
Activated Protein C Protects against Murine Contact Dermatitis by Suppressing Protease-Activated Receptor 2

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
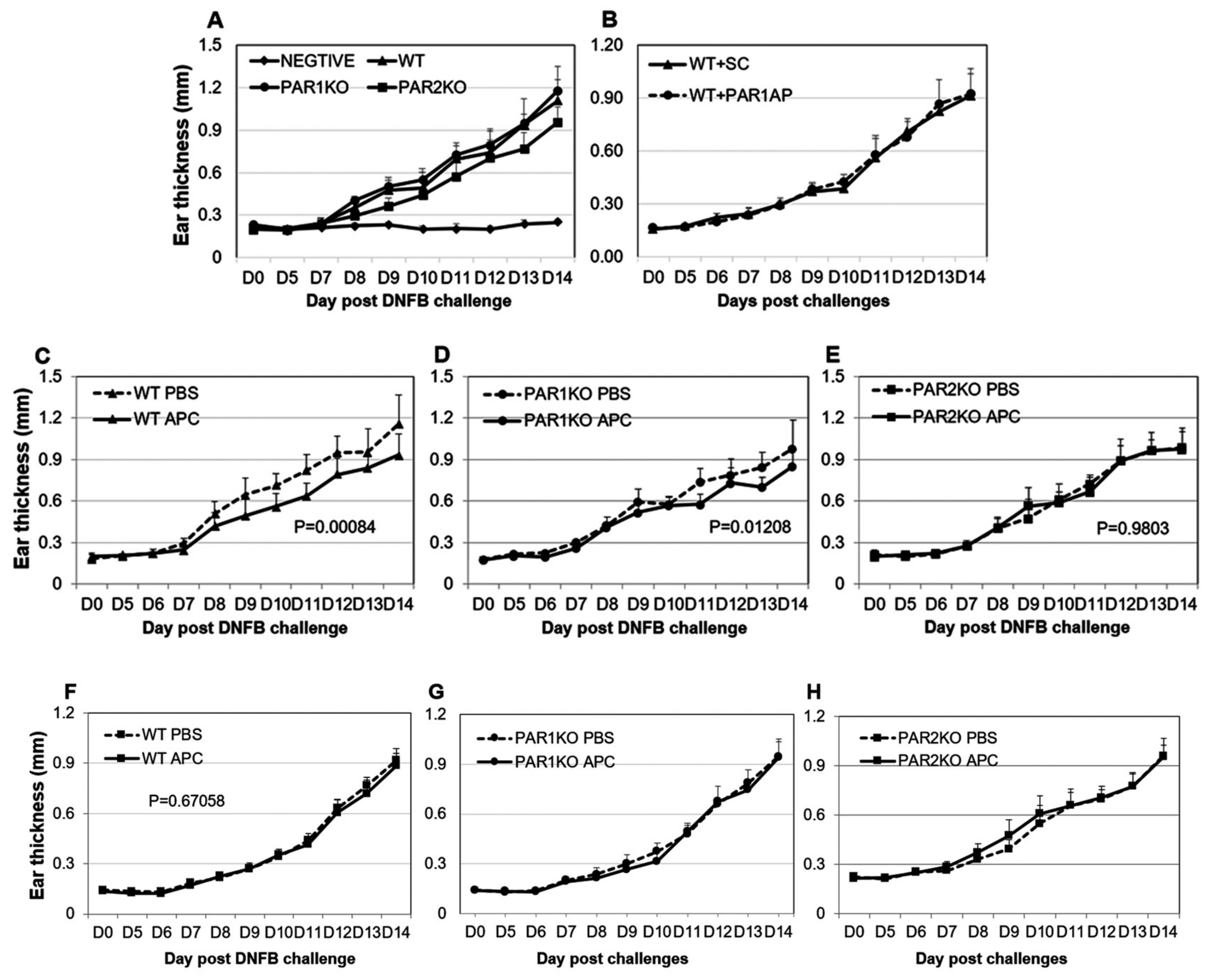
2.1. PAR2KO Mice Display Less Severe CHS and APC Mitigates CHS in WT and PAR1KO but Not PAR2KO Mice
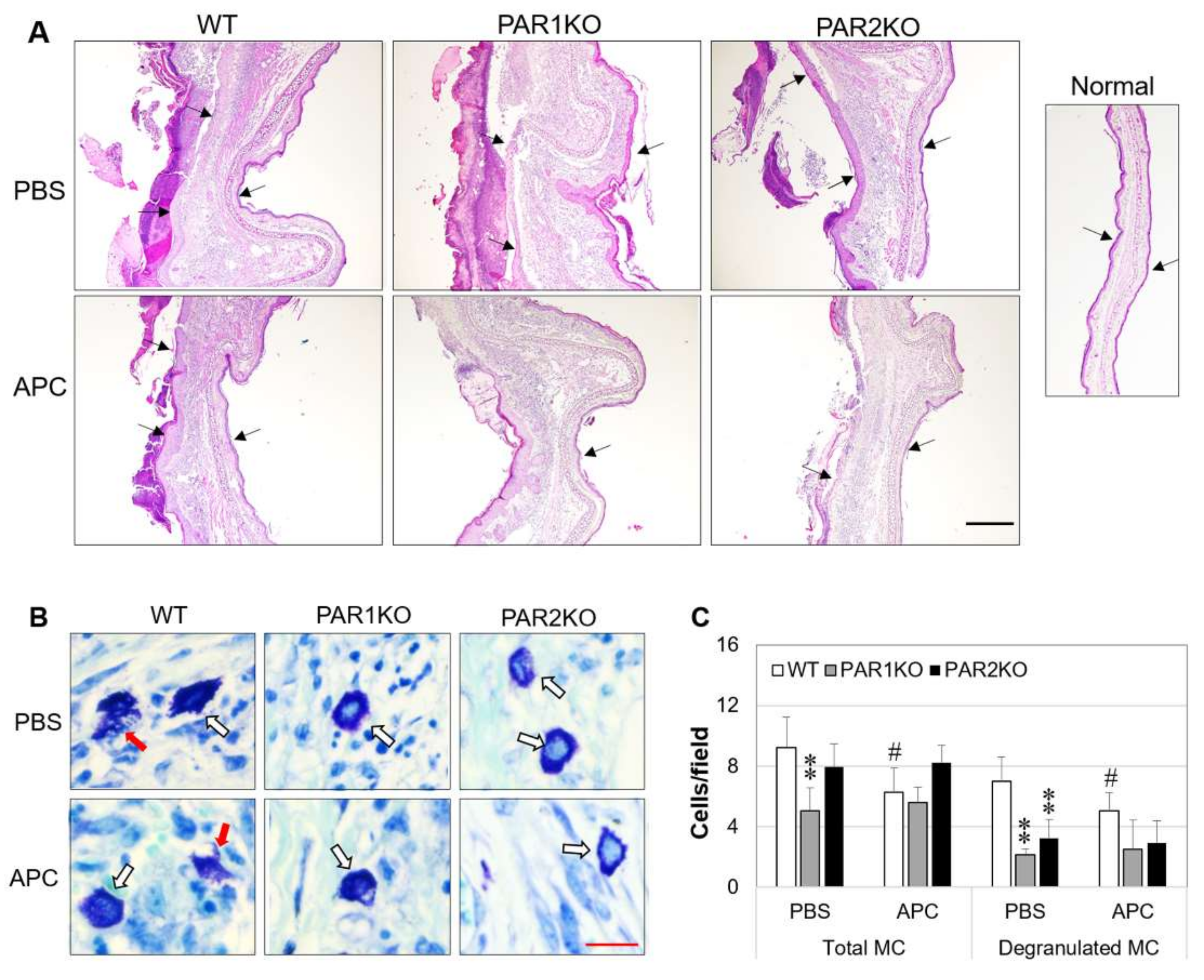
2.2. Effect of APC on Histopathological Features of CHS in Mice
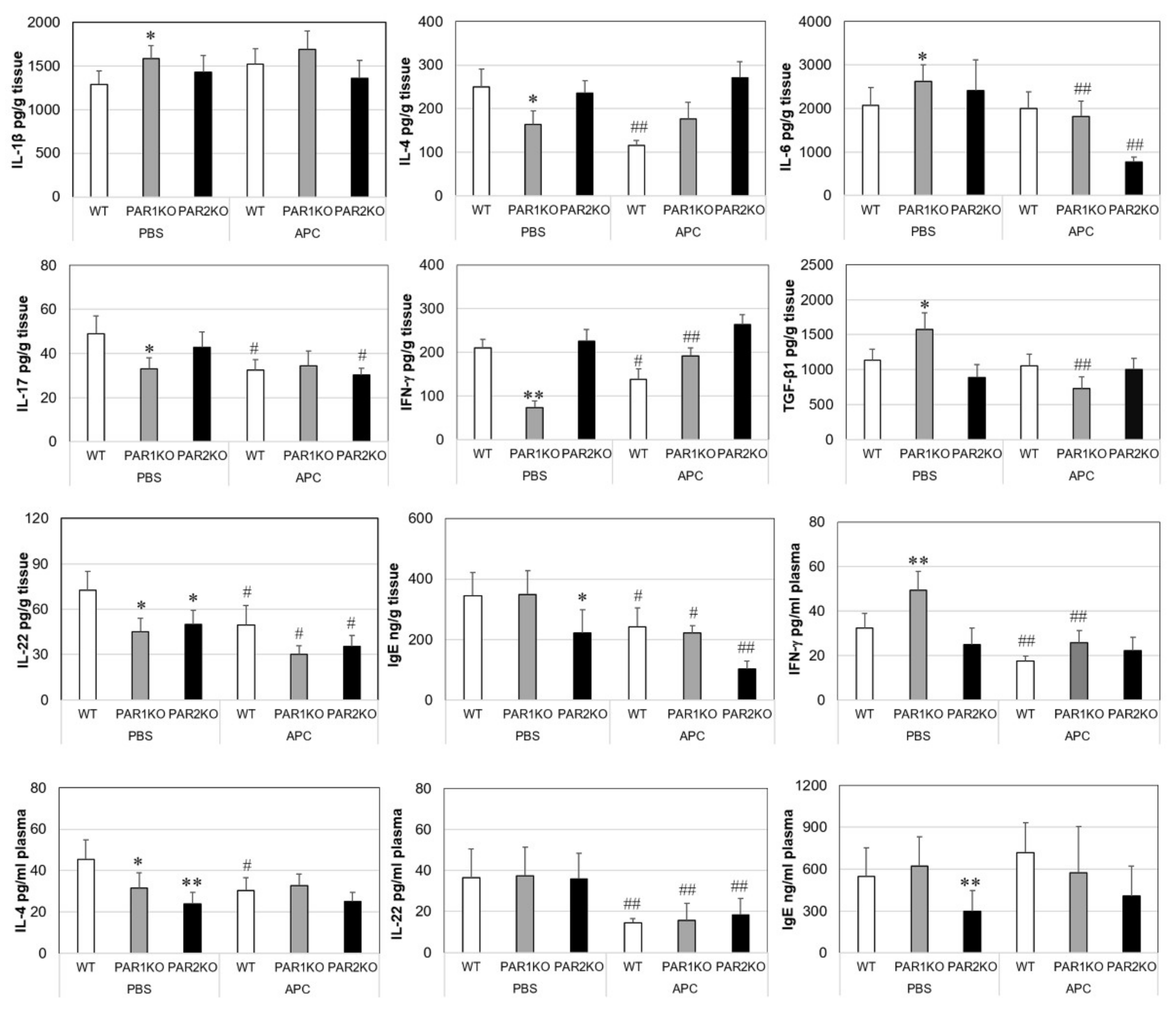
2.3. Effects of APC on Inflammatory Mediators in Mice with CHS
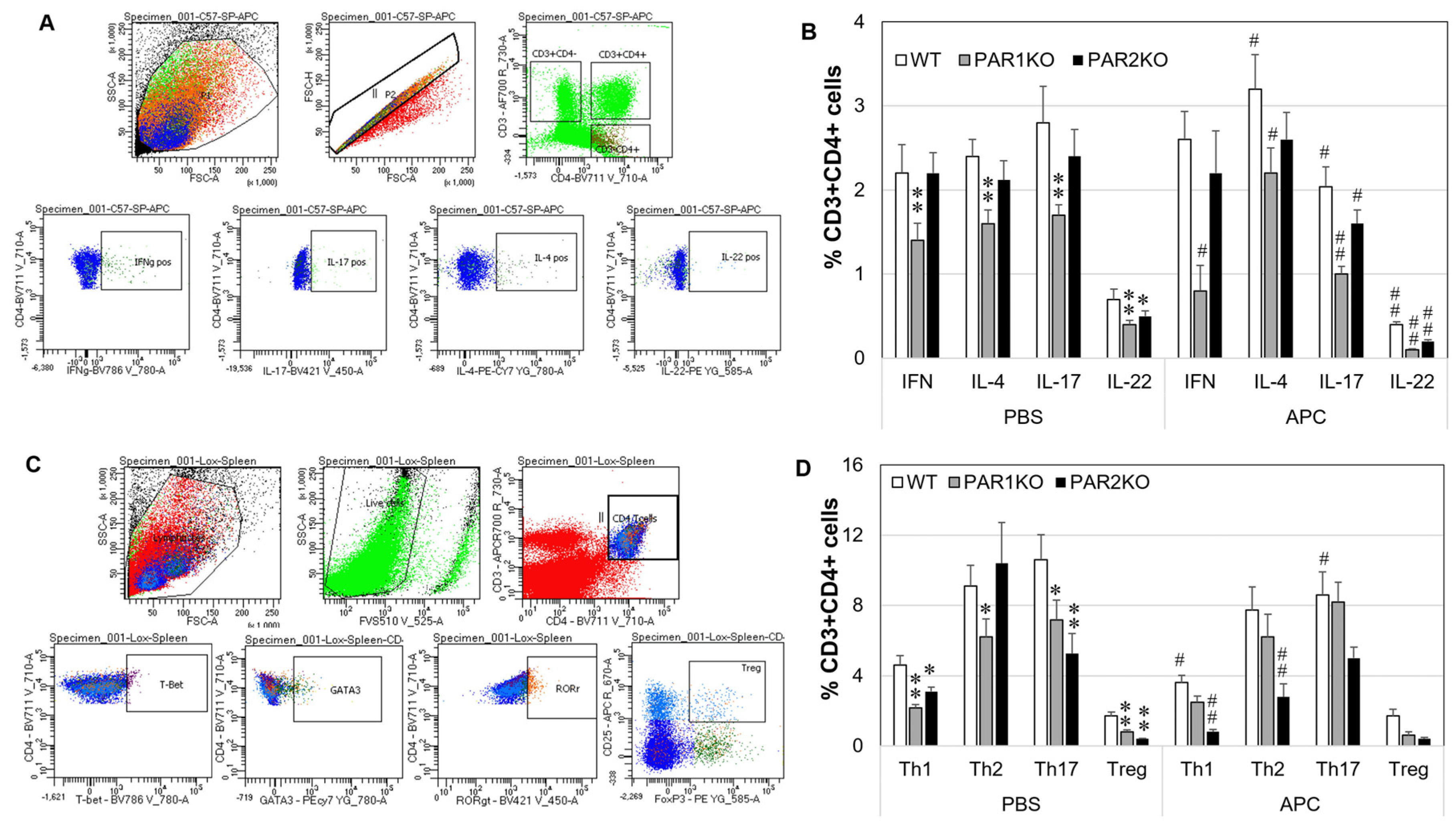
2.4. Effect of APC on Th1/Th2/Th17/Th22/Treg Phenotypes in Mouse Spleen Cells In Vitro
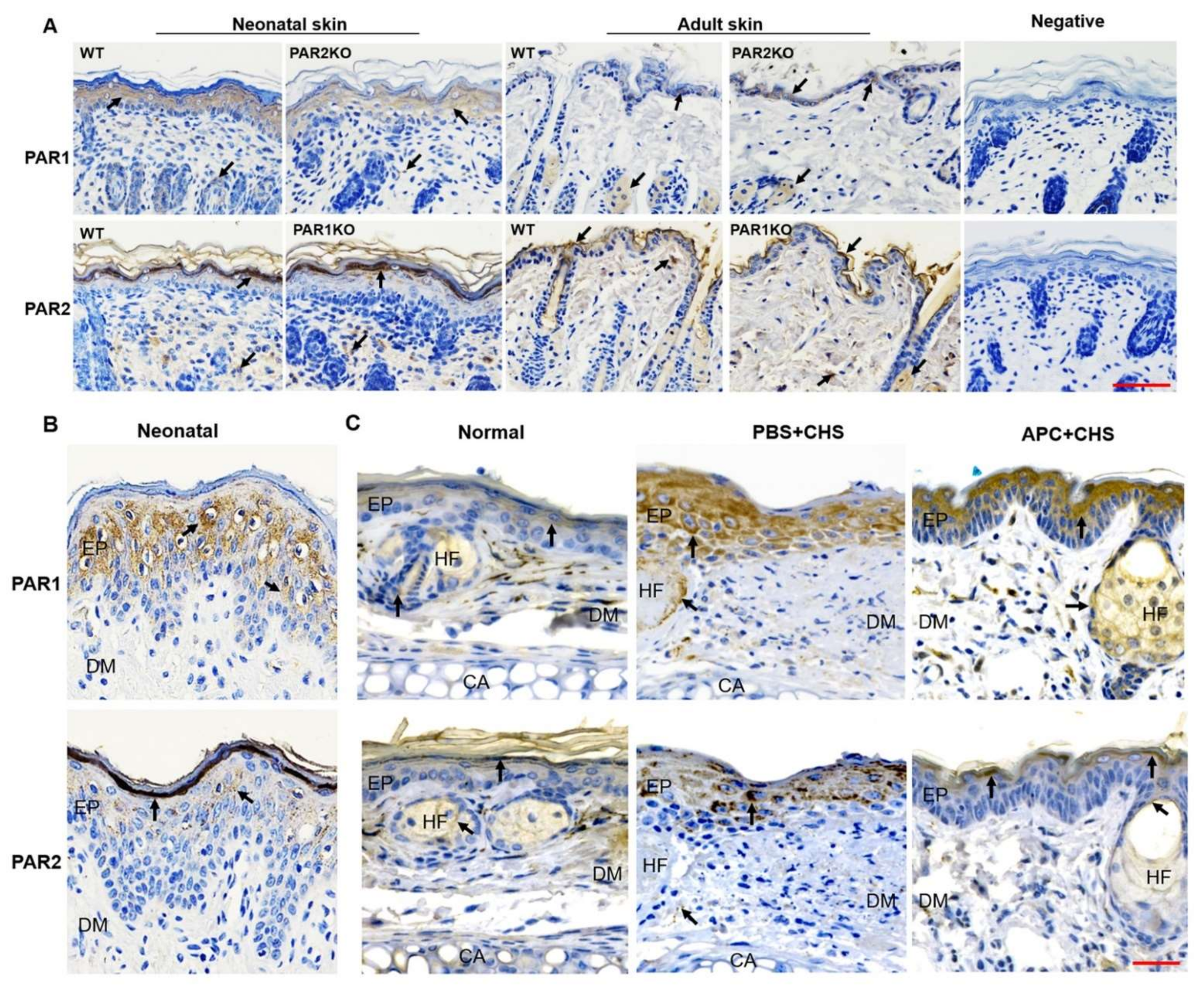
2.5. The Expression of PAR1 and PAR2 in Skin Epidermis
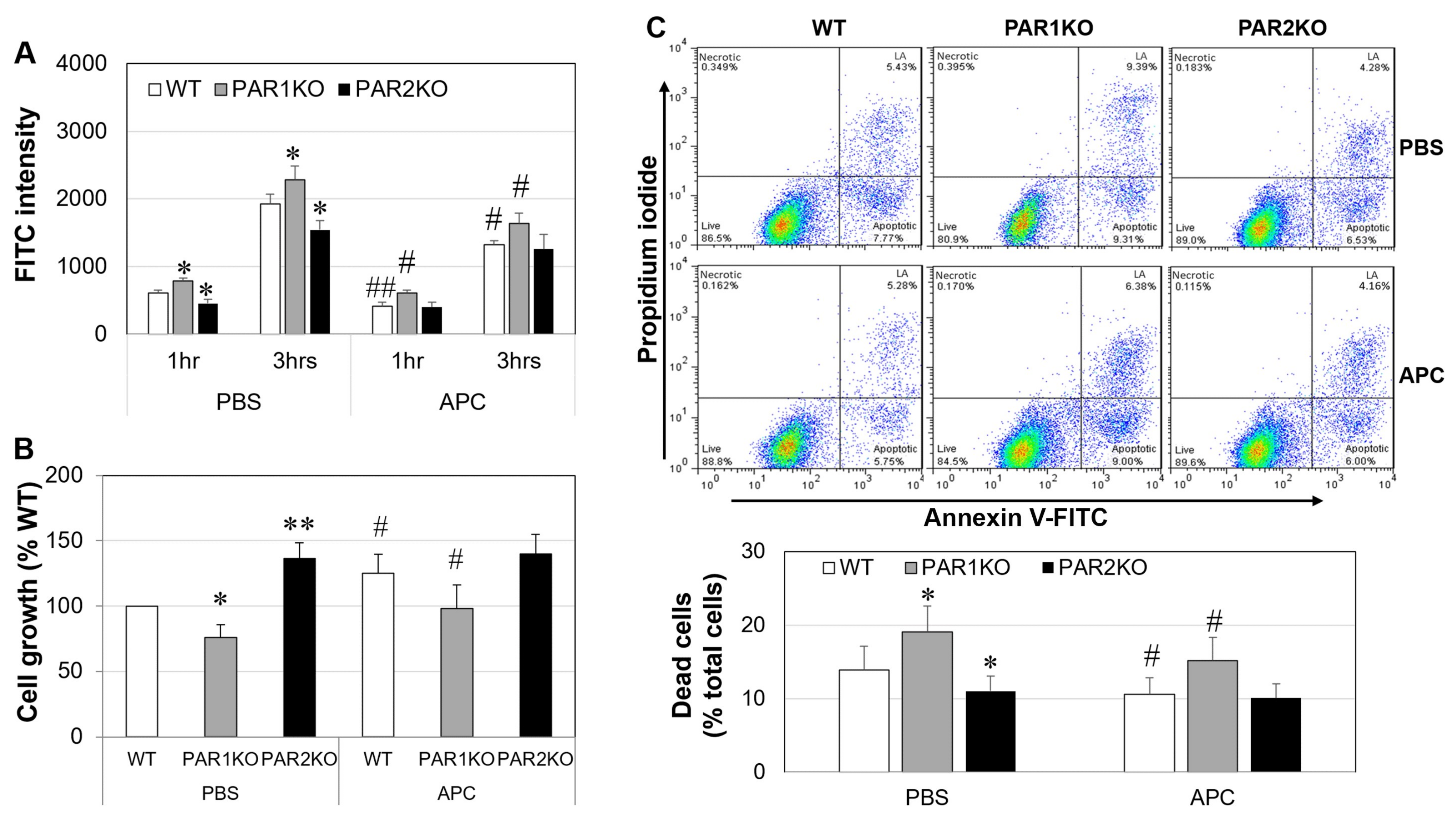
2.6. The Effect of APC on Mouse Keratinocytes In Vitro
3. Discussion
4. Materials and Methods
4.1. Animals and CHS Induction
4.2. Treatment
4.3. Clinical Evaluation of CHS
4.4. Histological Examination
4.5. Th Cytokine and Th Cell Phenotype Detection
4.6. Cytokines and IgE detection
4.7. Keratinocyte Isolation and Apoptosis Detection
4.8. Immunohistochemical Staining
4.9. Cell Proliferation and Apoptosis
4.10. Measurement of Flux of FITC-Dextran
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lloyd-Lavery, A.; Solman, L.; Grindlay, D.J.C.; Rogers, N.K.; Thomas, K.S.; Harman, K.E. What’s new in atopic eczema? An analysis of systematic reviews published in 2016. Part 2: Epidemiology, aetiology and risk factors. Clin. Exp. Derm. 2019, 44, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Nassau, S.; Fonacier, L. Allergic Contact Dermatitis. Med. Clin. N. Am. 2020, 104, 61–76. [Google Scholar] [CrossRef]
- Antoniak, S.; Owens, A.P., 3rd; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithauser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Investig. 2013, 123, 1310–1322. [Google Scholar] [CrossRef] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Eichenfield, L.F.; Ahluwalia, J.; Waldman, A.; Borok, J.; Udkoff, J.; Boguniewicz, M. Current guidelines for the evaluation and management of atopic dermatitis: A comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J. Allergy Clin. Immunol. 2017, 139, S49–S57. [Google Scholar] [CrossRef] [Green Version]
- Esmon, C.T. Protein C anticoagulant system–Anti-inflammatory effects. Semin. Immunopathol. 2012, 34, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lay, A.J.; Donahue, D.; Tsai, M.J.; Castellino, F.J. Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood 2007, 109, 1984–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated protein C: Biased for translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuda, H.; Adachi, Y.; Taguchi, O.; Gabazza, E.C.; Hataji, O.; Fujimoto, H.; Tamaki, S.; Nishikubo, K.; Fukudome, K.; D’Alessandro-Gabazza, C.N.; et al. Activated protein C inhibits bronchial hyperresponsiveness and Th2 cytokine expression in mice. Blood 2004, 103, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.D.; Berger, M.; Majoor, C.J.; Kager, L.M.; Meijers, J.C.; Terpstra, S.; Nieuwland, R.; Boing, A.N.; Lutter, R.; Wouters, D.; et al. Activated protein C inhibits neutrophil migration in allergic asthma: A randomised trial. Eur. Respir. J. 2015, 46, 1636–1644. [Google Scholar] [CrossRef]
- Xue, M.; Dervish, S.; Harrison, L.C.; Fulcher, G.; Jackson, C.J. Activated protein C inhibits pancreatic islet inflammation, stimulates T regulatory cells and prevents diabetes in NOD mice. J. Biol. Chem. 2012, 287, 16356–16364. [Google Scholar] [CrossRef] [Green Version]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002, 296, 1880–1882. [Google Scholar] [CrossRef] [PubMed]
- Moraes, T.J.; Martin, R.; Plumb, J.D.; Vachon, E.; Cameron, C.M.; Danesh, A.; Kelvin, D.J.; Ruf, W.; Downey, G.P. Role of PAR2 in murine pulmonary pseudomonal infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L368–L377. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Lin, H.; Liang, H.P.H.; McKelvey, K.; Zhao, R.; March, L.; Jackson, C. Deficiency of protease-activated receptor (PAR) 1 and PAR2 exacerbates collagen-induced arthritis in mice via differing mechanisms. Rheumatology 2021, 60, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Algermissen, B.; Sitzmann, J.; Nurnberg, W.; Laubscher, J.C.; Henz, B.M.; Bauer, F. Distribution and potential biologic function of the thrombin receptor PAR-1 on human keratinocytes. Arch.Dermatol.Res. 2000, 292, 488–495. [Google Scholar] [CrossRef]
- Steinhoff, M.; Corvera, C.U.; Thoma, M.S.; Kong, W.; McAlpine, B.E.; Caughey, G.H.; Ansel, J.C.; Bunnett, N.W. Proteinase-activated receptor-2 in human skin: Tissue distribution and activation of keratinocytes by mast cell tryptase. Exp. Derm. 1999, 8, 282–294. [Google Scholar] [CrossRef]
- Xue, M.; Campbell, D.; Sambrook, P.N.; Fukudome, K.; Jackson, C.J. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J. Investig. Dermatol. 2005, 125, 1279–1285. [Google Scholar] [CrossRef] [Green Version]
- Duitman, J.; Ruela-de-Sousa, R.R.; Shi, K.; de Boer, O.J.; Borensztajn, K.S.; Florquin, S.; Peppelenbosch, M.P.; Spek, C.A. Protease activated receptor-1 deficiency diminishes bleomycin-induced skin fibrosis. Mol. Med. 2014, 20, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Frateschi, S.; Camerer, E.; Crisante, G.; Rieser, S.; Membrez, M.; Charles, R.P.; Beermann, F.; Stehle, J.C.; Breiden, B.; Sandhoff, K.; et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat. Commun. 2011, 2, 161. [Google Scholar] [CrossRef] [Green Version]
- Tsujii, K.; Andoh, T.; Ui, H.; Lee, J.B.; Kuraishi, Y. Involvement of Tryptase and Proteinase-Activated Receptor-2 in Spontaneous Itch-Associated Response in Mice With Atopy-like Dermatitis. J. Pharmacol. Sci. 2009, 109, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Shpacovitch, V.; Feld, M.; Bunnett, N.W.; Steinhoff, M. Protease-activated receptors: Novel PARtners in innate immunity. Trends Immunol. 2007, 28, 541–550. [Google Scholar] [CrossRef]
- Demerjian, M.; Hachem, J.P.; Tschachler, E.; Denecker, G.; Declercq, W.; Vandenabeele, P.; Mauro, T.; Hupe, M.; Crumrine, D.; Roelandt, T.; et al. Acute modulations in permeability barrier function regulate epidermal cornification: Role of caspase-14 and the protease-activated receptor type 2. Am. J. Pathol. 2008, 172, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.E.; Jeong, S.K.; Lee, S.H. Protease and protease-activated receptor-2 signaling in the pathogenesis of atopic dermatitis. Yonsei Med. J. 2010, 51, 808–822. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.D.; Cottrell, G.S.; Amadesi, S.; Trevisani, M.; Nicoletti, P.; Materazzi, S.; Altier, C.; Cenac, N.; Zamponi, G.W.; Bautista-Cruz, F.; et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J. Physiol. 2007, 578, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.P.; Garzia, C.; Guha, S.; Fletcher, E.K.; Nguyen, N.; Wieschhaus, A.J.; Ferrer, L.; Covic, L.; Kuliopulos, A. PAR2 Pepducin-Based Suppression of Inflammation and Itch in Atopic Dermatitis Models. J. Investig. Derm. 2019, 139, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Rose, L.; Schneider, C.; Stock, C.; Zollner, T.M.; Docke, W.D. Extended DNFB-induced contact hypersensitivity models display characteristics of chronic inflammatory dermatoses. Exp. Derm. 2012, 21, 25–31. [Google Scholar] [CrossRef]
- Bir, N.; Lafargue, M.; Howard, M.; Goolaerts, A.; Roux, J.; Carles, M.; Cohen, M.J.; Iles, K.E.; Fernandez, J.A.; Griffin, J.H.; et al. Cytoprotective-selective Activated Protein C Attenuates P. aeruginosa-induced Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2011, 45, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Dervish, S.; McKelvey, K.J.; March, L.; Wang, F.; Little, C.B.; Jackson, C.J. Activated protein C targets immune cells and rheumatoid synovial fibroblasts to prevent inflammatory arthritis in mice. Rheumatology 2019, 58, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.M.; Tam, V.; Lam, R.; Walsh, K.A.; Tatarczuch, L.; Pagel, C.N.; Reynolds, E.C.; O’Brien-Simpson, N.M.; Mackie, E.J.; Pike, R.N. Protease-activated receptor 2 has pivotal roles in cellular mechanisms involved in experimental periodontitis. Infect. Immun. 2010, 78, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billi, A.C.; Ludwig, J.E.; Fritz, Y.; Rozic, R.; Swindell, W.R.; Tsoi, L.C.; Gruzska, D.; Abdollahi-Roodsaz, S.; Xing, X.; Diaconu, D.; et al. KLK6 expression in skin induces PAR1-mediated psoriasiform dermatitis and inflammatory joint disease. J. Clin. Investig. 2020, 130, 3151–3157. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Krueger, J.G.; Lebwohl, M.G. Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Exp. Derm. 2018, 27, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, H.; Leonard, H.; Chen, D.; Lawrence, T.; Robson, M.; Goossens, P.; McVey, J.H.; Dorling, A. PAR-1 signaling on macrophages is required for effective in vivo delayed-type hypersensitivity responses. iScience. 2021, 24, 101981. [Google Scholar] [CrossRef]
- Christensen, A.D.; Skov, S.; Haase, C. Local and systemic effects of co-stimulatory blockade using cytotoxic T lymphocyte antigen-4-immunoglobulin in dinitrofluorobenzene- and oxazolone-induced contact hypersensitivity in mice. Clin. Exp. Immunol. 2013, 171, 220–230. [Google Scholar] [CrossRef]
- Saeed, M.A.; Ng, G.Z.; Dabritz, J.; Wagner, J.; Judd, L.; Han, J.X.; Dhar, P.; Kirkwood, C.D.; Sutton, P. Protease-activated receptor 1 plays a proinflammatory role in colitis by promoting Th17-related immunity. Inflamm. Bowel Dis. 2017, 23, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.G.; Teng, Y.S.; Shan, Z.G.; Cheng, P.; Hao, C.J.; Lv, Y.P.; Mao, F.Y.; Yang, S.M.; Chen, W.; Zhao, Y.L.; et al. Arrestin domain containing 3 promotes Helicobacter pylori-associated gastritis by regulating protease-activated receptor 1. JCI Insight 2020, 5, e135849. [Google Scholar] [CrossRef]
- Hollenberg, M.D.; Mihara, K.; Polley, D.; Suen, J.Y.; Han, A.; Fairlie, D.P.; Ramachandran, R. Biased signalling and proteinase-activated receptors (PARs): Targeting inflammatory disease. Br. J. Pharm. 2014, 171, 1180–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, A.C.; Vergnolle, N.; MacNaughton, W.K.; Wallace, J.L.; Hollenberg, M.D.; Buret, A.G. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc. Natl. Acad. Sci. USA 2003, 100, 11104–11109. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Chow, S.O.; Dervish, S.; Chan, Y.K.; Julovi, S.M.; Jackson, C.J. Activated protein C enhances human keratinocyte barrier integrity via sequential activation of epidermal growth factor receptor and Tie2. J. Biol. Chem. 2011, 286, 6742–6750. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Lieu, T.; Barlow, N.; Metcalf, M.; Veldhuis, N.A.; Jensen, D.D.; Kocan, M.; Sostegni, S.; Haerteis, S.; Baraznenok, V.; et al. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J. Biol. Chem. 2014, 289, 27215–27234. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.S.; Yang, L.; Rezaie, A.R. Factor X/Xa elicits protective signaling responses in endothelial cells directly via PAR-2 and indirectly via endothelial protein C receptor-dependent recruitment of PAR-1. J. Biol. Chem. 2010, 285, 34803–34812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, A.A.; Rosenfeldt, L.; Mureb, D.; Shafer, J.; Sharma, B.K.; Lane, A.; Crowther, R.R.; McKell, M.C.; Whitt, J.; Alenghat, T.; et al. Cell type-specific mechanisms coupling protease-activated receptor-1 to infectious colitis pathogenesis. J. Thromb. Haemost. 2020, 18, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.F. Induction of contact hypersensitivity in the mouse model. Methods Mol. Biol. 2013, 961, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Hijnen, D.J. Shifting paradigms in the immunology of atopic dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.K.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J. Allergy Clin. Immunol. 2020, 145, 1406–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.T.; Goodarzi, H.; Chen, H.Y. IgE, mast cells, and eosinophils in atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Suender, C.A.; Leist, M.; Abrink, M.; Valentin, P.; Geldmacher, A.; Steinhoff, M.; Metz, M.; Maurer, M. Mast cells are critical for the limitation of thrombin-induced skin inflammation. Exp. Derm. 2018, 27, 50–57. [Google Scholar] [CrossRef]
- Wilson, S.R.; Thé, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.; Gatault, S.; Casals-Diaz, L.; Kelly, P.A.; Camerer, E.; Metais, C.; Knaus, U.G.; Eissner, G.; Steinhoff, M. House dust mite-treated PAR2 over-expressor mouse: A novel model of atopic dermatitis. Exp. Derm. 2019, 28, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hoffert, U.; Rogalski, C.; Seifert, S.; Schmeling, G.; Wingertszahn, J.; Proksch, E.; Wiedow, O. Trypsin induces epidermal proliferation and inflammation in murine skin. Exp. Derm. 2004, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.M.; Rusanova, A.V.; Gorbacheva, L.R.; Umarova, B.A.; Strukova, S.M. Effect of activated protein C on secretory activity of rat peritoneal mast cells. Bull. Exp. Biol. Med. 2006, 142, 403–405. [Google Scholar] [CrossRef]
- Matsumoto, T.; Matsushima, Y.; Toda, M.; Roeen, Z.; D’Alessandro-Gabazza, C.N.; Hinneh, J.A.; Harada, E.; Yasuma, T.; Yano, Y.; Urawa, M.; et al. Activated protein C modulates the proinflammatory activity of dendritic cells. J. Asthma Allergy 2015, 8, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, W.; Yao, J.; Cheng, H.; Sun, X.; Li, L. Correlation of Blood FoxP3+ Regulatory T Cells and Disease Activity of Atopic Dermatitis. J. Immunol. Res. 2019, 2019, 1820182. [Google Scholar] [CrossRef]
- Xue, M.; Campbell, D.; Jackson, C.J. Protein C is an autocrine growth factor for human skin keratinocytes. J. Biol. Chem. 2007, 282, 13610–13616. [Google Scholar] [CrossRef] [Green Version]
- Buhl, T.; Ikoma, A.; Kempkes, C.; Cevikbas, F.; Sulk, M.; Buddenkotte, J.; Akiyama, T.; Crumrine, D.; Camerer, E.; Carstens, E.; et al. Protease-Activated Receptor-2 Regulates Neuro-Epidermal Communication in Atopic Dermatitis. Front. Immunol. 2020, 11, 1740. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, S.; Goihl, A.; Kohli, S.; Gadi, I.; Pierau, M.; Shahzad, K.; Gupta, D.; Bock, F.; Wang, H.; Shaikh, H.; et al. Activated protein C protects from GvHD via PAR2/PAR3 signalling in regulatory T-cells. Nat. Commun. 2017, 8, 311. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Murphy, C.M.; Chan, B.; Kolind, M.; Cheng, T.L.; Mikulec, K.; Peacock, L.; Xue, M.; Park, S.Y.; Little, D.G.; et al. Activated protein C (APC) can increase bone anabolism via a protease-activated receptor (PAR)1/2 dependent mechanism. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2014, 32, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Lohman, R.J.; Cotterell, A.J.; Barry, G.D.; Liu, L.; Suen, J.Y.; Vesey, D.A.; Fairlie, D.P. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012, 26, 2877–2887. [Google Scholar] [CrossRef]
- Li, F.; Adase, C.A.; Zhang, L.J. Isolation and Culture of Primary Mouse Keratinocytes from Neonatal and Adult Mouse Skin. J. Vis. Exp. JOVE 2017, 125, 56027. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, M.; Lin, H.; Zhao, R.; Fryer, C.; March, L.; Jackson, C.J. Activated Protein C Protects against Murine Contact Dermatitis by Suppressing Protease-Activated Receptor 2. Int. J. Mol. Sci. 2022, 23, 516. https://doi.org/10.3390/ijms23010516
Xue M, Lin H, Zhao R, Fryer C, March L, Jackson CJ. Activated Protein C Protects against Murine Contact Dermatitis by Suppressing Protease-Activated Receptor 2. International Journal of Molecular Sciences. 2022; 23(1):516. https://doi.org/10.3390/ijms23010516
Chicago/Turabian StyleXue, Meilang, Haiyan Lin, Ruilong Zhao, Callum Fryer, Lyn March, and Christopher J. Jackson. 2022. "Activated Protein C Protects against Murine Contact Dermatitis by Suppressing Protease-Activated Receptor 2" International Journal of Molecular Sciences 23, no. 1: 516. https://doi.org/10.3390/ijms23010516