
Heat Shock Protein 60 Restricts Release of Mitochondrial dsRNA to Suppress Hepatic Inflammation and Ameliorate Non-Alcoholic Fatty Liver Disease in Mice

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
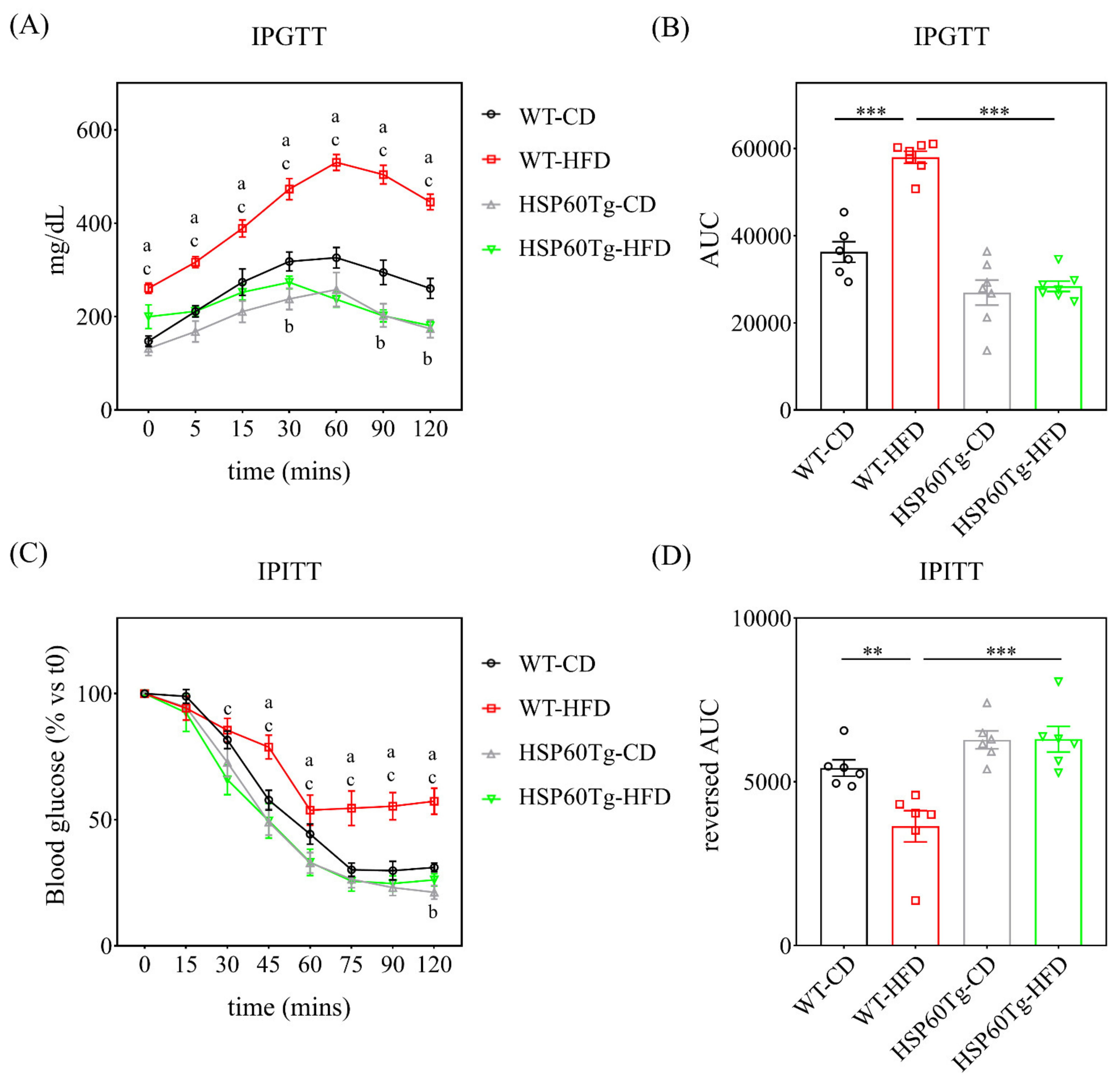
2.1. Overexpression of HSP60 Significantly Reduces Weight Gain and Fat Accumulation in Adipose Tissue and Improves Glucose Tolerance and Insulin Resistance
2.2. Overexpression of HSP60 Reduces Hepatocellular Steatosis in the Context of Chronic HFD
2.3. Overexpression of HSP60 Represses the Release of mt-dsRNA and Inflammation in the Context of Chronic HFD
2.4. Overexpression of HSP60 Modulates HFD-Caused Perturbation of Mitochondrial DNA Content in the Liver
2.5. HSP60 Restricts the Release of Mitochondrial dsRNA In Vitro
2.6. Gain of HSP60 Suppresses TLR3 and MDA5 In Vitro
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Animal Protocol
4.2. Construction and Breeding of the HSP60 Transgenic Mice
4.3. Blood Biochemical Analysis
4.4. Assessment of IPITT and IPGTT
4.5. Cell Culture and Transfection
4.6. Hematoxylin and Eosin Stain Staining
4.7. Immunohistochemistry and Immunofluorescence Staining
4.8. RNA Isolation and Quantitative Real-Time PCR
4.9. Western Blot
4.10. Quantification of mtDNA Copy Number by Real-Time PCR
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chiu, W.H.; Chen, R.C.; Chen, F.L.; Tung, T.H. The clinical investigation of disparity of nonalcoholic fatty liver disease in a Chinese occupational population in Taipei, Taiwan: Experience at a teaching hospital. Asia Pac. J. Public Health Asia Pac. Acad. Consort. Public Health 2015, 27, NP1793–NP1804. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, B.; Qiu, W.; Hao, Y.; Zhang, Z.; Yang, B.; Li, N.; Cheng, S.; Lin, Z.; Rui, Y.C.; et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat. Commun. 2019, 10, 3391. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Cardio-Metabolic Disorders in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; Fargion, S.; Fracanzani, A.L. Brain involvement in non-alcoholic fatty liver disease (NAFLD): A systematic review. Dig. Liver Dis. 2019, 51, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Younes, R.; Bugianesi, E. A spotlight on pathogenesis, interactions and novel therapeutic options in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 80–82. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. [Google Scholar] [CrossRef]
- Urbauer, E.; Rath, E.; Haller, D. Mitochondrial Metabolism in the Intestinal Stem Cell Niche-Sensing and Signaling in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 602814. [Google Scholar] [CrossRef]
- Yi, H.S. Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases. Endocrinol. Metab. 2019, 34, 39–46. [Google Scholar] [CrossRef]
- Rodriguez, A.; Von Salzen, D.; Holguin, B.A.; Bernal, R.A. Complex Destabilization in the Mitochondrial Chaperonin Hsp60 Leads to Disease. Front. Mol. Biosci. 2020, 7, 159. [Google Scholar] [CrossRef]
- Finka, A.; Mattoo, R.U.; Goloubinoff, P. Experimental Milestones in the Discovery of Molecular Chaperones as Polypeptide Unfolding Enzymes. Annu. Rev. Biochem. 2016, 85, 715–742. [Google Scholar] [CrossRef] [Green Version]
- Itoh, H.; Kobayashi, R.; Wakui, H.; Komatsuda, A.; Ohtani, H.; Miura, A.B.; Otaka, M.; Masamune, O.; Andoh, H.; Koyama, K.; et al. Mammalian 60-kDa stress protein (chaperonin homolog). Identification, biochemical properties, and localization. J. Biol. Chem. 1995, 270, 13429–13435. [Google Scholar] [CrossRef] [Green Version]
- Fourman, S. Effects of aminoproprionitrile on glaucoma filter blebs in rabbits. Ophthalmic. Surg. 1988, 19, 649–652. [Google Scholar] [CrossRef]
- Fan, F.; Duan, Y.; Yang, F.; Trexler, C.; Wang, H.; Huang, L.; Li, Y.; Tang, H.; Wang, G.; Fang, X.; et al. Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 2020, 27, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Magnoni, R.; Palmfeldt, J.; Hansen, J.; Christensen, J.H.; Corydon, T.J.; Bross, P. The Hsp60 folding machinery is crucial for manganese superoxide dismutase folding and function. Free Radic. Res. 2014, 48, 168–179. [Google Scholar] [CrossRef]
- Almaani, W.S.; Awidi, A.S. Spontaneous intracranial hemorrhage secondary to von Willebrand’s disease. Surg. Neurol. 1986, 26, 457–460. [Google Scholar] [CrossRef]
- Lin, H.Y.; Wang, F.S.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.L.; Kuo, H.C.; Wang, F.S.; Huang, Y.H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Yang, Y.L.; Wang, P.W.; Wang, F.S.; Huang, Y.H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef]
- Yang, Y.L.; Wang, P.W.; Wang, F.S.; Lin, H.Y.; Huang, Y.H. miR-29a Modulates GSK3beta/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 6884. [Google Scholar] [CrossRef]
- Yang, Y.L.; Wang, F.S.; Lin, H.Y.; Huang, Y.H. Exogenous Therapeutics of Microrna-29a Attenuates Development of Hepatic Fibrosis in Cholestatic Animal Model through Regulation of Phosphoinositide 3-Kinase p85 Alpha. Int. J. Mol. Sci. 2020, 21, 3636. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Lee, J.H.; Shim, Y.R.; Seo, W.; Kim, M.H.; Choi, W.M.; Kim, H.H.; Kim, Y.E.; Yang, K.; Ryu, T.; Jeong, J.M.; et al. Mitochondrial Double-Stranded RNA in Exosome Promotes Interleukin-17 Production Through Toll-Like Receptor 3 in Alcohol-associated Liver Injury. Hepatology 2020, 72, 609–625. [Google Scholar] [CrossRef]
- Bollmann, F.; Dohrke, J.N.; Wurm, C.A.; Jans, D.C.; Jakobs, S. Mitochondrial Protein Abundance Gradients Require the Distribution of Separated Mitochondria. Biology 2021, 10, 572. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Lonardo, A.; Leoni, S.; Alswat, K.A.; Fouad, Y. History of Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 5888. [Google Scholar] [CrossRef]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef]
- Tiao, M.M.; Wang, F.S.; Huang, L.T.; Chuang, J.H.; Kuo, H.C.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis 2014, 19, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Grochowska, J.; Czerwinska, J.; Borowski, L.S.; Szczesny, R.J. Mitochondrial RNA, a new trigger of the innate immune system. Wiley Interdiscip. Rev. RNA 2021, e1690. [Google Scholar] [CrossRef] [PubMed]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanin-Zhorov, A.; Bruck, R.; Tal, G.; Oren, S.; Aeed, H.; Hershkoviz, R.; Cohen, I.R.; Lider, O. Heat shock protein 60 inhibits Th1-mediated hepatitis model via innate regulation of Th1/Th2 transcription factors and cytokines. J. Immunol. 2005, 174, 3227–3236. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.C.; Chen, Y.J.; Yu, H.R.; Huang, L.T.; Tain, Y.L.; Lin, I.C.; Sheen, J.M.; Wang, P.W.; Tiao, M.M. Long term N-acetylcysteine administration rescues liver steatosis via endoplasmic reticulum stress with unfolded protein response in mice. Lipids Health Dis. 2020, 19, 105. [Google Scholar] [CrossRef]
- Ko, J.Y.; Sun, Y.C.; Li, W.C.; Wang, F.S. Chaperonin 60 regulation of SOX9 ubiquitination mitigates the development of knee osteoarthritis. J. Mol. Med. 2016, 94, 755–769. [Google Scholar] [CrossRef]
- Zhang, G.; Li, R.; Li, W.; Yang, S.; Sun, Q.; Yin, H.; Wang, C.; Hou, B.; Wang, H.; Yu, L.; et al. Toll-like receptor 3 ablation prevented high-fat diet-induced obesity and metabolic disorder. J. Nutr. Biochem. 2021, 95, 108761. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Sakuraba, H.; Horiuchi, M.; Ding, J.; Matsumiya, T.; Seya, K.; Iino, C.; Endo, T.; Kikuchi, H.; Yoshida, S.; et al. Hepatic Macrophages Express Melanoma Differentiation-Associated Gene 5 in Nonalcoholic Steatohepatitis. Inflammation 2021, 1–17. [Google Scholar] [CrossRef]
- Qiao, J.T.; Cui, C.; Qing, L.; Wang, L.S.; He, T.Y.; Yan, F.; Liu, F.Q.; Shen, Y.H.; Hou, X.G.; Chen, L. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metab. Clin. Exp. 2018, 81, 13–24. [Google Scholar] [CrossRef]
- Wang, F.S.; Lian, W.S.; Lee, M.S.; Weng, W.T.; Huang, Y.H.; Chen, Y.S.; Sun, Y.C.; Wu, S.L.; Chuang, P.C.; Ko, J.Y. Histone demethylase UTX counteracts glucocorticoid deregulation of osteogenesis by modulating histone-dependent and -independent pathways. J. Mol. Med. 2017, 95, 499–512. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT-CD | WT-HFD | HSP60Tg-CD | HSP60Tg-HFD | |
|---|---|---|---|---|
| Body weight (g) | 35.01 ± 0.65 | 46.74 ± 1.04 a | 30.26 ± 0.67 a | 33.87 ± 0.97 bc |
| GPT (dU/L) | 20.6 ± 6.1 | 58.1 ± 7.1 a | 16.7 ± 4.2 | 28.8 ± 7.1 c |
| Triglycerides (mg/dL) | 99 ± 16.2 | 155.3 ± 18.4 a | 125.7 ± 17.7 | 127.6 ± 20.3 |
| Total Cholesterol (mg/dL) | 183.3 ± 7.5 | 202.9 ± 7.5 a | 189.4 ± 2.4 | 197.8 ± 7.0 |
| Subcutaneous fat (g) | 1.09 ± 0.12 | 3.6 ± 0.34 a | 0.69 ± 0.09 | 1.24 ± 0.37 c |
| Visceral fat (g) | 1.11 ± 0.10 | 1.85 ± 0.12 a | 0.56 ± 0.09 a | 1.14 ± 0.2 bc |
| Mesenteric (g) | 0.47 ± 0.06 | 0.86 ± 0.07 a | 0.14 ± 0.04 a | 0.24 ± 0.07 c |
| Liver (g) | 1.65 ± 0.06 | 2.14 ± 0.17 a | 1.5 ± 0.08 | 1.55 ± 0.03 c |
| Pancreas (g) | 0.3 ± 0.02 | 0.25 ± 0.03 | 0.23 ± 0.02 a | 0.24 ± 0.02 |
| Spleen (g) | 0.09 ± 0.01 | 0.11 ± 0.02 | 0.09 ± 0.03 | 0.13 ± 0.02 |
| Kidney (g) | 0.55 ± 0.04 | 0.64 ± 0.04 | 0.48 ± 0.03 | 0.49 ± 0.02 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-H.; Wang, F.-S.; Wang, P.-W.; Lin, H.-Y.; Luo, S.-D.; Yang, Y.-L. Heat Shock Protein 60 Restricts Release of Mitochondrial dsRNA to Suppress Hepatic Inflammation and Ameliorate Non-Alcoholic Fatty Liver Disease in Mice. Int. J. Mol. Sci. 2022, 23, 577. https://doi.org/10.3390/ijms23010577
Huang Y-H, Wang F-S, Wang P-W, Lin H-Y, Luo S-D, Yang Y-L. Heat Shock Protein 60 Restricts Release of Mitochondrial dsRNA to Suppress Hepatic Inflammation and Ameliorate Non-Alcoholic Fatty Liver Disease in Mice. International Journal of Molecular Sciences. 2022; 23(1):577. https://doi.org/10.3390/ijms23010577
Chicago/Turabian StyleHuang, Ying-Hsien, Feng-Sheng Wang, Pei-Wen Wang, Hung-Yu Lin, Sheng-Dean Luo, and Ya-Ling Yang. 2022. "Heat Shock Protein 60 Restricts Release of Mitochondrial dsRNA to Suppress Hepatic Inflammation and Ameliorate Non-Alcoholic Fatty Liver Disease in Mice" International Journal of Molecular Sciences 23, no. 1: 577. https://doi.org/10.3390/ijms23010577