
Biological Evaluation and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives of 4,6-Dimethyl-2-sulfanylpyridine-3-carboxamide

 , ,
, ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Tests
2.2.1. Cyclooxygenase Inhibition Assay
2.2.2. MTT Assay
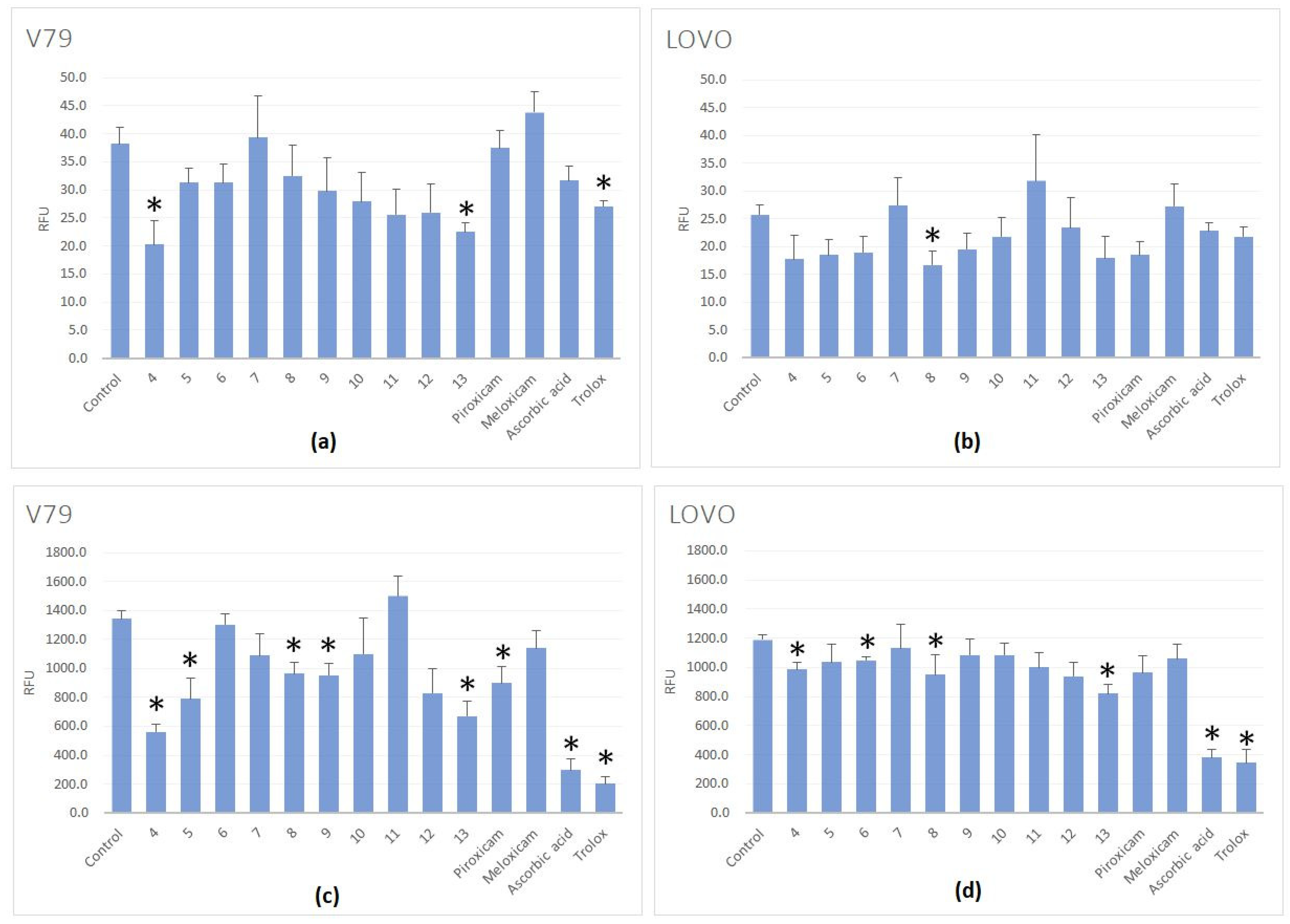
2.2.3. Evaluation of Reactive Oxygen Species (ROS) Level Inside the Cells
2.3. Static Density Functional Theory (DFT) Models and Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. Instruments and Chemicals
3.1.2. Preparation and Experimental Properties of Compounds 2–13
3.2. Biological Section
3.2.1. Cell Lines
3.2.2. Cell Culture Conditions
3.2.3. Cyclooxygenase Inhibitory Activity
3.2.4. MTT Assay
3.2.5. Estimation of Intracellular ROS Level
3.2.6. Statistics
3.3. Molecular Modeling—Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meshram, M.A.; Bhise, U.O.; Makhal, P.N.; Kaki, V.R. Synthetically-Tailored and Nature-Derived Dual COX-2/5-LOX Inhibitors: Structural Aspects and SAR. Eur. J. Med. Chem. 2021, 225, 113804. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Wiatrak, B.; Jawień, P.; Marciniak, A.; Kotynia, A.; Świątek, P. New N-Substituted-1,2,4-Triazole Derivatives of Pyrrolo[3,4-d]Pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies. Int. J. Mol. Sci. 2021, 22, 11235. [Google Scholar] [CrossRef]
- Glomb, T.; Wiatrak, B.; Gębczak, K.; Gębarowski, T.; Bodetko, D.; Czyżnikowska, Ż.; Świątek, P. New 1,3,4-Oxadiazole Derivatives of Pyridothiazine-1,1-Dioxide with Anti-Inflammatory Activity. Int. J. Mol. Sci. 2020, 21, 9122. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive Lipids, Inflammation and Chronic Diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Arulselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of Antioxidants and Natural Products in Inflammation. Oxidative Med. Cell. Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Khan, S.; Andrews, K.L.; Chin-Dusting, J.P.F. Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory? Int. J. Mol. Sci. 2019, 20, 4262. [Google Scholar] [CrossRef] [Green Version]
- Süleyman, H.; Demircan, B.; Karagöz, Y. Anti-Inflammatory and Side Effects of Cyclooxygenase Inhibitors. Pharmacol. Rep. 2007, 59, 247–258. [Google Scholar]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Organ Damage: A Current Perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Campos, D.C.O.; Costa, A.S.; Luz, P.B.; Soares, P.M.G.; Alencar, N.M.N.; Oliveira, H.D. Morinda Citrifolia Lipid Transfer Protein 1 Exhibits Anti-Inflammatory Activity by Modulation of pro- and Anti-Inflammatory Cytokines. Int. J. Biol. Macromol. 2017, 103, 1121–1129. [Google Scholar] [CrossRef]
- Wang, M.T.; Honn, K.V.; Nie, D. Cyclooxygenases, Prostanoids, and Tumor Progression. Cancer Metastasis Rev. 2007, 26, 525–534. [Google Scholar] [CrossRef]
- Setia, S.; Vaish, V.; Sanyal, S.N. Chemopreventive Effects of NSAIDs as Inhibitors of Cyclooxygenase-2 and Inducers of Apoptosis in Experimental Lung Carcinogenesis. Mol. Cell. Biochem. 2012, 366, 89–99. [Google Scholar] [CrossRef]
- Umezawa, S.; Higurashi, T.; Komiya, Y.; Arimoto, J.; Horita, N.; Kaneko, T.; Iwasaki, M.; Nakagama, H.; Nakajima, A. Chemoprevention of Colorectal Cancer: Past, Present, and Future. Cancer Sci. 2019, 110, 3018–3026. [Google Scholar] [CrossRef]
- Kajal, A.; Bala, S.; Sharma, N.; Kamboj, S.; Saini, V. Therapeutic Potential of Hydrazones as Anti-Inflammatory Agents. Int. J. Med. Chem. 2014, 2014. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; de Sena Murteira Pinheiro, P.; Lima, L.M.; Fraga, C.A.M.; Barreiro, E.J. N-Acylhydrazones as Drugs. Bioorg. Med. Chem. Lett. 2018, 28, 2797–2806. [Google Scholar] [CrossRef]
- Popiołek, Ł. Hydrazide-Hydrazones as Potential Antimicrobial Agents: Overview of the Literature since 2010. Med. Chem. Res. 2017, 26, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Nasr, T.; Bondock, S.; Rashed, H.M.; Fayad, W.; Youns, M.; Sakr, T.M. Novel Hydrazide-Hydrazone and Amide Substituted Coumarin Derivatives: Synthesis, Cytotoxicity Screening, Microarray, Radiolabeling and in Vivo Pharmacokinetic Studies. Eur. J. Med. Chem. 2018, 151, 723–739. [Google Scholar] [CrossRef]
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Datta, S.; Ghosh Dastidar, D.; Kumar, D.; Chakrabarti, G. NMK-BH2, a Novel Microtubule-Depolymerising Bis (Indolyl)-Hydrazide-Hydrazone, Induces Apoptotic and Autophagic Cell Death in Cervical Cancer Cells by Binding to Tubulin at Colchicine—Site. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118762. [Google Scholar] [CrossRef]
- Świątek, P.; Saczko, J.; Rembiałkowska, N.; Kulbacka, J. Synthesis of New Hydrazone Derivatives and Evaluation of Their Efficacy as Proliferation Inhibitors in Human Cancer Cells. Med. Chem. 2019, 15, 903–910. [Google Scholar] [CrossRef]
- Horchani, M.; Della Sala, G.; Caso, A.; D’Aria, F.; Esposito, G.; Laurenzana, I.; Giancola, C.; Costantino, V.; Jannet, H.B.; Romdhane, A. Molecular Docking and Biophysical Studies for Antiproliferative Assessment of Synthetic Pyrazolo-Pyrimidinones Tethered with Hydrazide-Hydrazones. Int. J. Mol. Sci. 2021, 22, 2742. [Google Scholar] [CrossRef]
- Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Legoabe, L.J.; Khanye, S.D. Quinolone-Isoniazid Hybrids: Synthesis and Preliminary: In Vitro Cytotoxicity and Anti-Tuberculosis Evaluation. Medchemcomm 2019, 10, 326–331. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Saudi, M.N.; Hassan, A.M.M.; Fahmy, S.M.; Ibrahim, T.M.; Ghareeb, D.; El-Seidy, A.M.; Nasralla, S.N.; Bekhit, A.E.D.A. Synthesis, in Silico Experiments and Biological Evaluation of 1,3,4-Trisubstituted Pyrazole Derivatives as Antimalarial Agents. Eur. J. Med. Chem. 2019, 163, 353–366. [Google Scholar] [CrossRef]
- Popiołek, Ł. Updated Information on Antimicrobial Activity of Hydrazide–Hydrazones. Int. J. Mol. Sci. 2021, 22, 9389. [Google Scholar] [CrossRef]
- Senkardes, S.; Kaushik-Basu, N.; Durmaz, I.; Manvar, D.; Basu, A.; Atalay, R.; Guniz Kucukguzel, S. Synthesis of Novel Diflunisal Hydrazide-Hydrazones as Anti-Hepatitis C Virus Agents and Hepatocellular Carcinoma Inhibitors. In European Journal of Medicinal Chemistry; Elsevier Masson SAS: Issy-les-Moulineaux, France, 2016; Volume 108, pp. 301–308. [Google Scholar] [CrossRef]
- Angelova, V.; Karabeliov, V.; Andreeva-Gateva, P.A.; Tchekalarova, J. Recent Developments of Hydrazide/Hydrazone Derivatives and Their Analogs as Anticonvulsant Agents in Animal Models. Drug Dev. Res. 2016, 77, 379–392. [Google Scholar] [CrossRef]
- Zeeshan, S.; Naveed, M.; Khan, A.; Atiq, A.; Arif, M.; Ahmed, M.N.; Kim, Y.S.; Khan, S. N-Pyrazoloyl and N-Thiopheneacetyl Hydrazone of Isatin Exhibited Potent Anti-Inflammatory and Anti-Nociceptive Properties through Suppression of NF-ΚB, MAPK and Oxidative Stress Signaling in Animal Models of Inflammation. Inflamm. Res. 2019, 68, 613–632. [Google Scholar] [CrossRef]
- Meira, C.S.; dos Santos Filho, J.M.; Sousa, C.C.; Anjos, P.S.; Cerqueira, J.V.; Dias Neto, H.A.; da Silveira, R.G.; Russo, H.M.; Wolfender, J.L.; Queiroz, E.F.; et al. Structural Design, Synthesis and Substituent Effect of Hydrazone-N-Acylhydrazones Reveal Potent Immunomodulatory Agents. Bioorg. Med. Chem. 2018, 26, 1971–1985. [Google Scholar] [CrossRef]
- Osmaniye, D.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis and Biological Evaluation of New N-Acyl Hydrazones with a Methyl Sulfonyl Moiety as Selective COX-2 Inhibitors. Chem. Biodivers. 2021, 18, e2100521. [Google Scholar] [CrossRef]
- De Oliveira Moraes, A.D.T.; de Miranda, M.D.S.; Jacob, Í.T.T.; da Cruz Amorim, C.A.; de Moura, R.O.; da Silva, S.Â.S.; Soares, M.B.P.; de Almeida, S.M.V.; de Lima Souza, T.R.C.; de Oliveira, J.F.; et al. Synthesis, in Vitro and in Vivo Biological Evaluation, COX-1/2 Inhibition and Molecular Docking Study of Indole-N-Acylhydrazone Derivatives. Bioorg. Med. Chem. 2018, 26, 5388–5396. [Google Scholar] [CrossRef]
- Moussa, Z.; Al-Mamary, M.; Al-Juhani, S.; Ahmed, S.A. Preparation and Biological Assessment of Some Aromatic Hydrazones Derived from Hydrazides of Phenolic Acids and Aromatic Aldehydes. Heliyon 2020, 6, e05019. [Google Scholar] [CrossRef]
- De Melo, T.R.F.; Chelucci, R.C.; Pires, M.E.L.; Dutra, L.A.; Barbieri, K.P.; Bosquesi, P.L.; Trossini, G.H.G.; Chung, M.C.; dos Santos, J.L. Pharmacological Evaluation and Preparation of Nonsteroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit. Int. J. Mol. Sci. 2014, 15, 5821–5837. [Google Scholar] [CrossRef] [Green Version]
- Mahy, J.P.; Gaspard, S.; Mansuy, D. Phenylhydrazones as New Good Substrates for the Dioxygenase and Peroxidase Reactions of Prostaglandin Synthase: Formation of Iron(III)-Sigma-Phenyl Complexes. Biochemistry 1993, 32, 4014–4021. [Google Scholar] [CrossRef]
- Rana, K.; Salahuddin; Sahu, J.K. Significance of 1,3,4-Oxadiazole Containing Compounds in New Drug Development. Curr. Drug Res. Rev. 2021, 13, 90–100. [Google Scholar] [CrossRef]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef]
- Boyd, S.A.; Fung, A.K.L.; Baker, W.R.; Mantei, R.A.; Stein, H.H.; Cohen, J.; Barlow, J.L.; Klinghofer, V.; Wessale, J.L.; Verburg, K.M.; et al. Nonpeptide Renin Inhibitors with Good Intraduodenal Bioavailability and Efficacy in Dog. J. Med. Chem. 1994, 37, 2991–3007. [Google Scholar] [CrossRef]
- Carroll, F.I.; Gray, J.L.; Abraham, P.; Kuzemko, M.A.; Lewin, A.H.; Boja, J.W.; Kuhar, M.J. 3-Aryl-2-(3′-Substituted-1′,2′,4′-Oxadiazol-5′-Yl)Tropane Analogues of Cocaine: Affinities at the Cocaine Binding Site at the Dopamine, Serotonin, and Norepinephrine Transporters. J. Med. Chem. 1993, 36, 2886–2890. [Google Scholar] [CrossRef]
- Street, L.J.; Baker, R.; Castro, J.L.; Chambers, M.S.; Guiblin, A.R.; Hobbs, S.C.; Matassa, V.G.; Reeve, A.J.; Beer, M.S.; Middlemiss, D.N.; et al. Synthesis and Serotonergic Activity of 5-(Oxadiazolyl)Tryptamines: Potent Agonists for 5-HT1D Receptors. J. Med. Chem. 1993, 36, 1529–1538. [Google Scholar] [CrossRef]
- Dunbar, P.G.; Durant, G.J.; Fang, Z.; Abuh, Y.F.; El-Assadi, A.A.; Ngur, D.O.; Periyasamy, S.; Hoss, W.P.; Messer, W.S. Design, Synthesis, and Neurochemical Evaluation of 5-(3-Alkyl-1,2,4-Oxadiazol-5-Yl)-1,4,5,6-Tetrahydropyrimidines as M1 Muscarinic Receptor Agonists. J. Med. Chem. 1993, 36, 842–847. [Google Scholar] [CrossRef]
- Adelstein, G.W.; Yen, C.H.; Dajani, E.Z.; Bianchi, R.G. 3,3-Diphenyl-3-(2-Alkyl-1,3,4-Oxadiazol-5-Yl)Propylcycloalkylamines, a Novel Series of Antidiarrheal Agents. J. Med. Chem. 1976, 19, 1221–1225. [Google Scholar] [CrossRef]
- Tully, W.R.; Gardner, C.R.; Gillespie, R.J.; Westwood, R. 2-(Oxadiazolyl)- and 2-(Thiazolyl)Imidazo[1,2-a]Pyrimidines as Agonists and Inverse Agonists at Benzodiazepine Receptors. J. Med. Chem. 1991, 34, 2060–2067. [Google Scholar] [CrossRef]
- Orlek, B.S.; Blaney, F.E.; Brown, F.; Clark, M.S.G.; Hadley, M.S.; Hatcher, J.; Riley, G.J.; Rosenberg, H.E.; Wadsworth, H.J.; Wyman, P. Comparison of Azabicyclic Esters and Oxadiazoles as Ligands for the Muscarinic Receptor. J. Med. Chem. 1991, 34, 2726–2735. [Google Scholar] [CrossRef]
- EL Mansouri, A.E.; Oubella, A.; Mehdi, A.; AitItto, M.Y.; Zahouily, M.; Morjani, H.; Lazrek, H.B. Design, Synthesis, Biological Evaluation and Molecular Docking of New 1,3,4-Oxadiazole Homonucleosides and Their Double-Headed Analogs as Antitumor Agents. Bioorg. Chem. 2021, 108, 104558. [Google Scholar] [CrossRef]
- Ananth, A.H.; Manikandan, N.; Rajan, R.K.; Elancheran, R.; Lakshmithendral, K.; Ramanathan, M.; Bhattacharjee, A.; Kabilan, S. Design, Synthesis, and Biological Evaluation of 2-(2-Bromo-3-nitrophenyl)-5-phenyl-1,3,4-oxadiazole Derivatives as Possible Anti-Breast Cancer Agents. Chem. Biodivers. 2020, 17, e1900659. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Mohamed, A.A.B.; Tawfik, S.S.; Hassan, H.M.; El-Emam, A.A. 1,3,4-Oxadiazole N-Mannich Bases: Synthesis, Antimicrobial, and Anti-Proliferative Activities. Molecules 2021, 26, 2110. [Google Scholar] [CrossRef]
- Bordei Telehoiu, A.T.; Nuță, D.C.; Căproiu, M.T.; Dumitrascu, F.; Zarafu, I.; Ioniță, P.; Bădiceanu, C.D.; Avram, S.; Chifiriuc, M.C.; Bleotu, C.; et al. Design, Synthesis and In Vitro Characterization of Novel Antimicrobial Agents Based on 6-Chloro-9H-Carbazol Derivatives and 1,3,4-Oxadiazole Scaffolds. Molecules 2020, 25, 266. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.L.; Zhu, Y.; Liu, J.R.; Guo, S.K.; Gu, Y.C.; Han, X.; Dong, H.Q.; Sun, Q.; Zhang, W.H.; Zhang, M.Z. Diversity-Oriented Synthesis and Antifungal Activities of Novel Pimprinine Derivative Bearing a 1,3,4-Oxadiazole-5-Thioether Moiety. Mol. Divers. 2020, 25, 205–221. [Google Scholar] [CrossRef]
- Glomb, T.; Świątek, P. Antimicrobial Activity of 1,3,4-Oxadiazole Derivatives. Int. J. Mol. Sci. 2021, 22, 6979. [Google Scholar] [CrossRef]
- Tantray, M.A.; Khan, I.; Hamid, H.; Alam, M.S.; Dhulap, A.; Kalam, A. Synthesis of Benzimidazole-Linked-1,3,4-Oxadiazole Carboxamides as GSK-3β Inhibitors with in Vivo Antidepressant Activity. Bioorg. Chem. 2018, 77, 393–401. [Google Scholar] [CrossRef]
- Khokra, S.L.; Kaur, S.; Banwala, S.; Wadhwa, K.; Husain, A. Synthesis, Molecular Docking, and Biological Evaluation of Some Novel 2-(5-Substituted 1,3,4-Oxadiazole-2-Yl)-1,3-Benzothiazole Derivatives as Anticonvulsant Agents. Cent. Nerv. Syst. Agents Med. Chem. 2021, 21, 130–141. [Google Scholar] [CrossRef]
- Kaur, J.; Soto-Velasquez, M.; Ding, Z.; Ghanbarpour, A.; Lill, M.A.; van Rijn, R.M.; Watts, V.J.; Flaherty, D.P. Optimization of a 1,3,4-Oxadiazole Series for Inhibition of Ca2+/Calmodulin-Stimulated Activity of Adenylyl Cyclases 1 and 8 for the Treatment of Chronic Pain. Eur. J. Med. Chem. 2019, 162, 568–585. [Google Scholar] [CrossRef]
- Dewangan, D.; Nakhate, K.T.; Verma, V.S.; Nagori, K.; Badwaik, H.; Nair, N.; Tripathi, D.K.; Mishra, A. Synthesis and Molecular Docking Study of Novel Hybrids of 1,3,4-Oxadiazoles and Quinoxaline as a Potential Analgesic and Anti-Inflammatory Agents. J. Heterocycl. Chem. 2018, 55, 2901–2910. [Google Scholar] [CrossRef]
- Gulnaz, A.R.; Mohammed, Y.H.E.; Khanum, S.A. Design, Synthesis and Molecular Docking of Benzophenone Conjugated with Oxadiazole Sulphur Bridge Pyrazole Pharmacophores as Anti Inflammatory and Analgesic Agents. Bioorg. Chem. 2019, 92, 103220. [Google Scholar] [CrossRef]
- Puttaswamy, N.; Malojiao, V.H.; Mohammed, Y.H.E.; Sherapura, A.; Prabhakar, B.T.; Khanum, S.A. Synthesis and Amelioration of Inflammatory Paw Edema by Novel Benzophenone Appended Oxadiazole Derivatives by Exhibiting Cyclooxygenase-2 Antagonist Activity. Biomed. Pharmacother. 2018, 103, 1446–1455. [Google Scholar] [CrossRef]
- Kashid, B.B.; Salunkhe, P.H.; Dongare, B.B.; More, K.R.; Khedkar, V.M.; Ghanwat, A.A. Synthesis of Novel of 2,5-Disubstituted 1, 3, 4- Oxadiazole Derivatives and Their in Vitro Anti-Inflammatory, Anti-Oxidant Evaluation, and Molecular Docking Study. Bioorg. Med. Chem. Lett. 2020, 30, 127136. [Google Scholar] [CrossRef]
- Świątek, P.; Gębczak, K.; Gębarowski, T.; Urniaz, R. Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives. Molecules 2019, 24, 1093. [Google Scholar] [CrossRef] [Green Version]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 Inhibition Activities and Molecular Docking Study of Isothiazolopyridine Derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef]
- Vicini, P.; Incerti, M.; Doytchinova, I.A.; La Colla, P.; Busonera, B.; Loddo, R. Synthesis and Antiproliferative Activity of Benzo[d]Isothiazole Hydrazones. Eur. J. Med. Chem. 2006, 41, 624–632. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational Behaviour and E/Z Isomerization of N-Acyl and N-Aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Wyrzykiewicz, E.; Prukała, D. New Isomeric N -Substituted Hydrazones of 2-, 3-and 4-Pyridinecarboxaldehydes. J. Heterocycl. Chem. 1998, 35, 381–387. [Google Scholar] [CrossRef]
- Galić, N.; Perić, B.; Kojić-Prodić, B.; Cimerman, Z. Structural and Spectroscopic Characteristics of Aroylhydrazones Derived from Nicotinic Acid Hydrazide. J. Mol. Struct. 2001, 559, 187–194. [Google Scholar] [CrossRef]
- Tesei, A.; Ulivi, P.; Fabbri, F.; Rosetti, M.; Leonetti, C.; Scarsella, M.; Zupi, G.; Amadori, D.; Bolla, M.; Zoli, W. In Vitro and In Vivo Evaluation of NCX 4040 Cytotoxic Activity in Human Colon Cancer Cell Lines. J. Transl. Med. 2005, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.Y.; Xiao, W.; Zhu, M.X.; Pan, X.J.; Yang, Z.H.; Zhou, S.Y. Inhibition of Cyclooxygenase-2 by Tetramethylpyrazine and Its Effects on A549 Cell Invasion and Metastasis. Int. J. Oncol. 2012, 40, 2029–2037. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.X.; Du, Y.Y.; Zhang, Y.; Pan, Y.Y. Synergistic Effects of Exemestane and Aspirin on MCF-7 Human Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2012, 13, 5903–5908. [Google Scholar] [CrossRef] [Green Version]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. In Cancer Cell Culture. Methods in Molecular Biology (Methods and Protocols); Cree, I.A., Ed.; Humana Press: Clifton, NJ, USA, 2011; Volume 731, pp. 237–245. [Google Scholar] [CrossRef]
- A549 Cell Line: Cell Culture and Transfection Protocol. Available online: https://www.a549.com/ (accessed on 25 November 2021).
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic Inflammation and Oxidative Stress in Human Carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Ferrari, G.V.; Natera, J.; Paulina Montaña, M.; Muñoz, V.; Gutiérrez, E.L.; Massad, W.; Miskoski, S.; García, N.A. Scavenging of Photogenerated ROS by Oxicams. Possible Biological and Environmental Implications. J. Photochem. Photobiol. B Biol. 2015, 153, 233–239. [Google Scholar] [CrossRef]
- Van Antwerpen, P.; Nève, J. In Vitro Comparative Assessment of the Scavenging Activity against Three Reactive Oxygen Species of Non-Steroidal Anti-Inflammatory Drugs from the Oxicam and Sulfoanilide Families. Eur. J. Pharmacol. 2004, 496, 55–61. [Google Scholar] [CrossRef]
- García, N.A.; Bregliani, M.; Pajares, A. Interaction of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) with Reactive Oxygen Species (ROS): Possible Biomedical Implications. In Pain Relief—From Analgesics to Alternative Therapies; Maldonado, C., Ed.; InTechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.H.; Lee, I.S. Cytoprotective Effect of Artemisia Capillaris Fractions on Oxidative Stress-Induced Apoptosis in V79 Cells. BioFactors 2009, 35, 380–388. [Google Scholar] [CrossRef]
- Chubatsu, L.S.; Gennari, M.; Meneghini, R. Glutathione Is the Antioxidant Responsible for Resistance to Oxidative Stress in V79 Chinese Hamster Fibroblasts Rendered Resistant to Cadmium. Chem. Biol. Interact. 1992, 82, 99–110. [Google Scholar] [CrossRef]
- Boojar, M.M.A.; Shockravi, A. On the Cytotoxicity and Status of Oxidative Stress of Two Novel Synthesized Tri-Aza Macrocyclic Diamides as Studied in the V79 Cell Lines. Bioorg. Med. Chem. 2007, 15, 3437–3444. [Google Scholar] [CrossRef]
- Speit, G.; Haupter, S.; Schütz, P.; Kreis, P. Comparative Evaluation of the Genotoxic Properties of Potassium Bromate and Potassium Superoxide in V79 Chinese Hamster Cells. Mutat. Res.-Genet. Toxicol. Environ. Mutagen. 1999, 439, 213–221. [Google Scholar] [CrossRef]
- Szczȩśniak-Siega, B.; Gȩbczak, K.; Gȩbarowski, T.; Maniewska, J. Synthesis, COX-1/2 Inhibition and Antioxidant Activities of New Oxicam Analogues Designed as Potential Chemopreventive Agents. Acta Biochim. Pol. 2018, 65, 199–207. [Google Scholar] [CrossRef]
- Miciaccia, M.; Belviso, B.D.; Iaselli, M.; Cingolani, G.; Ferorelli, S.; Cappellari, M.; Loguercio Polosa, P.; Perrone, M.G.; Caliandro, R.; Scilimati, A. Three-Dimensional Structure of Human Cyclooxygenase (HCOX)-1. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site via a Two-Water-Mediated h-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [Green Version]
- Orlando, B.J.; Malkowski, M.G. Crystal Structure of Rofecoxib Bound to Human Cyclooxygenase-2. Acta Crystallogr. Sect. Struct. Biol. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef] [Green Version]
- DSV: Discover Studio Visualiser, V21.1.0.20298; BIOVIA, Dassault Systèmes: San Diego, SA, USA, 2020.
- Eruslanov, E.; Kusmartsev, S. Identification of ROS Using Oxidized DCFDA and Flow-Cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Loo, A.E.K.; Halliwell, B. Effects of Hydrogen Peroxide in a Keratinocyte-Fibroblast Co-Culture Model of Wound Healing. Biochem. Biophys. Res. Commun. 2012, 423, 253–258. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A Pre- and Post-Processing Program for Molecular and Electronic Structures. J. Comput. Aided. Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 16 Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1697. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- PyMOL: The PyMOL Molecular Graphics System, Version 2.3.0; Schrödinger LLC.: New York, NY, USA, 2020.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] ± SD | COX-2/COX-1 Selectivity Ratio | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| 4 | 135.20 ± 5.99 | NA | - |
| 5 | 73.91 ± 3.03 | 186.95 ± 15.25 | 2.53 |
| 6 | 144.79 ± 20.88 | 173.93 ± 5.84 | 1.20 |
| 7 | 147.55 ± 19.81 | 184.43 ± 18.84 | 1.25 |
| 8 | 156.65 ± 12.12 | NA | - |
| 9 | 153.89 ± 6.29 | 102.58 ± 10.61 | 0.67 |
| 10 | NA | 127.54 ± 1.87 | - |
| 11 | 86.19 ± 2.60 | 169.75 ± 19.14 | 1.97 |
| 12 | 66.84 ± 5.19 | 90.71 ± 19.61 | 1.36 |
| 13 | 84.37 ± 5.89 | 73.23 ± 5.66 | 0.80 |
| Piroxicam | 87.44 ± 4.17 | 80.11 ± 1.26 | 0.92 |
| Meloxicam | 128.76 ± 4.72 | 76.38 ± 6.46 | 0.59 |
| Compound | IC50 [µM] ± SD | ||||||
|---|---|---|---|---|---|---|---|
| NHDF | VERO | V79 | A549 | MCF-7 | LoVo | LoVo/Dx | |
| 4 | 57.46 ± 1.22 | 77.44 ± 5.84 | 71.70 ± 5.80 | 3.84 ± 0.30 | 9.63 ± 1.66 | 3.01 ± 0.20 | 5.73 ± 3.84 |
| 5 | 58.97 ± 1.49 | 100.1 ± 13.49 | 84.30 ± 3.40 | 3.62 ± 0.43 | 6.37 ± 1.09 | 3.13 ± 0.17 | 5.69 ± 1.03 |
| 6 | 58.21 ± 0.92 | 59.62 ± 5.42 | 80.70 ± 5.60 | 4.05 ± 0.26 | 6.37 ± 0.19 | 3.20 ± 0.26 | 7.07 ± 2.48 |
| 7 | 58.23 ± 2.00 | 68.31 ± 3.24 | 47.70 ± 1.73 | 3.72 ± 0.54 | 6.33 ± 0.56 | 3.46 ± 0.39 | 7.89 ± 1.78 |
| 8 | 58.69 ± 1.94 | 50.38 ± 2.45 | 71.10 ± 2.40 | 4.34 ± 0.56 | 9.41 ± 4.80 | 3.60 ± 0.55 | 21.00 ± 5.92 |
| 9 | 59.44 ± 1.90 | 89.76 ± 8.43 | 77.70 ± 4.41 | 3.36 ± 0.20 | 8.22 ± 5.98 | 3.24 ± 0.10 | 6.87 ± 1.45 |
| 10 | 59.90 ± 2.04 | 129.8 ± 9.50 | 56.10 ± 5.42 | 3.52 ± 0.19 | 6.19 ± 0.19 | 3.12 ± 0.23 | 22.31 ± 2.10 |
| 11 | 58.99 ± 3.62 | 104.5 ± 4.98 | 55.20 ± 2.75 | 3.72 ± 0.24 | 8.29 ± 4.45 | 3.12 ± 0.31 | 17.50 ± 8.73 |
| 12 | 58.73 ± 2.21 | 79.2 ± 14.44 | 52.80 ± 7.20 | 3.88 ± 0.50 | 9.04 ± 2.84 | 3.07 ± 0.21 | 22.56 ± 8.19 |
| 13 | 59.26 ± 3.43 | 55.66 ± 1.84 | 65.40 ± 4.04 | 3.16 ± 0.21 | 7.23 ± 0.49 | 2.89 ± 0.08 | 9.40 ± 5.97 |
| Piroxicam | 162.23 ± 22.85 | NA | 213.10 ± 23.25 | 110.12 ± 28.23 | NA | 122.16 ± 10.20 | 125.50 ± 11.23 |
| Meloxicam | 195.66 ± 35.22 | NA | 200.80 ± 17.80 | 148.30 ± 27.58 | NA | 129.56 ± 8.80 | 142.30 ± 9.46 |
| Compound | Therapeutic Index | |||
|---|---|---|---|---|
| NHDF/A549 | NHDF/MCF-7 | NHDF/LoVo | NHDF/LoVo/Dx | |
| 4 | 14.40 ± 0.70 a | 6.16 ± 0.98 a | 18.26 ± 1.20 a | 8.97 ± 3.21 |
| 5 | 15.82 ± 1.78 a | 9.29 ± 1.29 a | 16.93 ± 1.34 a | 7.60 ± 1.50 a |
| 6 | 13.64 ± 1.32 a | 9.21 ± 0.03 a | 16.51 ± 2.01 a | 2.94 ± 0.77 a |
| 7 | 17.71 ± 0.49 a | 9.42 ± 0.54 a | 18.35 ± 0.02 a | 8.88 ± 1.63 a |
| 8 | 17.03 ± 0.34 a | 7.76 ± 4.27 | 19.24 ± 0.77 a | 2.70 ± 0.16 a |
| 9 | 15.86 ± 0.05 a | 12.10 ± 5.01 | 18.96 ± 0.73 a | 4.02 ± 2.04 |
| 10 | 15.26 ± 1.41 a | 9.49 ± 0.07 a | 19.16 ± 0.59 a | 2.84 ± 1.00 |
| 11 | 18.76 ± 0.16 a | 8.87 ± 5.03 | 20.49 ± 0.62 a | 8.89 ± 3.50 |
| 12 | 1.50 ± 0.18 a | 18.67 ± 3.51 a | 1.32 ± 0.07 a | 1.29 ± 0.07 a |
| 13 | 1.32 ± 0.01 a | 26.92 ± 3.05 a | 1.50 ± 0.17 a | 1.37 ± 0.16 |
| Piroxicam | 15.01 ± 0.86 a | - | 19.13 ± 0.87 a | 15.31 ± 6.67 |
| Meloxicam | 16.41 ± 1.55 a | - | 18.86 ± 0.55 a | 10.57 ± 1.68 a |
| Compound | without H2O2 | with H2O2 |
|---|---|---|
| Mean E/E0 ± SD | ||
| 4 | 0.53 ± 0.22 a | 0.42 ± 0.08 a |
| 5 | 0.82 ± 0.13 | 0.59 ± 0.21 a |
| 6 | 0.82 ± 0.17 | 0.97 ± 0.11 |
| 7 | 1.03 ± 0.39 | 0.81 ± 0.23 |
| 8 | 0.85 ± 0.29 | 0.72 ± 0.11 a |
| 9 | 0.78 ± 0.31 | 0.71 ± 0.12 a |
| 10 | 0.73 ± 0.27 | 0.82 ± 0.37 |
| 11 | 0.67 ± 0.24 | 1.12 ± 0.20 |
| 12 | 0.68 ± 0.27 | 0.62 ± 0.25 |
| 13 | 0.59 ± 0.08 a | 0.50 ± 0.15 a |
| Piroxicam | 0.98 ± 0.17 | 0.67 ± 0.17 a |
| Meloxicam | 1.15 ± 0.19 | 0.85 ± 0.18 |
| Ascorbic acid | 0.83 ± 0.13 | 0.22 ± 0.12 a |
| Trolox | 0.71 ± 0.05 a | 0.15 ± 0.08 a |
| Compound | without H2O2 | with H2O2 |
|---|---|---|
| Mean E/E0 ± SD | ||
| 4 | 0.69 ± 0.33 | 0.83 ± 0.08 a |
| 5 | 0.72 ± 0.21 | 0.87 ± 0.21 |
| 6 | 0.74 ± 0.22 | 0.88 ± 0.04 a |
| 7 | 1.07 ± 0.38 | 0.95 ± 0.27 |
| 8 | 0.65 ± 0.20 a | 0.80 ± 0.22 |
| 9 | 0.76 ± 0.22 | 0.91 ± 0.19 |
| 10 | 0.85 ± 0.27 | 0.91 ± 0.14 |
| 11 | 1.24 ± 0.65 | 0.84 ± 0.17 |
| 12 | 0.91 ± 0.43 | 0.79 ± 0.16 |
| 13 | 0.70 ± 0.30 | 0.69 ± 0.10a |
| Piroxicam | 0.72 ± 0.19 | 0.81 ± 0.19 |
| Meloxicam | 1.06 ± 0.32 | 0.89 ± 0.16 |
| Ascorbic acid | 0.89 ± 0.11 | 0.32 ± 0.09 a |
| Trolox | 0.85 ± 0.13 | 0.29 ± 0.15 a |
| COX-1 [75] | |||
|---|---|---|---|
| Compound | Affinity Energy [kcal/mol] | Compound | Affinity Energy [kcal/mol] |
| 4(E) | −6.7 | 4(S) | −6.0 |
| 5(E) | −7.5 | 5(S) | −8.2 |
| 6(E) | −7.0 | 6(S) | −8.0 |
| 7(E) | −7.0 | 7(S) | −8.2 |
| 8(E) | −7.6 | 8(S) | −7.8 |
| 9(E) | −6.8 | 9(S) | −8.3 |
| 10(E) | −7.8 | 10(S) | −7.9 |
| 11(E) | −8.7 | 11(S) | −8.5 |
| 12(E) | −7.0 | 12(S) | −8.2 |
| 13(E) | −8.3 | 13(S) | −8.3 |
| RCX/MXM | −8.1/−6.4 | Piroxicam | −7.5 |
| COX-1 [76] | |||
|---|---|---|---|
| Compound | Affinity Energy [kcal/mol] | Compound | Affinity Energy [kcal/mol] |
| 4(E) | −8.4 | 4(S) | −8.0 |
| 5(E) | −10.1 | 5(S) | −10.2 |
| 6(E) | −10.2 | 6(S) | −10.4 |
| 7(E) | −10.7 | 7(S) | −10.6 |
| 8(E) | −9.8 | 8(S) | −9.5 |
| 9(E) | −10.4 | 9(S) | −10.7 |
| 10(E) | −8.6 | 10(S) | −9.5 |
| 11(E) | −9.8 | 11(S) | −10.3 |
| 12(E) | −7.9 | 12(S) | −8.3 |
| 13(E) | −8.7 | 13(S) | −8.3 |
| RCX/MXM | −9.5/−9.1 | Piroxicam | −9.7 |
| COX-2 [76] | |||
|---|---|---|---|
| Compound | Affinity Energy [kcal/mol] | Compound | Affinity Energy [kcal/mol] |
| 4(E) | −8.8 | 4(S) | −9.0 |
| 5(E) | −10.0 | 5(S) | −10.1 |
| 6(E) | −10.6 | 6(S) | −10.4 |
| 7(E) | −10.7 | 7(S) | −10.6 |
| 8(E) | −10.1 | 8(S) | −10.1 |
| 9(E) | −10.6 | 9(S) | −10.9 |
| 10(E) | −10.3 | 10(S) | −10.8 |
| 11(E) | −11.5 | 11(S) | −10.8 |
| 12(E) | −10.2 | 12(S) | −10.6 |
| 13(E) | −10.4 | 13(S) | −10.6 |
| RCX/MXM | −10.3/−10.0 | Piroxicam | −10.4 |
| COX-2 [77] | |||
|---|---|---|---|
| Compound | Affinity Energy [kcal/mol] | Compound | Affinity Energy [kcal/mol] |
| 4(E) | −9.3 | 4(S) | −9.4 |
| 5(E) | −10.8 | 5(S) | −9.4 |
| 6(E) | −9.8 | 6(S) | −10.2 |
| 7(E) | −10.0 | 7(S) | −9.5 |
| 8(E) | −9.6 | 8(S) | −9.1 |
| 9(E) | −10.1 | 9(S) | −9.6 |
| 10(E) | −10.0 | 10(S) | −9.4 |
| 11(E) | −11.4 | 11(S) | −10.2 |
| 12(E) | −10.2 | 12(S) | −9.7 |
| 13(E) | −10.8 | 13(S) | −10.9 |
| RCX/MXM | −10.9/−9.9 | Piroxicam | −9.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świątek, P.; Glomb, T.; Dobosz, A.; Gębarowski, T.; Wojtkowiak, K.; Jezierska, A.; Panek, J.J.; Świątek, M.; Strzelecka, M. Biological Evaluation and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives of 4,6-Dimethyl-2-sulfanylpyridine-3-carboxamide. Int. J. Mol. Sci. 2022, 23, 549. https://doi.org/10.3390/ijms23010549
Świątek P, Glomb T, Dobosz A, Gębarowski T, Wojtkowiak K, Jezierska A, Panek JJ, Świątek M, Strzelecka M. Biological Evaluation and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives of 4,6-Dimethyl-2-sulfanylpyridine-3-carboxamide. International Journal of Molecular Sciences. 2022; 23(1):549. https://doi.org/10.3390/ijms23010549
Chicago/Turabian StyleŚwiątek, Piotr, Teresa Glomb, Agnieszka Dobosz, Tomasz Gębarowski, Kamil Wojtkowiak, Aneta Jezierska, Jarosław J. Panek, Małgorzata Świątek, and Małgorzata Strzelecka. 2022. "Biological Evaluation and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives of 4,6-Dimethyl-2-sulfanylpyridine-3-carboxamide" International Journal of Molecular Sciences 23, no. 1: 549. https://doi.org/10.3390/ijms23010549