.; Kanda, S.; Taylor, R.; Tang, W.; Tang, M.; et al. A Phase 1 study of gefitinib combined with durvalumab in EGFR TKI-naive patients with EGFR mutation-positive locally advanced/metastatic non-small-cell lung cancer. Br. J. Cancer 2020, 124, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Socinski, M.A.; Villaruz, L.C. Immune Checkpoint Blockade in Oncogene-Driven Non-Small-Cell Lung Cancer. Drugs 2020, 80, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, S.; Yang, Z.; Algazi, A.P.; Lomeli, S.H.; Wang, Y.; Othus, M.; Hong, A.; Wang, X.; Randolph, C.E.; et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell 2021, 39, 1375–1387.e6. [Google Scholar] [CrossRef]
- Traynor, A.M.; Schiller, J.H.; Stabile, L.P.; Kolesar, J.M.; Eickhoff, J.C.; Dacic, S.; Hoang, T.; Dubey, S.; Marcotte, S.M.; Siegfried, J.M. Pilot study of gefitinib and fulvestrant in the treatment of post-menopausal women with advanced non-small cell lung cancer. Lung Cancer 2009, 64, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garon, E.B.; Siegfried, J.M.; Stabile, L.P.; Young, P.A.; Marquez-Garban, D.C.; Park, D.J.; Patel, R.; Hu, E.H.; Sadeghi, S.; Parikh, R.J.; et al. Randomized phase II study of fulvestrant and erlotinib compared with erlotinib alone in patients with advanced or metastatic non-small cell lung cancer. Lung Cancer 2018, 123, 91–98. [Google Scholar] [CrossRef]
- Tan, Y.S.; Lei, Y.L. Isolation of Tumor-Infiltrating Lymphocytes by Ficoll-Paque Density Gradient Centrifugation. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; pp. 93–99. [Google Scholar]
- Chen, Y.; Guggisberg, N.; Jorda, M.; Gonzalez-Angulo, A.; Hennessy, B.; Mills, G.B.; Tan, C.-K.; Slingerland, J.M. Combined Src and Aromatase Inhibition Impairs Human Breast Cancer Growth In vivo and Bypass Pathways Are Activated in AZD0530-Resistant Tumors. Clin. Cancer Res. 2009, 15, 3396–3405. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almotlak, A.A.; Farooqui, M.; Soloff, A.C.; Siegfried, J.M.; Stabile, L.P. Targeting the ERβ/HER Oncogenic Network in KRAS Mutant Lung Cancer Modulates the Tumor Microenvironment and Is Synergistic with Sequential Immunotherapy. Int. J. Mol. Sci. 2022, 23, 81. https://doi.org/10.3390/ijms23010081
Almotlak AA, Farooqui M, Soloff AC, Siegfried JM, Stabile LP. Targeting the ERβ/HER Oncogenic Network in KRAS Mutant Lung Cancer Modulates the Tumor Microenvironment and Is Synergistic with Sequential Immunotherapy. International Journal of Molecular Sciences. 2022; 23(1):81. https://doi.org/10.3390/ijms23010081
Chicago/Turabian StyleAlmotlak, Abdulaziz A., Mariya Farooqui, Adam C. Soloff, Jill M. Siegfried, and Laura P. Stabile. 2022. "Targeting the ERβ/HER Oncogenic Network in KRAS Mutant Lung Cancer Modulates the Tumor Microenvironment and Is Synergistic with Sequential Immunotherapy" International Journal of Molecular Sciences 23, no. 1: 81. https://doi.org/10.3390/ijms23010081