
Specific Mutations in Aph1 Cause γ-Secretase Activation


 ,
,
Abstract
:1. Introduction
2. Results
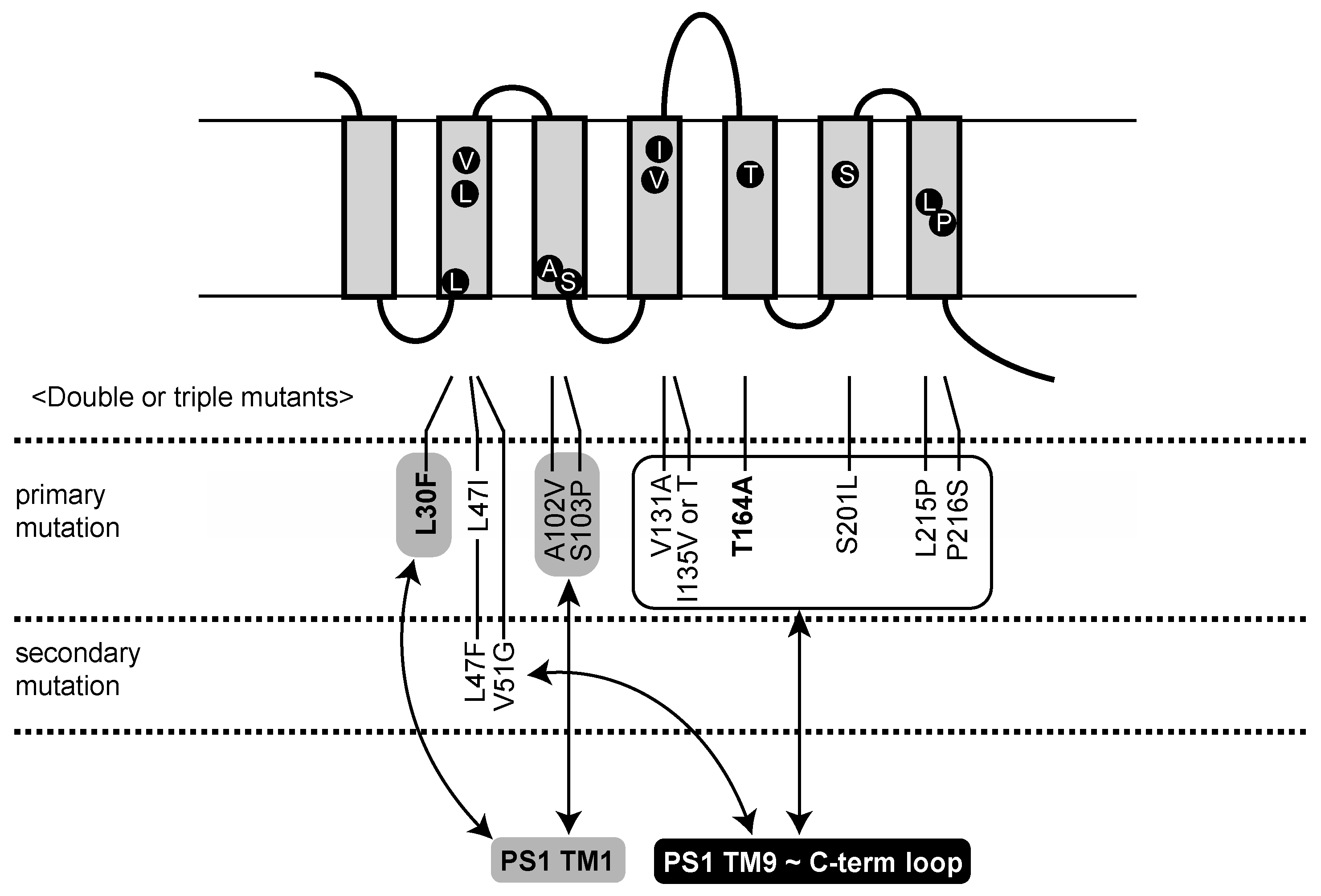
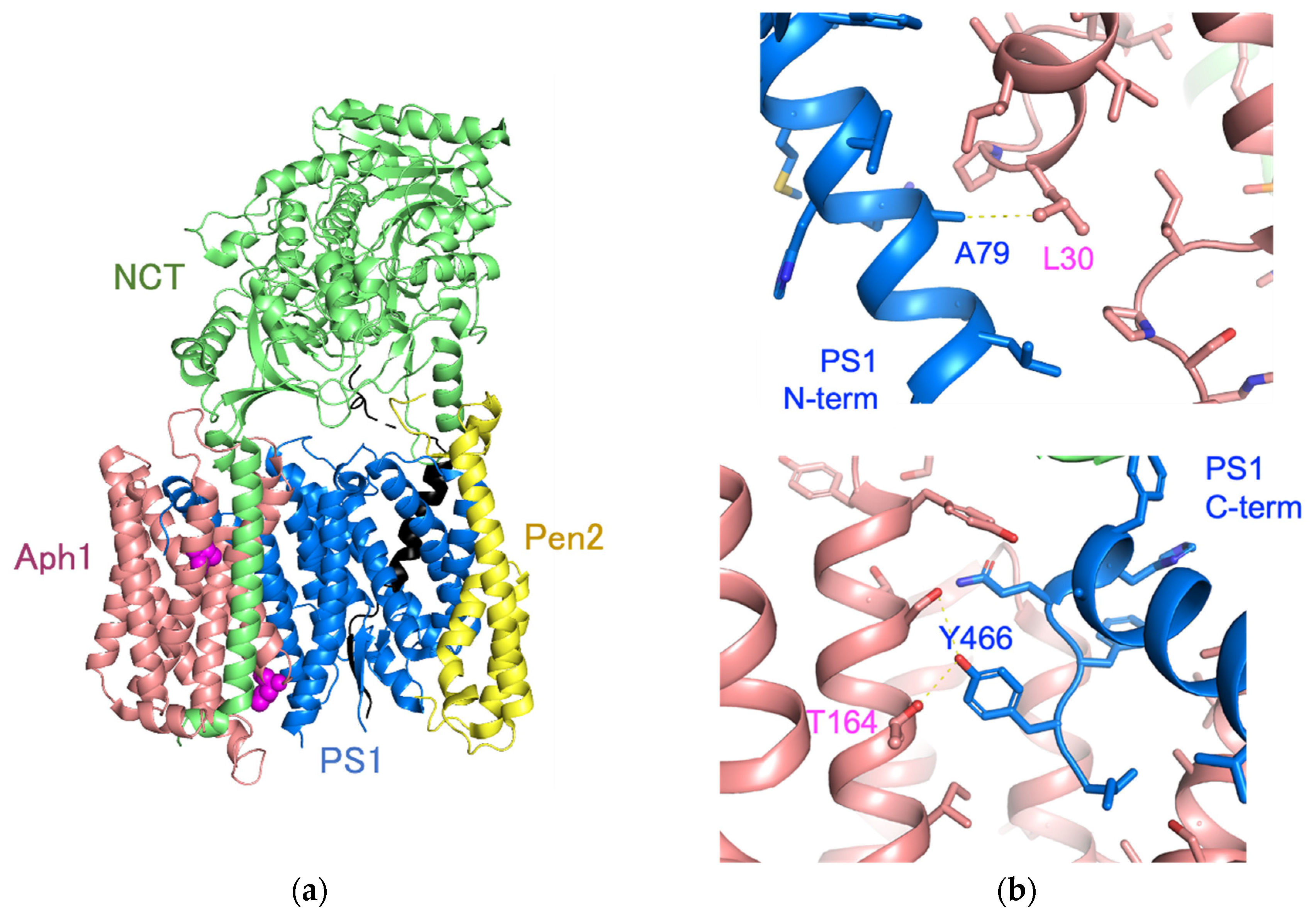
2.1. Identification of Aph1 Mutations That Activate γ-Secretase in the Absence of NCT
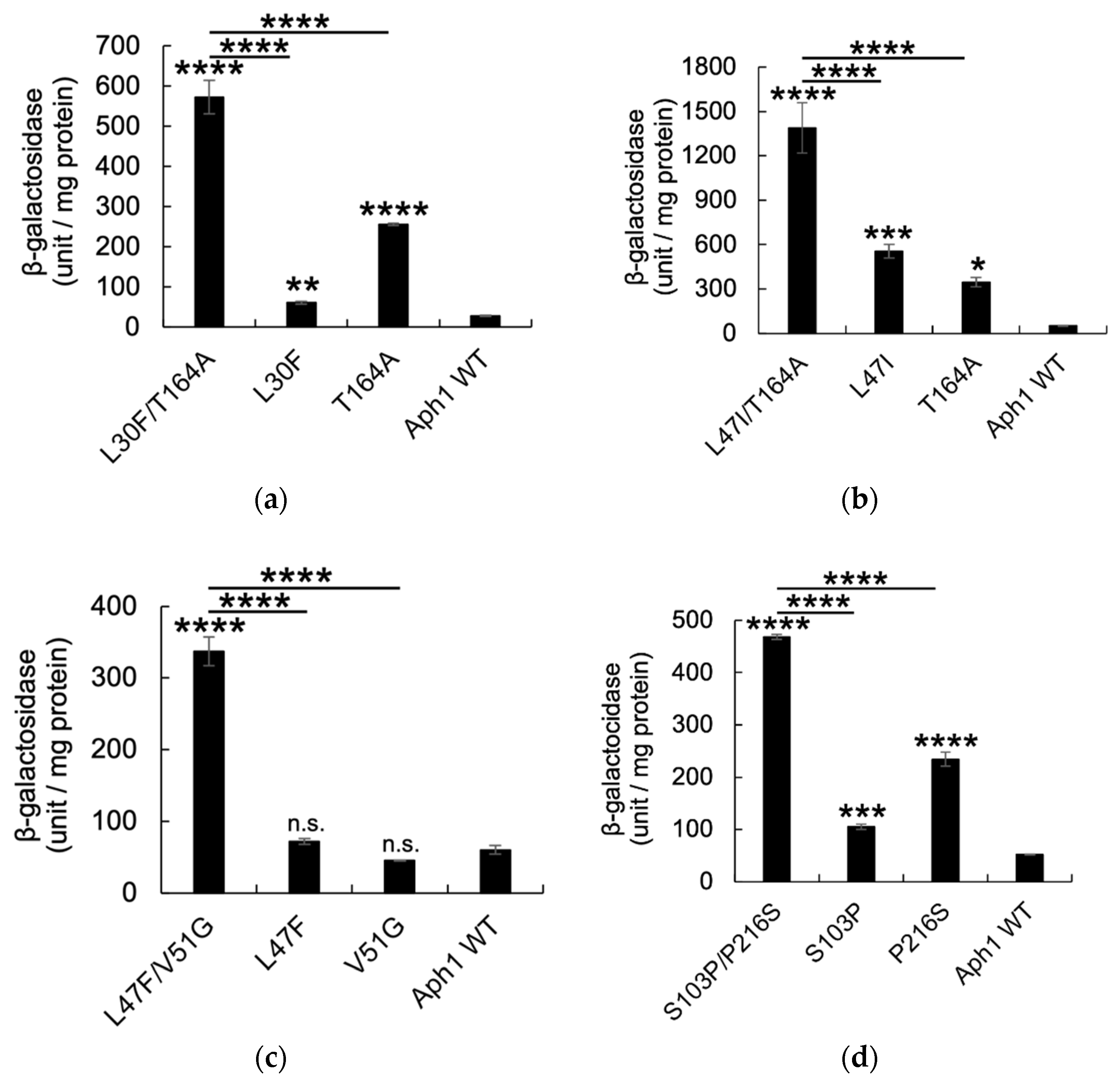
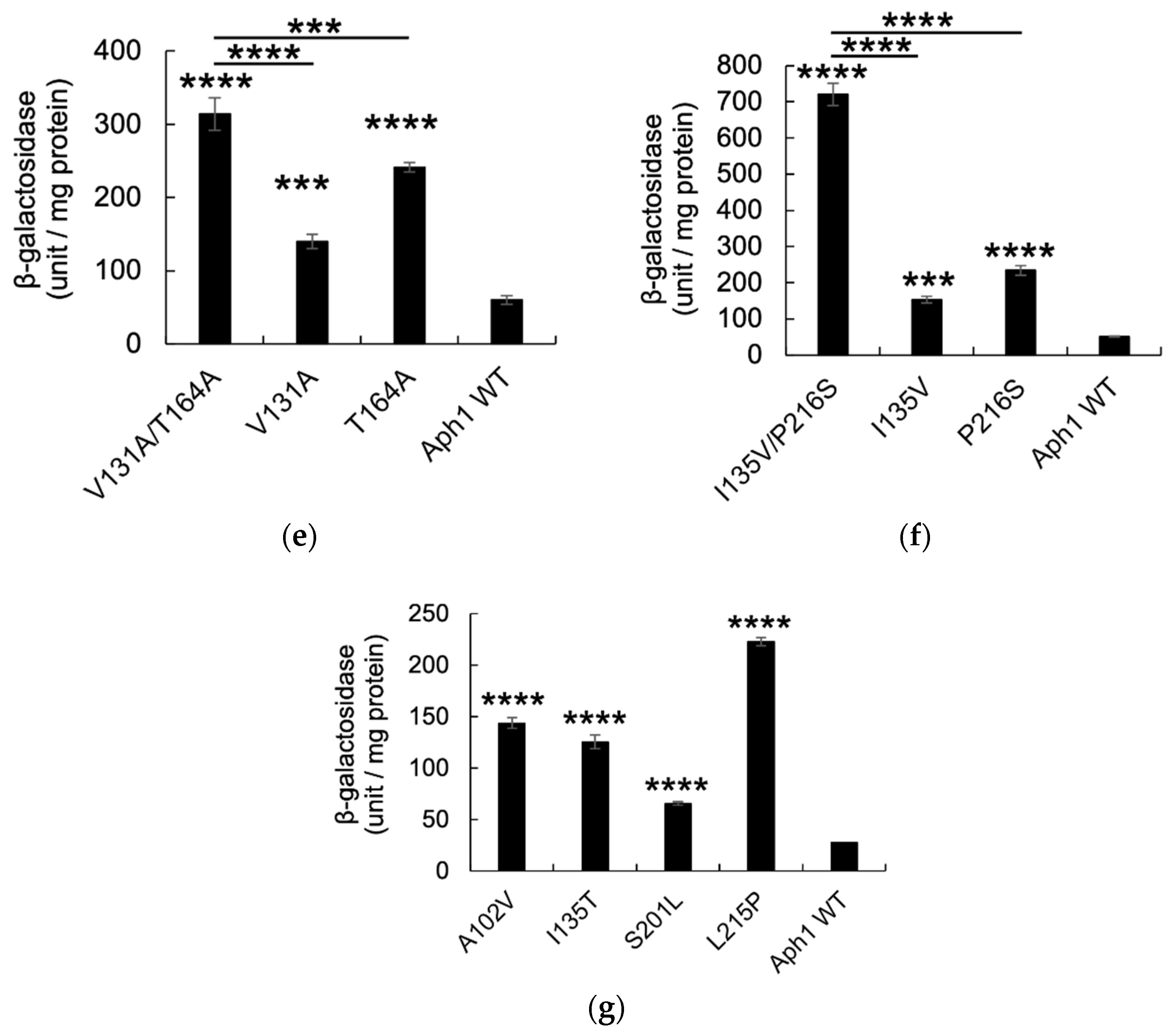
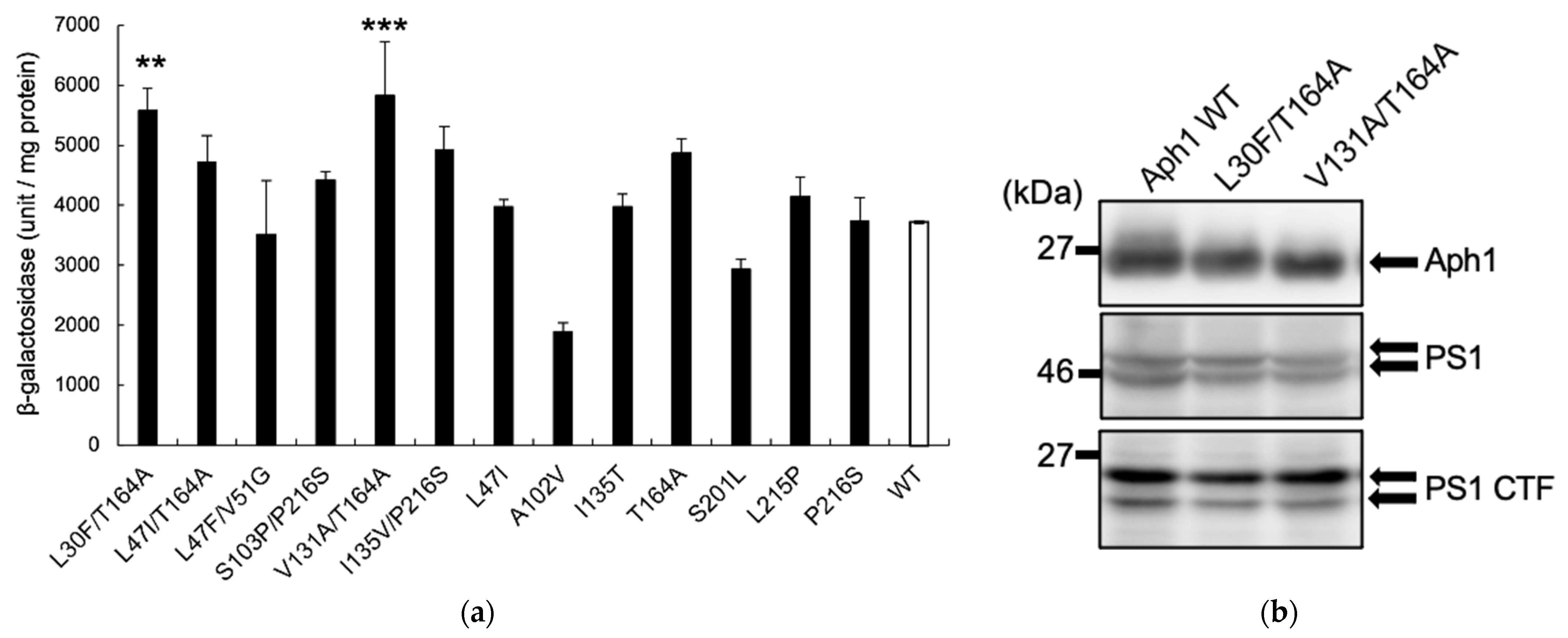
2.2. Aph1 Mutations Activate γ-Secretase Activities
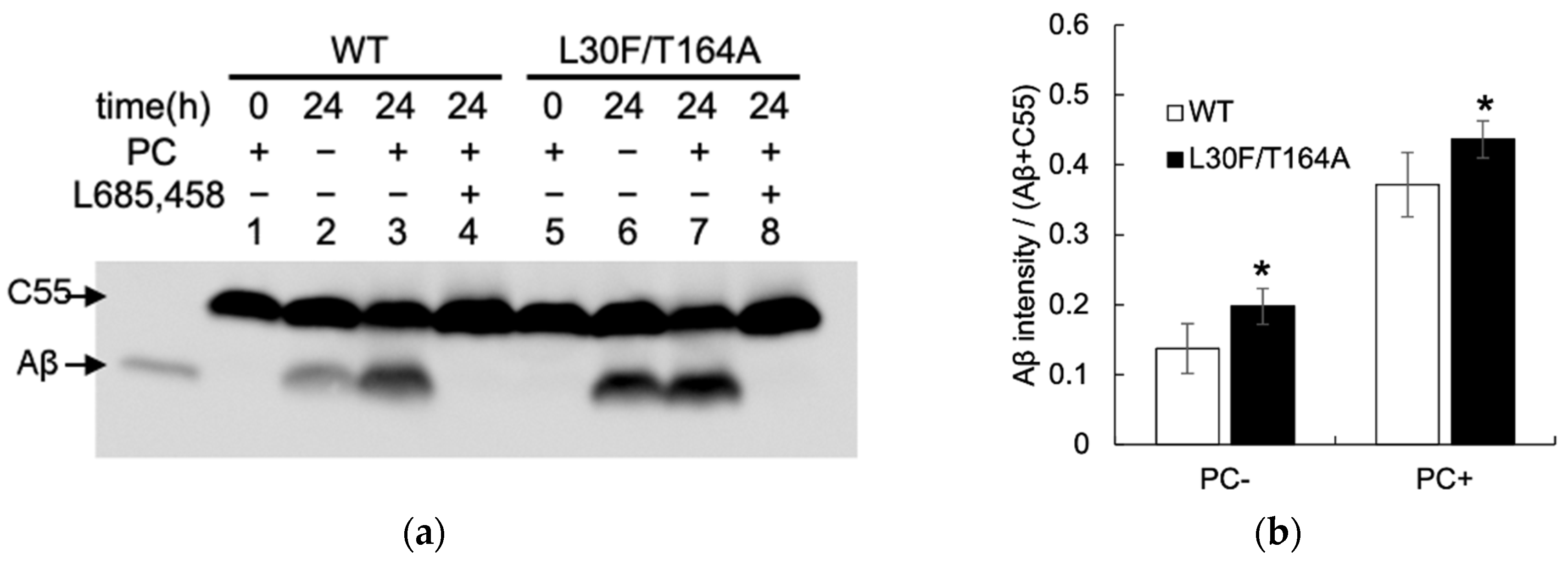
2.3. Aβ Production by Mutant Aph1 in Yeast Microsomes
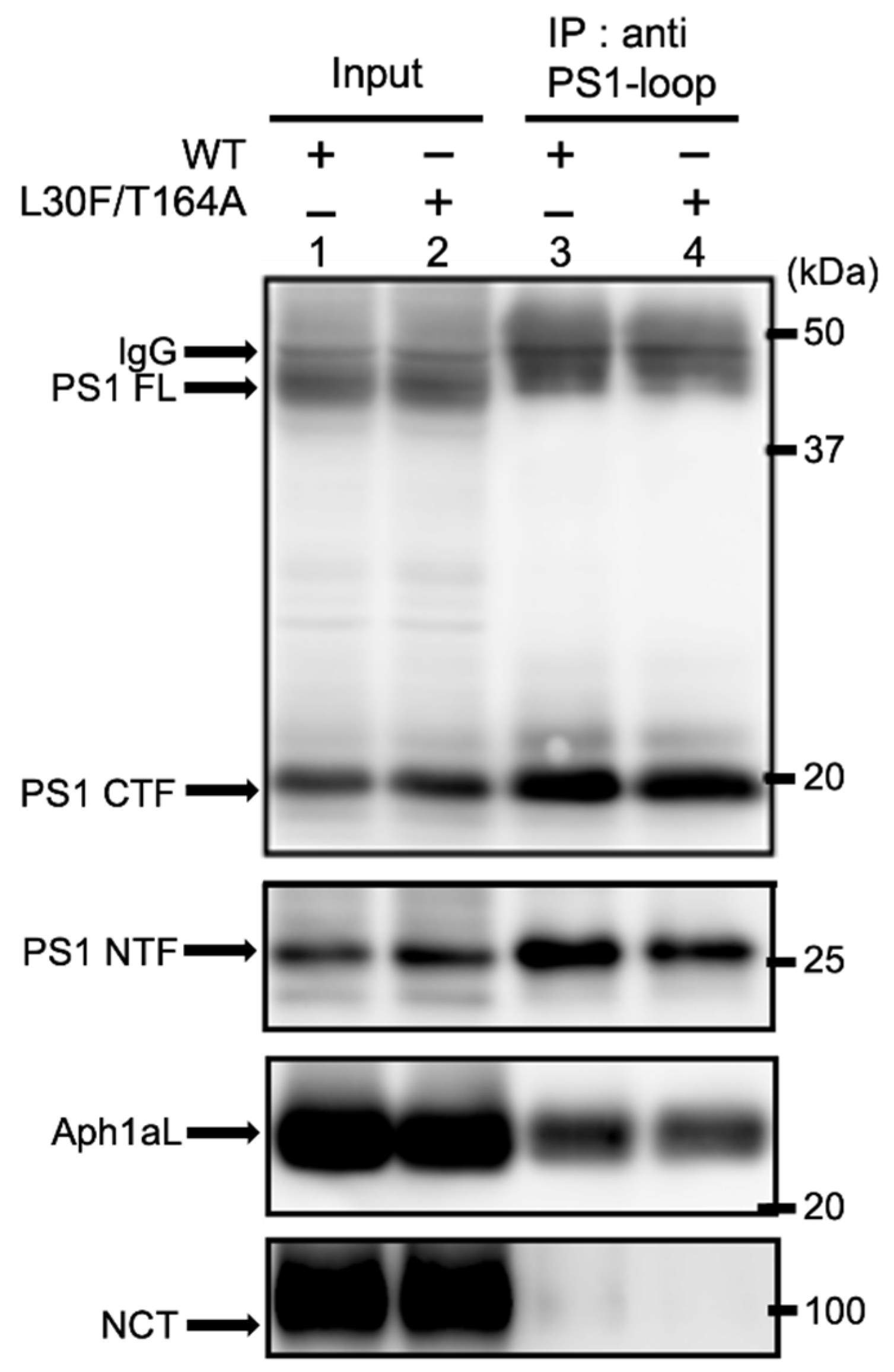
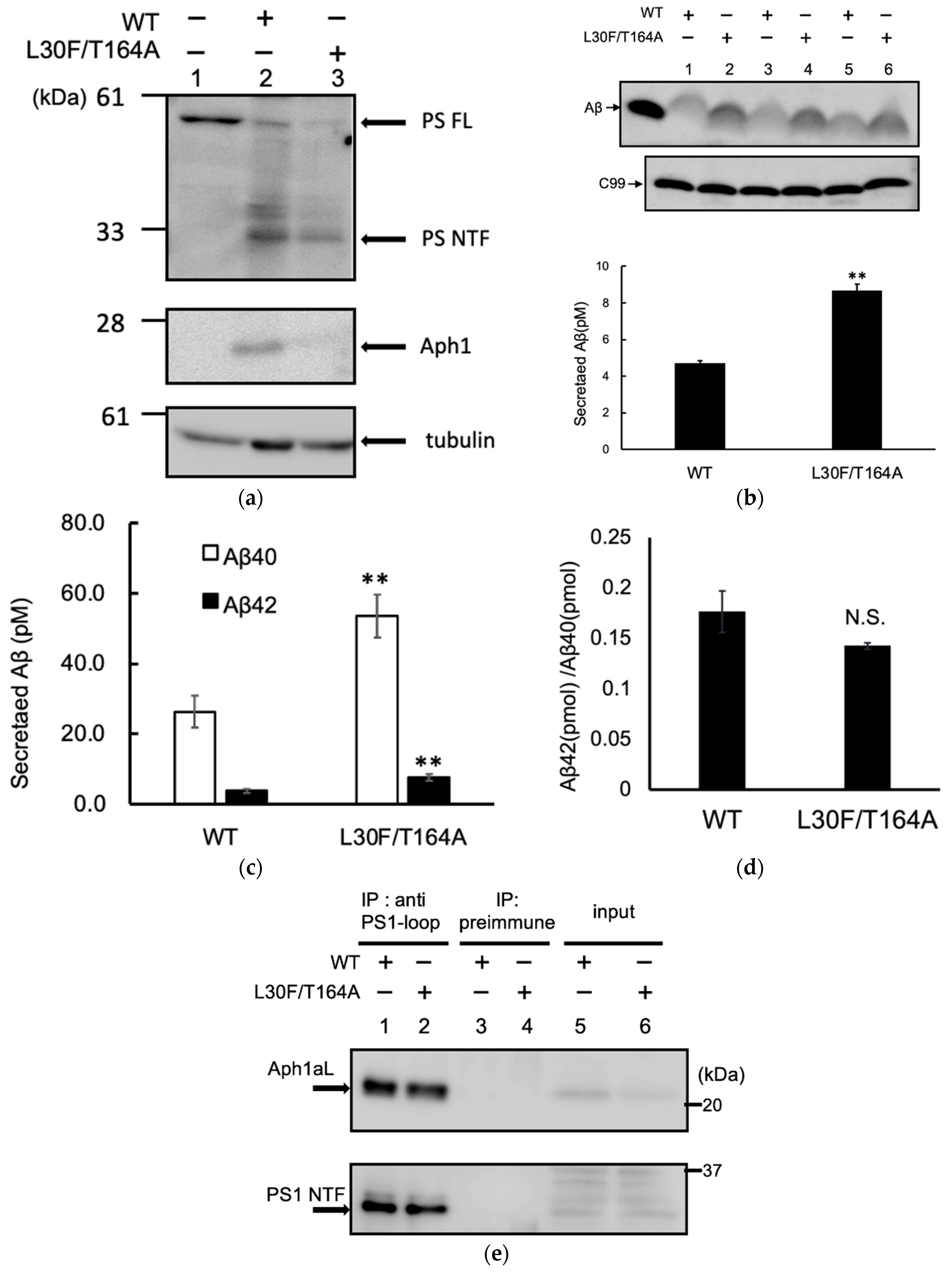
2.4. Aph1 Mutations Activate γ-Secretase in Mouse Fibroblasts
3. Discussion
4. Materials and Methods
4.1. γ-Secretase Reconstitution and Reporter Assays
4.2. In Vitro γ-Secretase Assays Using Yeast Microsomes
4.3. Aβ Production in Mouse Embryonic Fibroblasts
4.4. Immunoprecipitation of γ-Secretase
4.5. Antibodies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretase: Enzymes with therapeutic potential in Alzeimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Tomita, T. Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 2014, 156, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishita-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase. eLife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury Jr, P.T. The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 1993, 695, 144–148. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initinally deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N.S.; Lashley, T.; Fox, N.C.; Murayama, S.; et al. Qualitative changes in human γ-secretase under-lie familial Alzheimer’s disease. J. Exp. Med. 2015, 212, 2003–2013. [Google Scholar] [CrossRef]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in pre-senilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Laudon, H.; Hansson, E.M.; Melén, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; von Heijne, G.; Näslund, J. A Nine-transmembrane domain topology for Presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2016, 114, E476–E485. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Kelleher, R.J. 3rd The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Fortna, R.R.; Crystal, A.S.; Morais, V.A.; Pijak, D.S.; Lee, V.M.Y.; Doms, R.W. Membrane topology and Nicas-trin-enhances endoproteolysis of Aph-1, a component of the γ-secretase complex. J. Biol. Chem. 2004, 279, 3685–3693. [Google Scholar] [CrossRef] [Green Version]
- Serneels, L.; Van Biervliet, J.; Craessaerts, K.; Dejaegere, T.; Horre, K.; Van Houtvin, T.; Esselmann, H.; Paul, S.; Schafer, M.K.; Berezovska, O.; et al. γ-Secretase heterogeneity in the Aph1 subunit: Relevance for Alzheimer’s disease. Science 2009, 324, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Morohashi, Y.; Tomita, T.; Iwatsubo, T. Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 2006, 26, 12081–12088. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Takagi, S.; Tomita, T.; Iwatsubo, T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J. Neurosci. 2008, 28, 6264–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Tominaga, A.; Sato, C.; Tomita, T.; Iwatsubo, T. Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the γ-secretase. J. Neurosci. 2010, 30, 15943–15950. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.; Laudon, H.; Winblad, B.; Lundkvist, J.; Näslund, J. The extreme C terminus of presenilin 1 is essential for gamma-secretase complex assembly and activity. J. Biol. Chem. 2004, 279, 45564–45572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornilova, A.Y.; Bihel, F.; Das, C.; Wolfe, M.S. The initial substrate-binding site of gamma-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3230–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Lee, S.; Tabuchi, K.; Hao, Y.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E., 3rd; Südhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin functions to sterically hinder γ-secretase–substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. USA 2015, 113, E509–E518. [Google Scholar] [CrossRef] [Green Version]
- Takagi-Niidome, S.; Sasaki, T.; Osawa, S.; Sato, T.; Morishima, K.; Cai, T.; Iwatsubo, T.; Tomita, T. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of γ-secretase. J. Neurosci. 2015, 35, 2646–2656. [Google Scholar] [CrossRef] [Green Version]
- Fukumori, A.; Steiner, H. Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 2016, 35, 1628–1643. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Guo, X.; Yan, C.; Lei, J.; Shi, Y. Structural basis of γ-secretase inhibition by small molecule drugs. Cell 2019, 184, 521–533. [Google Scholar] [CrossRef]
- Futai, E.; Yagishita, S.; Ishiura, S. Nicastrin is dispensable for gamma-secretase protease activity in the presence of specific presenilin mutations. J. Biol. Chem. 2009, 284, 13013–13022. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, S.; Futai, E.; Ishiura, S. In vitro reconstitution of gamma-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 2008, 377, 141–145. [Google Scholar] [CrossRef]
- Futai, E.; Osawa, S.; Cai, T.; Fujisawa, T.; Ishiura, S.; Tomita, T. Suppressor mutations for presenilin 1 familial Alzheimer disease mutants modulate γ-secretase activities. J. Biol. Chem. 2016, 291, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Cai, T.; Yoshida, C.; Tomita, T.; Futai, E. Specific mutations in presenilin 1 cause conformational changes in γ-secretase to modulate amyloid β trimming. J. Biochem. 2018, 165, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.F.; Shah, S.; Yu, C.; Wigley, W.C.; Li, H.; Lim, M.; Pedersen, K.; Han, W.; Thomas, P.; Lundkvist, J.; et al. A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J. Biol. Chem. 2004, 279, 4144–4152. [Google Scholar] [CrossRef] [Green Version]
- LaVoie, M.J.; Fraering, P.C.; Ostaszewski, B.L.; Ye, W.; Kimberly, W.T.; Wolfe, M.S.; Selkoe, D.J. Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J. Biol. Chem. 2003, 278, 37213–37222. [Google Scholar] [CrossRef] [Green Version]
- Niimura, M.; Isoo, N.; Takasugi, N.; Tsuruoka, M.; Ui-Tei, K.; Saigo, K.; Morohashi, Y.; Tomita, T.; Iwatsubo, T. Aph-1 contributes to the stabilization and trafficking of the γ-secretase complex through mechanisms involving intermolecular and intramolecular interactions. J. Biol. Chem. 2005, 280, 12967–12975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.; Yonaga, M.; Tomita, T. Activation of γ-secretase trimming activity by topological changes of transmembrane domain 1 of presenilin 1. J. Neurosci. 2017, 37, 12272–12280. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021, 53, 392–402. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, J. Association between poromoter polymorphism in anterior pharynx-defective-1a and sporadic Alz-heimer’s disease in the North Chinese Han population. Neurosci. Lett. 2009, 455, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutation in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef] [PubMed]
- Futai, E. Advanced yeast models of familial Alzheimer disease expressing FAD-linked presenilin to screen mutations and γ-secretase modulators. Methods Mol. Biol. 2019, 2049, 403–417. [Google Scholar] [PubMed]
- Miller, C.A., 3rd; Martinat, M.A.; Hyman, L.E. Assessment of aryl hydrocarbon receptor complex interactions using pBEVY plasmids: Expressionvectors with bi-directional promoters for use in Saccharomyces cerevisiae. Nucleic. Acids. Res. 1998, 26, 3577–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- James, P.; Halladay, J.; Craig, E.A. Genomic libraries and a host strain designed for highly efficient Two-Hybrid selection in yeast. Genetics 1996, 144, 1425–1436. [Google Scholar] [CrossRef]
- Qi-Takahara, Y.; Morishima-Kawashima, M.; Tanimura, Y.; Dolios, G.; Hirotani, N.; Horikoshi, Y.; Kametani, F.; Maeda, M.; Saido, T.C.; Wang, R.; et al. Longer forms of amyloid beta protein: Implications for the mechanism of intramem-brane cleavage by gamma-secretase. J. Neurosci. 2005, 25, 436–445. [Google Scholar] [CrossRef]
- Kitamura, T.; Koshino, Y.; Shibata, F.; Oki, T.; Nakajima, H.; Nosaka, T.; Kumagai, H. Retrovirus-mediated gene transfer and expression cloning: Powerful tools in functional genomics. Exp. Hematol. 2003, 31, 1007–1014. [Google Scholar] [CrossRef]
- Morita, S.; Kojima, T.; Kitamura, T. Plat-E: An efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000, 7, 1063–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, P.-M.; Fortna, R.R.; Price, D.L.; Li, T.; Wong, P.C. Specific domains in anterior pharynx-defective 1 determine its intramembrane interactions with nicastrin and presenilin. Neurobiol. Aging 2012, 33, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asami-Odaka, A.; Ishibashi, Y.; Kikuchi, T.; Kitada, C.; Suzuki, N. Long Amyloid beta-protein secreted from wild-type human neuroblastoma IMR-32 Cells. Biochemistry 1995, 34, 10272–10278. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aph1 Mutants | Growth with PS1S438P | Growth with PS1WT | No. of Clones |
|---|---|---|---|
| L30F/T164A | +++ | - | 1 |
| L47I | +++ | - | 4 |
| L47I/T164A | +++ | - | 1 |
| L47F/V51G | +++ | - | 1 |
| A102V | +++ | - | 1 |
| S103P/P216S | +++ | - | 1 |
| V131A/T164A | +++ | - | 1 |
| I135V/P216S | +++ | - | 1 |
| I135T | ++ | - | 1 |
| T164A | +++ | - | 3 |
| S201L | ++ | - | 1 |
| L215P | +++ | - | 2 |
| P216S | +++ | - | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, H.; Yoshida, C.; Hidaka, M.; Ogawa, T.; Tomita, T.; Futai, E. Specific Mutations in Aph1 Cause γ-Secretase Activation. Int. J. Mol. Sci. 2022, 23, 507. https://doi.org/10.3390/ijms23010507
Watanabe H, Yoshida C, Hidaka M, Ogawa T, Tomita T, Futai E. Specific Mutations in Aph1 Cause γ-Secretase Activation. International Journal of Molecular Sciences. 2022; 23(1):507. https://doi.org/10.3390/ijms23010507
Chicago/Turabian StyleWatanabe, Hikari, Chika Yoshida, Masafumi Hidaka, Tomohisa Ogawa, Taisuke Tomita, and Eugene Futai. 2022. "Specific Mutations in Aph1 Cause γ-Secretase Activation" International Journal of Molecular Sciences 23, no. 1: 507. https://doi.org/10.3390/ijms23010507