
The (Poly)Pharmacology of Cannabidiol in Neurological and Neuropsychiatric Disorders: Molecular Mechanisms and Targets


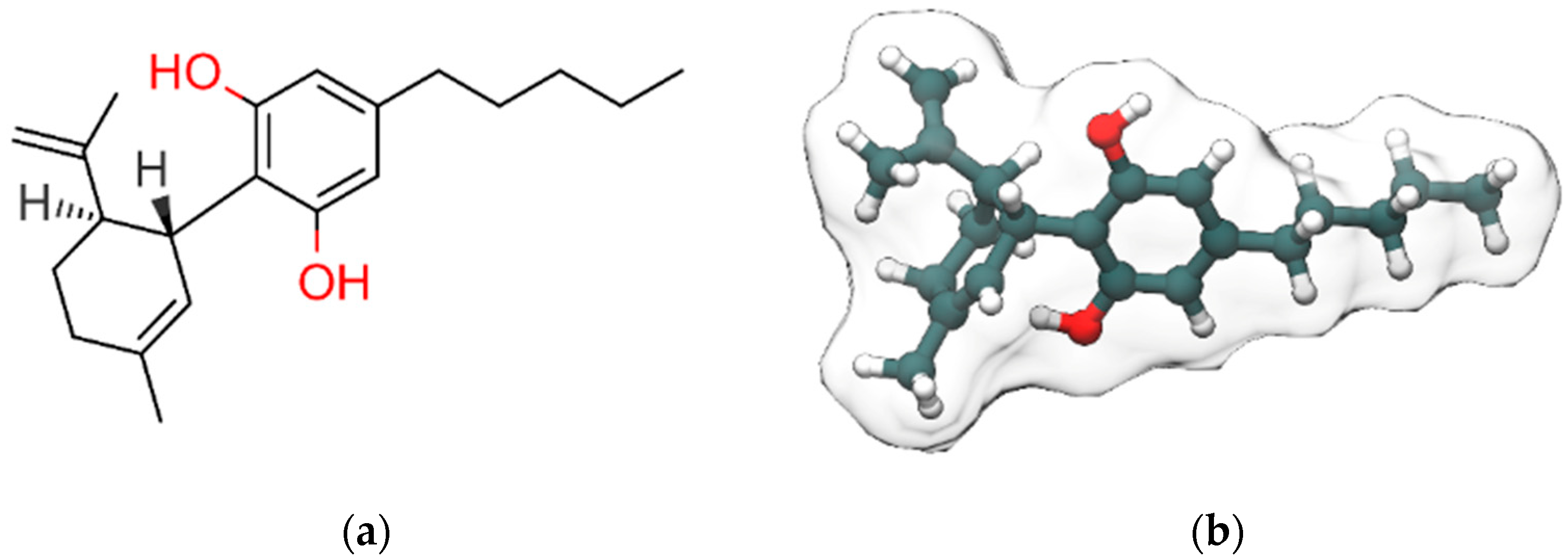{kind=link}
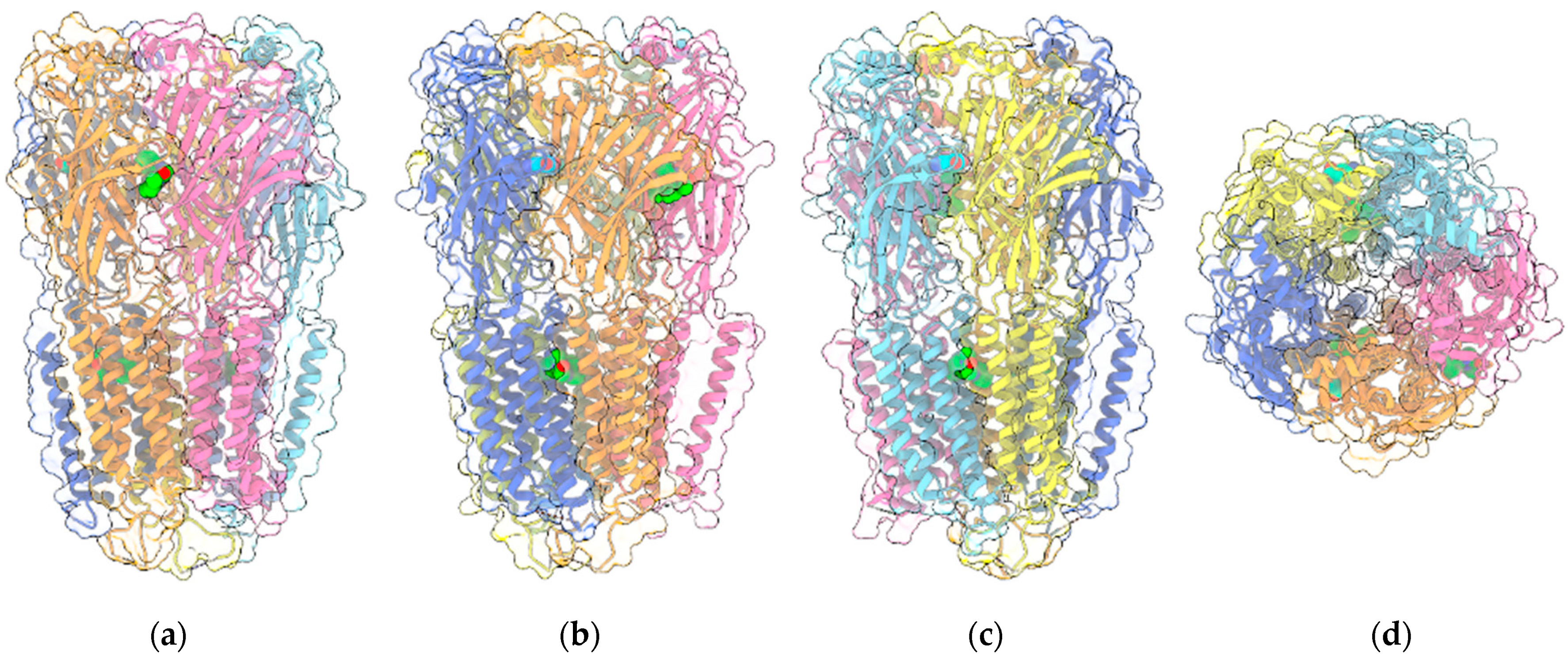{kind=link}
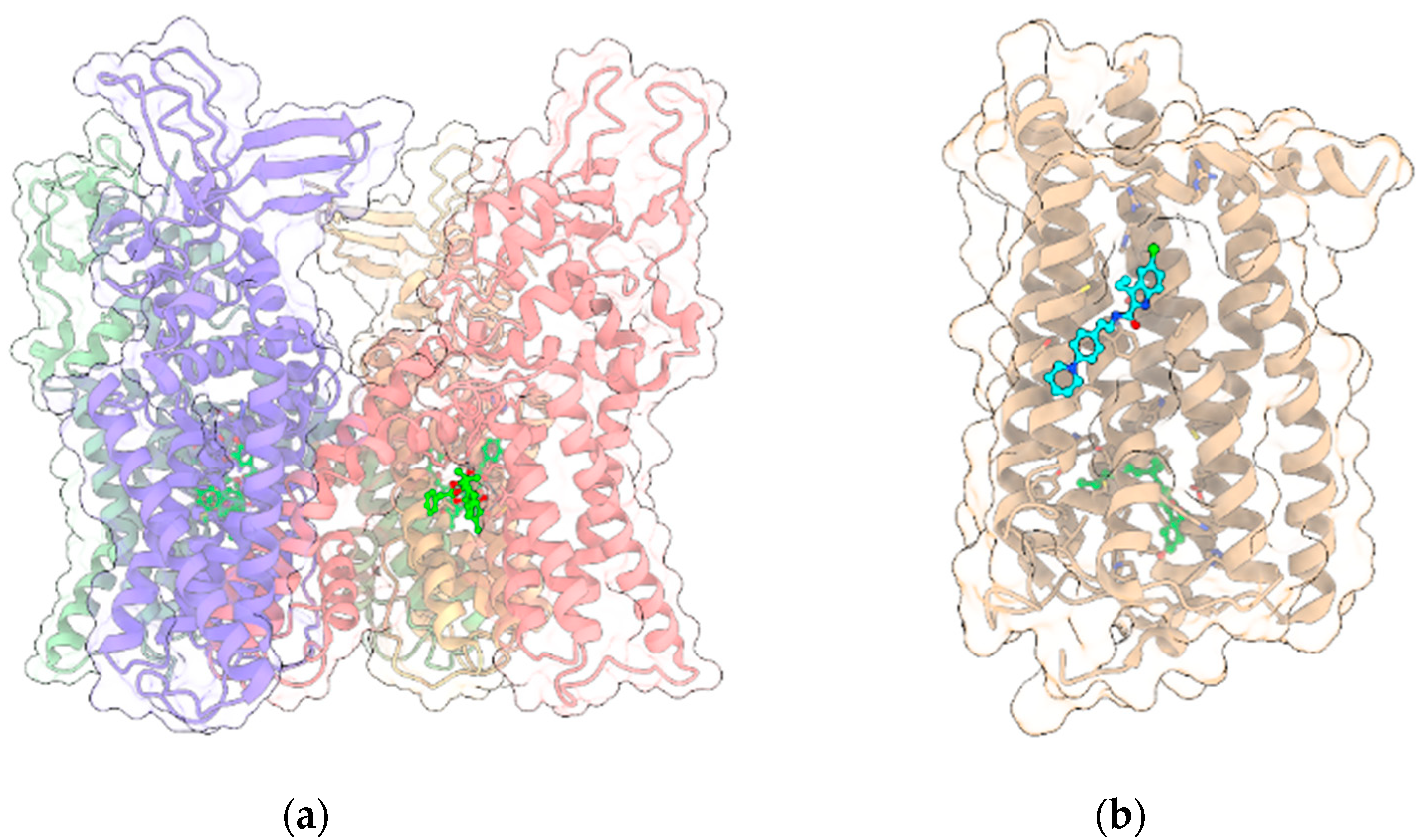{kind=link}
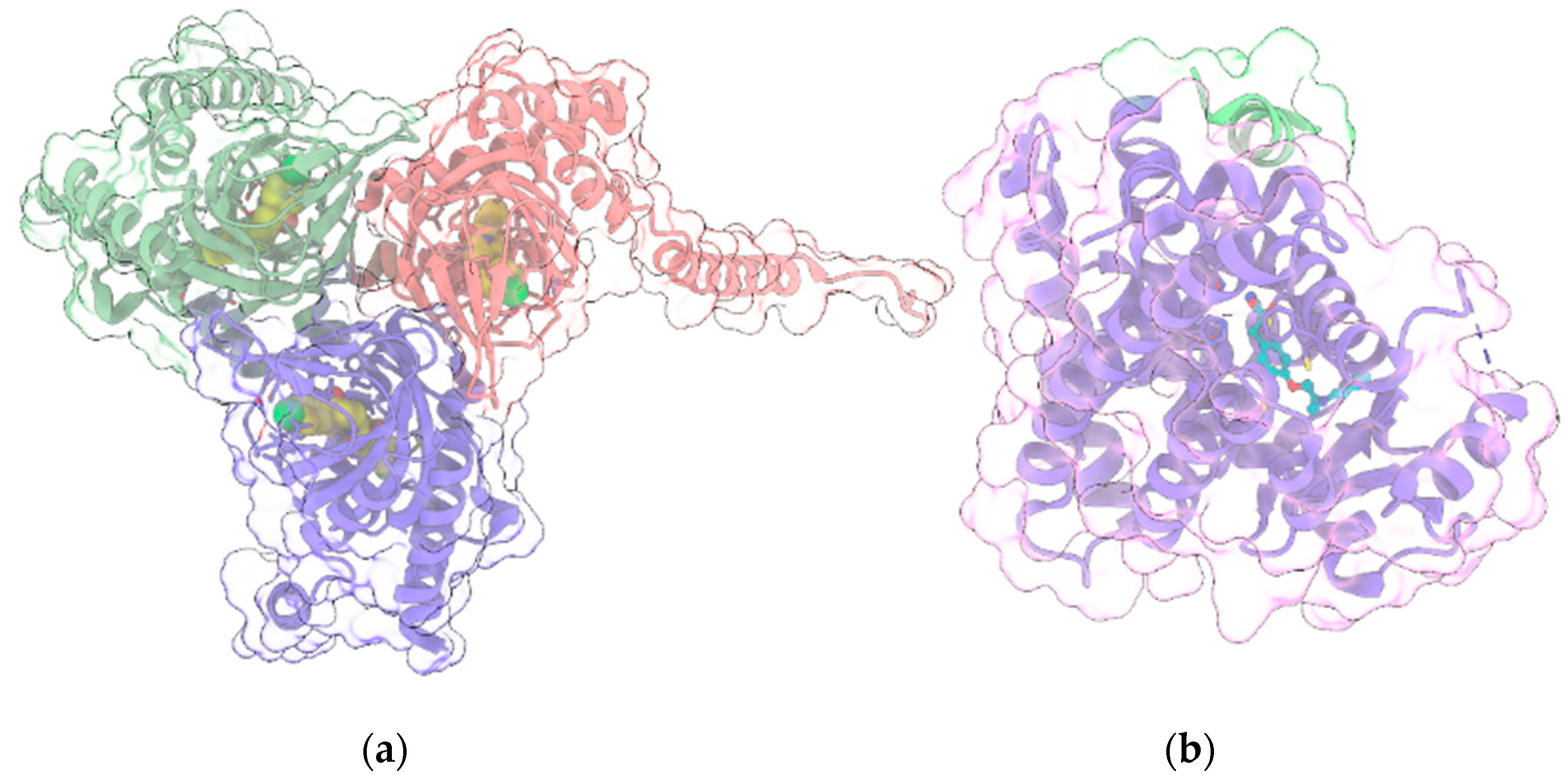{kind=link}
{kind=link}
Abstract
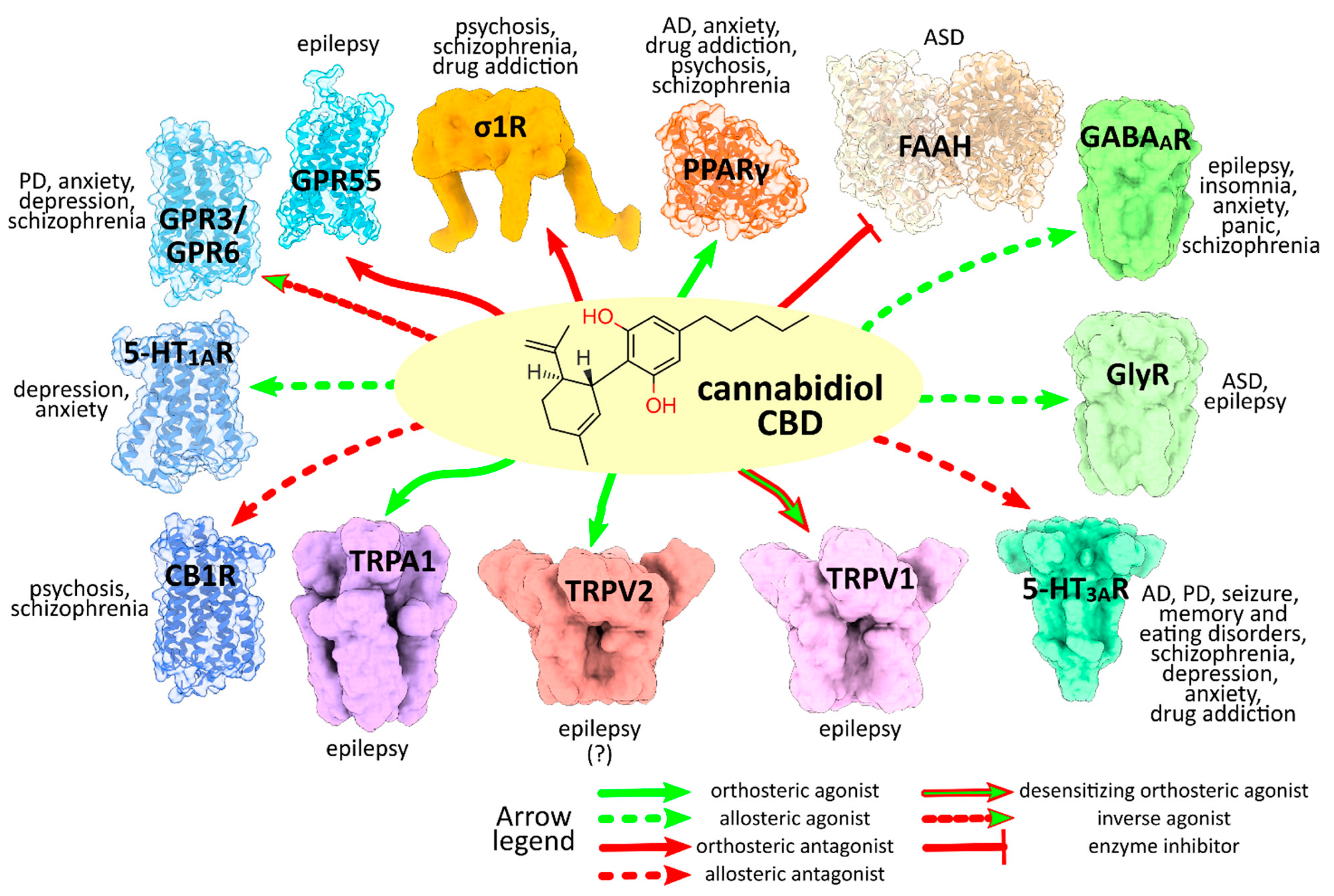
:1. Cannabidiol
2. Neurological and Neuropsychiatric Disorders Potentially Affected by CBD Treatment
2.1. Epilepsy and Cannabidiol
2.2. Alzheimer’s Disease and Cannabidiol
2.3. Parkinson’s Disease and Cannabidiol
2.4. Depression and Cannabidiol
2.5. Anxiety Disorders and Cannabidiol
2.6. Drug Addiction and Cannabidiol
2.7. Autism Spectrum Disorder and Cannabidiol
2.8. Psychotic Disorders and Cannabidiol
3. CBD Molecular Targets and Mechanisms Involved in the Neurological and Neuropsychiatric Disorders
3.1. Cys-Loop Superfamily of Ligand-Gated Ion Channels
3.1.1. GABAARs
3.1.2. GlyRs
3.1.3. 5-HT3Rs
3.2. TRP Channels
3.2.1. TRPV1
3.2.2. Other TRP Channels
3.3. GPCRs
3.3.1. CB1R
3.3.2. 5-HT1AR
3.3.3. GPR3 and GPR6
3.3.4. GPR55
3.4. σ1R
3.5. PPARγ
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoyl glycerol |
| 5-HT | 5-hydroxytryptamine/serotonin |
| 5-HTnRs | 5-hydroxytryptamine subtype n receptors (n = 1A, 3) |
| 8-OH-DPAT | 8-hydroxy-2-(di-n-propylamino)tetralin |
| AD | Alzheimer’s disease |
| AEA | N-arachidonoylethanolamine/anandamide |
| AEDs | antiepileptic drugs |
| ARDs | ankyrin repeat domains |
| ASD | Autism spectrum disorder |
| BDNF | brain-derived neurotrophic factor |
| BDZ | benzodiazepine |
| CaM-Kinase II | Ca2+/calmodulin-dependent protein kinase II |
| cAMP | cyclic adenosine monophosphate |
| CB1R | cannabinoid receptor 1 |
| CBD | Cannabidiol |
| CBDV | cannabidivarin |
| CNS | central nervous system |
| DRE | drug-resistant epilepsy |
| DS | Dravet syndrome |
| DZP | diazepam |
| ECD | extracellular N-terminal domain |
| EMA | European Medicines Agency |
| FAAH | fatty acid amide hydrolase |
| FDA | US Food and Drug Administration |
| FSK | forskolin |
| GABA | γ-aminobutyric acid |
| GABANRs | GABA N receptors (N = A,B) |
| GlyRs | glycine receptors |
| GPCRs | G-protein coupled receptors |
| GTPγS | guanosine 5′-O-[γ-thio]triphosphate |
| HINT1 | histidine triad nucleotide-binding protein 1 |
| HMGB1 | high mobility group box 1 |
| IL-n | interleukin n (n = 1β, 6) |
| LGS | Lennox-Gastaut syndrome |
| LTD | long-term depression |
| NAM | negative allosteric modulator |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| NMR | nuclear magnetic resonance |
| PD | Parkinson’s disease |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PPAR | Peroxisome proliferator-activated receptor |
| PTZ | pentylenetetrazol |
| TCA | trans-cinnamaldehyde |
| TLE | temporal lobe epilepsy |
| TMD | transmembrane domain |
| TNF | tumor necrosis factor |
| TRPA1 | transient receptor potential ankyrin subtype 1 protein |
| TRPVn | transient receptor potential cation channel subfamily V member n (n = 1–6) |
| VSLD | voltage-sensor like domain |
| Δ9-THC | Δ9-tetrahydrocannabinol |
References
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB 1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [Green Version]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Hurd, Y.L.; Yoon, M.; Manini, A.F.; Hernandez, S.; Olmedo, R.; Ostman, M.; Jutras-Aswad, D. Early Phase in the Development of Cannabidiol as a Treatment for Addiction: Opioid Relapse Takes Initial Center Stage. Neurotherapeutics 2015, 12, 807–815. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish—I. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Thiele, E.A.; Marsh, E.D.; French, J.A.; Mazurkiewicz-Beldzinska, M.; Benbadis, S.R.; Joshi, C.; Lyons, P.D.; Taylor, A.; Roberts, C.; Sommerville, K.; et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 1085–1096. [Google Scholar] [CrossRef]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-Resistant Epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilio, M.R.; Thiele, E.A.; Devinsky, O. The case for assessing cannabidiol in epilepsy. Epilepsia 2014, 55, 787–790. [Google Scholar] [CrossRef]
- Silvestro, S.; Mammana, S.; Cavalli, E.; Bramanti, P.; Mazzon, E. Use of Cannabidiol in the Treatment of Epilepsy: Efficacy and Security in Clinical Trials. Molecules 2019, 24, 1459. [Google Scholar] [CrossRef] [Green Version]
- Lazarini-Lopes, W.; Do Val-da Silva, R.A.; da Silva-Júnior, R.M.P.; Leite, J.P.; Garcia-Cairasco, N. The anticonvulsant effects of cannabidiol in experimental models of epileptic seizures: From behavior and mechanisms to clinical insights. Neurosci. Biobehav. Rev. 2020, 111, 166–182. [Google Scholar] [CrossRef]
- Abu-Sawwa, R.; Scutt, B.; Park, Y. Emerging Use of Epidiolex (Cannabidiol) in Epilepsy. J. Pediatr. Pharmacol. Ther. 2020, 25, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.; Karl, T. In vivo Evidence for Therapeutic Properties of Cannabidiol (CBD) for Alzheimer’s Disease. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Karl, T.; Garner, B.; Cheng, D. The therapeutic potential of the phytocannabinoid cannabidiol for Alzheimer’s disease. Behav. Pharmacol. 2017, 28, 142–160. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in β-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-κB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Horváth, B.; Bátkai, S.; Park, O.; Tanchian, G.; Gao, R.Y.; Patel, V.; Wink, D.A.; Liaudet, L.; et al. Cannabidiol protects against hepatic ischemia/reperfusion injury by attenuating inflammatory signaling and response, oxidative/nitrative stress, and cell death. Free Radic. Biol. Med. 2011, 50, 1368–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Janefjord, E.; Mååg, J.L.V.; Harvey, B.S.; Smid, S.D. Cannabinoid Effects on β Amyloid Fibril and Aggregate Formation, Neuronal and Microglial-Activated Neurotoxicity In Vitro. Cell. Mol. Neurobiol. 2014, 34, 31–42. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol Promotes Amyloid Precursor Protein Ubiquitination and Reduction of Beta Amyloid Expression in SHSY5Y(APP+) Cells Through PPARγ Involvement. Phytother. Res. 2013. [Google Scholar] [CrossRef]
- Antony, P.M.A.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [Green Version]
- Beitz, J.M. Parkinson’s disease a review. Front. Biosci. 2014, S6, S415. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Crippa, J.A.S.; Hallak, J.E.C.; Zuardi, A.W.; Guimarães, F.S.; Tumas, V.; dos Santos, R.G. Is cannabidiol the ideal drug to treat non-motor Parkinson’s disease symptoms? Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 121–133. [Google Scholar] [CrossRef]
- Schier, A.; Ribeiro, N.; Coutinho, D.; Machado, S.; Arias-Carrion, O.; Crippa, J.; Zuardi, A.; Nardi, A.; Silva, A. Antidepressant-Like and Anxiolytic-Like Effects of Cannabidiol: A Chemical Compound of Cannabis sativa. CNS Neurol. Disord. Drug Targets 2014, 13, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réus, G.Z.; Stringari, R.B.; Ribeiro, K.F.; Luft, T.; Abelaira, H.M.; Fries, G.R.; Aguiar, B.W.; Kapczinski, F.; Hallak, J.E.; Zuardi, A.W.; et al. Administration of cannabidiol and imipramine induces antidepressant-like effects in the forced swimming test and increases brain-derived neurotrophic factor levels in the rat amygdala. Acta Neuropsychiatr. 2011, 23, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A.; Lewis, D.R.; Wiegand, S.J.; Lindsay, R.M. Antidepressant-Like Effect of Brain-derived Neurotrophic Factor (BDNF). Pharmacol. Biochem. Behav. 1997, 56, 131–137. [Google Scholar] [CrossRef]
- Siuciak, J.A.; Clark, M.S.; Rind, H.B.; Whittemore, S.R.; Russo, A.F. BDNF induction of tryptophan hydroxylase mRNA levels in the rat brain. J. Neurosci. Res. 1998, 52, 149–158. [Google Scholar] [CrossRef]
- Goggi, J.; Pullar, I.A.; Carney, S.L.; Bradford, H.F. Modulation of neurotransmitter release induced by brain-derived neurotrophic factor in rat brain striatal slices in vitro. Brain Res. 2002, 941, 34–42. [Google Scholar] [CrossRef]
- Martinowich, K.; Lu, B. Interaction between BDNF and Serotonin: Role in Mood Disorders. Neuropsychopharmacology 2008, 33, 73–83. [Google Scholar] [CrossRef]
- di Giacomo, V.; Chiavaroli, A.; Recinella, L.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Ronci, M.; Leone, S.; Brunetti, L.; et al. Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. Int. J. Mol. Sci. 2020, 21, 3575. [Google Scholar] [CrossRef]
- Zwanzger, P. Pharmakotherapie bei Angsterkrankungen. Fortschritte der Neurol. · Psychiatr. 2016, 84, 306–314. [Google Scholar] [CrossRef]
- Blessing, E.M.; Steenkamp, M.M.; Manzanares, J.; Marmar, C.R. Cannabidiol as a Potential Treatment for Anxiety Disorders. Neurotherapeutics 2015, 12, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Shannon, S. Cannabidiol in Anxiety and Sleep: A Large Case Series. Perm. J. 2019, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, F.; Aracil-Fernández, A.; Manzanares, J. Cannabidiol regulates behavioural alterations and gene expression changes induced by spontaneous cannabinoid withdrawal. Br. J. Pharmacol. 2018, 175, 2676–2688. [Google Scholar] [CrossRef] [Green Version]
- Hurd, Y.L. Cannabidiol: Swinging the Marijuana Pendulum From ‘Weed’ to Medication to Treat the Opioid Epidemic. Trends Neurosci. 2017, 40, 124–127. [Google Scholar] [CrossRef]
- Prud’homme, M.; Cata, R.; Jutras-Aswad, D. Cannabidiol as an Intervention for Addictive Behaviors: A Systematic Review of the Evidence. Subst. Abus. Res. Treat. 2015, 9, SART.S25081. [Google Scholar] [CrossRef] [Green Version]
- Sanchack, K.E.; Thomas, C.A. Autism Spectrum Disorder: Primary Care Principles. Am. Fam. Physician 2016, 94, 972–979; [Google Scholar]
- Talkowski, M.E.; Minikel, E.V.; Gusella, J.F. Autism Spectrum Disorder Genetics. Harv. Rev. Psychiatry 2014, 22, 65–75. [Google Scholar] [CrossRef]
- Mandy, W.; Lai, M.-C. Annual Research Review: The role of the environment in the developmental psychopathology of autism spectrum condition. J. Child Psychol. Psychiatry 2016, 57, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Zamberletti, E.; Gabaglio, M.; Parolaro, D. The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. Int. J. Mol. Sci. 2017, 18, 1916. [Google Scholar] [CrossRef]
- Karhson, D.S.; Krasinska, K.M.; Dallaire, J.A.; Libove, R.A.; Phillips, J.M.; Chien, A.S.; Garner, J.P.; Hardan, A.Y.; Parker, K.J. Plasma anandamide concentrations are lower in children with autism spectrum disorder. Mol. Autism 2018, 9, 18. [Google Scholar] [CrossRef]
- Ballaban-Gil, K.; Tuchman, R. Epilepsy and epileptiform EEG: Association with autism and language disorders. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 300–308. [Google Scholar] [CrossRef]
- Lee, B.H.; Smith, T.; Paciorkowski, A.R. Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy Behav. 2015, 47, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, A.; Cassuto, H.; Lubotzky, A.; Wattad, N.; Hazan, E. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems—A Retrospective Feasibility Study. J. Autism Dev. Disord. 2019, 49, 1284–1288. [Google Scholar] [CrossRef]
- Fleury-Teixeira, P.; Caixeta, F.V.; Ramires da Silva, L.C.; Brasil-Neto, J.P.; Malcher-Lopes, R. Effects of CBD-Enriched Cannabis sativa Extract on Autism Spectrum Disorder Symptoms: An Observational Study of 18 Participants Undergoing Compassionate Use. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Lieberman, J.A.; First, M.B. Psychotic Disorders. N. Engl. J. Med. 2018, 379, 270–280. [Google Scholar] [CrossRef] [PubMed]
- van Amsterdam, J.; Brunt, T.; van den Brink, W. The adverse health effects of synthetic cannabinoids with emphasis on psychosis-like effects. J. Psychopharmacol. 2015, 29, 254–263. [Google Scholar] [CrossRef]
- Sideli, L.; Quigley, H.; La Cascia, C.; Murray, R.M. Cannabis Use and the Risk for Psychosis and Affective Disorders. J. Dual Diagn. 2020, 16, 22–42. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: Implications in psychosis and schizophrenia. Front. Pharmacol. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javitt, D.C. Glutamate and Schizophrenia: Phencyclidine, N-Methyl-d-Aspartate Receptors, and Dopamine–Glutamate Interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar]
- Sieghart, W.; Sperk, G. Subunit Composition, Distribution and Function of GABA-A Receptor Subtypes. Curr. Top. Med. Chem. 2002, 2, 795–816. [Google Scholar] [CrossRef] [PubMed]
- Sigel, E.; Steinmann, M.E. Structure, Function, and Modulation of GABAA Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [Green Version]
- Brickley, S.G.; Mody, I. Extrasynaptic GABAA Receptors: Their Function in the CNS and Implications for Disease. Neuron 2012, 73, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MÖHLER, H. GABA A Receptors in Central Nervous System Disease: Anxiety, Epilepsy, and Insomnia. J. Recept. Signal Transduct. 2006, 26, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, U.; Knoflach, F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 2011, 10, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanovsky, Y.; Schubring, S.; Fleischer, W.; Gisselmann, G.; Zhu, X.-R.; Lübbert, H.; Hatt, H.; Rudolph, U.; Haas, H.L.; Sergeeva, O.A. GABAA receptors involved in sleep and anaesthesia: β1- versus β3-containing assemblies. Pflügers Arch. Eur. J. Physiol. 2012, 463, 187–199. [Google Scholar] [CrossRef]
- Olsen, R.W. Allosteric Ligands and Their Binding Sites Define γ-Aminobutyric Acid (GABA) Type a Receptor Subtypes. Adv. Pharmacol. 2015, 73, 167–202. [Google Scholar]
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y.; Howard, R.J.; Zhu, S.; Noviello, C.M.; Walsh, R.M.; Lindahl, E.; Hibbs, R.E. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABAA receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef]
- Sigel, E.; Baur, R.; Racz, I.; Marazzi, J.; Smart, T.G.; Zimmer, A.; Gertsch, J. The major central endocannabinoid directly acts at GABAA receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 18150–18155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakas, T.; van Nieuwenhuijzen, P.S.; Devenish, S.O.; McGregor, I.S.; Arnold, J.C.; Chebib, M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABA A receptors. Pharmacol. Res. 2017, 119, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Grudzinska, J.; Schemm, R.; Haeger, S.; Nicke, A.; Schmalzing, G.; Betz, H.; Laube, B. The β Subunit Determines the Ligand Binding Properties of Synaptic Glycine Receptors. Neuron 2005, 45, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, G.; Kirsch, J.; Betz, H.; Langosch, D. Identification of a gephyrin binding motif on the glycine receptor β subunit. Neuron 1995, 15, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Malosio, M.L.; Marquèze-Pouey, B.; Kuhse, J.; Betz, H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J. 1991, 10, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Pilorge, M.; Fassier, C.; Le Corronc, H.; Potey, A.; Bai, J.; De Gois, S.; Delaby, E.; Assouline, B.; Guinchat, V.; Devillard, F.; et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 2016, 21, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Winkelmann, A.; Maggio, N.; Eller, J.; Caliskan, G.; Semtner, M.; Häussler, U.; Jüttner, R.; Dugladze, T.; Smolinsky, B.; Kowalczyk, S.; et al. Changes in neural network homeostasis trigger neuropsychiatric symptoms. J. Clin. Investig. 2014, 124, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, J.; Demir, R.; Leuwer, M.; de la Roche, J.; Krampfl, K.; Foadi, N.; Karst, M.; Haeseler, G. The Nonpsychotropic Cannabinoid Cannabidiol Modulates and Directly Activates Alpha-1 and Alpha-1-Beta Glycine Receptor Function. Pharmacology 2009, 83, 217–222. [Google Scholar] [CrossRef]
- Xiong, W.; Cui, T.; Cheng, K.; Yang, F.; Chen, S.-R.; Willenbring, D.; Guan, Y.; Pan, H.-L.; Ren, K.; Xu, Y.; et al. Cannabinoids suppress inflammatory and neuropathic pain by targeting α3 glycine receptors. J. Exp. Med. 2012, 209, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Foadi, N.; Leuwer, M.; Demir, R.; Dengler, R.; Buchholz, V.; de la Roche, J.; Karst, M.; Haeseler, G.; Ahrens, J. Lack of positive allosteric modulation of mutated α1S267I glycine receptors by cannabinoids. Naunyn Schmiedeberg’s Arch. Pharmacol. 2010, 381, 477–482. [Google Scholar] [CrossRef]
- Fakhfouri, G.; Rahimian, R.; Dyhrfjeld-Johnsen, J.; Zirak, M.R.; Beaulieu, J.-M. 5-HT 3 Receptor Antagonists in Neurologic and Neuropsychiatric Disorders: The Iceberg Still Lies beneath the Surface. Pharmacol. Rev. 2019, 71, 383–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, C.; Cichon, S.; Mühleisen, T.W.; Haenisch, B.; Degenhardt, F.; Mattheisen, M.; Breuer, R.; Witt, S.H.; Strohmaier, J.; Oruc, L.; et al. Replication of functional serotonin receptor type 3A and B variants in bipolar affective disorder: A European multicenter study. Transl. Psychiatry 2012, 2, e103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juza, R.; Vlcek, P.; Mezeiova, E.; Musilek, K.; Soukup, O.; Korabecny, J. Recent advances with 5-HT 3 modulators for neuropsychiatric and gastrointestinal disorders. Med. Res. Rev. 2020, 40, 1593–1678. [Google Scholar] [CrossRef]
- Yang, K.-H.; Galadari, S.; Isaev, D.; Petroianu, G.; Shippenberg, T.S.; Oz, M. The Nonpsychoactive Cannabinoid Cannabidiol Inhibits 5-Hydroxytryptamine 3A Receptor-Mediated Currents in Xenopus laevis Oocytes. J. Pharmacol. Exp. Ther. 2010, 333, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Vitale, R.M.; Moriello, A.S.; De Petrocellis, L. Chapter 6. Natural Compounds and Synthetic Drugs Targeting the Ionotropic Cannabinoid Members of Transient Receptor Potential (TRP) Channels. In New Tools to Interrogate Endocannabinoid Signalling: From Natural Compounds to Synthetic Drugs; The Royal Society of Chemistry: London, UK, 2020; pp. 201–300. [Google Scholar]
- Gibson, H.E.; Edwards, J.G.; Page, R.S.; Van Hook, M.J.; Kauer, J.A. TRPV1 Channels Mediate Long-Term Depression at Synapses on Hippocampal Interneurons. Neuron 2008, 57, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.J.; Gereau, R.W. Hotheaded: TRPV1 as Mediator of Hippocampal Synaptic Plasticity. Neuron 2008, 57, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Nazıroğlu, M.; Taner, A.N.; Balbay, E.; Çiğ, B. Inhibitions of anandamide transport and FAAH synthesis decrease apoptosis and oxidative stress through inhibition of TRPV1 channel in an in vitro seizure model. Mol. Cell. Biochem. 2019, 453, 143–155. [Google Scholar] [CrossRef]
- Shirakawa, H.; Yamaoka, T.; Sanpei, K.; Sasaoka, H.; Nakagawa, T.; Kaneko, S. TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem. Biophys. Res. Commun. 2008, 377, 1211–1215. [Google Scholar] [CrossRef]
- Bhaskaran, M.D.; Smith, B.N. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol. 2010, 223, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.-L.; Min, J.-W.; Liu, Y.-L.; Li, J.-X.; He, X.-H.; Peng, B.-W. Role of TRPV1 in susceptibility to PTZ-induced seizure following repeated hyperthermia challenges in neonatal mice. Epilepsy Behav. 2014, 31, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-X.; Yu, F.; Sanchez, R.M.; Liu, Y.-Q.; Min, J.-W.; Hu, J.-J.; Bsoul, N.B.; Han, S.; Yin, J.; Liu, W.-H.; et al. TRPV1 promotes repetitive febrile seizures by pro-inflammatory cytokines in immature brain. Brain. Behav. Immun. 2015, 48, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Nazıroglu, M. TRPV1 Channel: A Potential Drug Target for Treating Epilepsy. Curr. Neuropharmacol. 2015, 13, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.-J.; Guo, W.; Zheng, D.-H.; Zhang, C.-Q.; Li, S.; Liu, S.-Y.; Yin, Q.; Yang, H.; Shu, H.-F. Increased Expression of TRPV1 in the Cortex and Hippocampus from Patients with Mesial Temporal Lobe Epilepsy. J. Mol. Neurosci. 2013, 49, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Cortright, D.N.; Szallasi, A. Biochemical pharmacology of the vanilloid receptor TRPV1. An update. Eur. J. Biochem. 2004, 271, 1814–1819. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günaydın, C.; Arslan, G.; Bilge, S.S. Proconvulsant effect of trans-cinnamaldehyde in pentylenetetrazole-induced kindling model of epilepsy: The role of TRPA1 channels. Neurosci. Lett. 2020, 721, 134823. [Google Scholar] [CrossRef]
- Iffland, K.; Grotenhermen, F. An Update on Safety and Side Effects of Cannabidiol: A Review of Clinical Data and Relevant Animal Studies. Cannabis Cannabinoid Res. 2017, 2, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Szabo, G.G.; Lenkey, N.; Holderith, N.; Andrasi, T.; Nusser, Z.; Hajos, N. Presynaptic Calcium Channel Inhibition Underlies CB1 Cannabinoid Receptor-Mediated Suppression of GABA Release. J. Neurosci. 2014, 34, 7958–7963. [Google Scholar] [CrossRef]
- Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [CrossRef] [Green Version]
- Gerdeman, G.; Lovinger, D.M. CB1 Cannabinoid Receptor Inhibits Synaptic Release of Glutamate in Rat Dorsolateral Striatum. J. Neurophysiol. 2001, 85, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Sánchez, A.; Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol. Brain 2013, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Khurana, L.; Mackie, K.; Piomelli, D.; Kendall, D.A. Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology 2017, 124, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Albert, P.R.; Vahid-Ansari, F. The 5-HT1A receptor: Signaling to behavior. Biochimie 2019, 161, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R. Transcriptional regulation of the 5-HT 1A receptor: Implications for mental illness. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2402–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Garcia, A.L.; Newman-Tancredi, A.; Leonardo, E.D. 5-HT1A receptors in mood and anxiety: Recent insights into autoreceptor versus heteroreceptor function. Psychopharmacology 2014, 231, 623–636. [Google Scholar] [CrossRef] [Green Version]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic Properties of Cannabidiol at 5-HT1a Receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Rock, E.; Bolognini, D.; Limebeer, C.; Cascio, M.; Anavi-Goffer, S.; Fletcher, P.; Mechoulam, R.; Pertwee, R.; Parker, L. Cannabidiol, a non-psychotropic component of cannabis, attenuates vomiting and nausea-like behaviour via indirect agonism of 5-HT1A somatodendritic autoreceptors in the dorsal raphe nucleus. Br. J. Pharmacol. 2012, 165, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Zanelati, T.; Biojone, C.; Moreira, F.; Guimarães, F.; Joca, S. Antidepressant-like effects of cannabidiol in mice: Possible involvement of 5-HT1A receptors. Br. J. Pharmacol. 2010, 159, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.C.; Guimarães, F.S. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology 2008, 199, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sartim, A.G.; Guimarães, F.S.; Joca, S.R.L. Antidepressant-like effect of cannabidiol injection into the ventral medial prefrontal cortex—Possible involvement of 5-HT1A and CB1 receptors. Behav. Brain Res. 2016, 303, 218–227. [Google Scholar] [CrossRef]
- Sales, A.J.; Crestani, C.C.; Guimarães, F.S.; Joca, S.R.L. Antidepressant-like effect induced by Cannabidiol is dependent on brain serotonin levels. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 86, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Norris, C.; Loureiro, M.; Kramar, C.; Zunder, J.; Renard, J.; Rushlow, W.; Laviolette, S.R. Cannabidiol Modulates Fear Memory Formation Through Interactions with Serotonergic Transmission in the Mesolimbic System. Neuropsychopharmacology 2016, 41, 2839–2850. [Google Scholar] [CrossRef]
- Laun, A.S.; Song, Z.-H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem. Biophys. Res. Commun. 2017, 490, 17–21. [Google Scholar] [CrossRef]
- Valverde, O.; Célérier, E.; Baranyi, M.; Vanderhaeghen, P.; Maldonado, R.; Sperlagh, B.; Vassart, G.; Ledent, C. GPR3 Receptor, a Novel Actor in the Emotional-Like Responses. PLoS ONE 2009, 4, e4704. [Google Scholar] [CrossRef] [Green Version]
- Oeckl, P.; Hengerer, B.; Ferger, B. G-protein coupled receptor 6 deficiency alters striatal dopamine and cAMP concentrations and reduces dyskinesia in a mouse model of Parkinson’s disease. Exp. Neurol. 2014, 257, 1–9. [Google Scholar] [CrossRef]
- Komatsu, H. Novel Therapeutic GPCRs for Psychiatric Disorders. Int. J. Mol. Sci. 2015, 16, 14109–14121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laun, A.S.; Shrader, S.H.; Song, Z.-H. Novel inverse agonists for the orphan G protein-coupled receptor 6. Heliyon 2018, 4, e00933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2009, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, L.; Shi, W.; Wang, L.; Zhou, S.; Guan, S.; Zhao, M.; Yang, Q. The novel cannabinoid receptor GPR55 mediates anxiolytic-like effects in the medial orbital cortex of mice with acute stress. Mol. Brain 2017, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Tai, C.; Jones, C.J.; Scheuer, T.; Catterall, W.A. Enhancement of Inhibitory Neurotransmission by GABA A Receptors Having α 2,3 -Subunits Ameliorates Behavioral Deficits in a Mouse Model of Autism. Neuron 2014, 81, 1282–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Ossa, F.; Schnell, J.R.; Ortega-Roldan, J.L. A Review of the Human Sigma-1 Receptor Structure. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology, vol 964; Smith, S., Su, T.P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 15–29. ISBN 978-3-319-50172-7. [Google Scholar]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Pozo-Rodrigálvarez, A.; Sánchez-Blázquez, P.; Garzón-Niño, J. The ON:OFF switch, σ1R-HINT1 protein, controls GPCR-NMDA receptor cross-regulation: Implications in neurological disorders. Oncotarget 2015, 6, 35458–35477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Muñoz, M.; Onetti, Y.; Cortés-Montero, E.; Garzón, J.; Sánchez-Blázquez, P. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol. Brain 2018, 11, 51. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Iannotti, F.A.; Vitale, R.M. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Domi, E.; Uhrig, S.; Soverchia, L.; Spanagel, R.; Hansson, A.C.; Barbier, E.; Heilig, M.; Ciccocioppo, R.; Ubaldi, M. Genetic Deletion of Neuronal PPARγ Enhances the Emotional Response to Acute Stress and Exacerbates Anxiety: An Effect Reversed by Rescue of Amygdala PPARγ Function. J. Neurosci. 2016, 36, 12611–12623. [Google Scholar] [CrossRef]
- de Guglielmo, G.; Melis, M.; De Luca, M.A.; Kallupi, M.; Li, H.W.; Niswender, K.; Giordano, A.; Senzacqua, M.; Somaini, L.; Cippitelli, A.; et al. PPARγ Activation Attenuates Opioid Consumption and Modulates Mesolimbic Dopamine Transmission. Neuropsychopharmacology 2015, 40, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopponi, S.; Somaini, L.; Cippitelli, A.; Cannella, N.; Braconi, S.; Kallupi, M.; Ruggeri, B.; Heilig, M.; Demopulos, G.; Gaitanaris, G.; et al. Activation of Nuclear PPARγ Receptors by the Antidiabetic Agent Pioglitazone Suppresses Alcohol Drinking and Relapse to Alcohol Seeking. Biol. Psychiatry 2011, 69, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Di Ciano, P.; Panlilio, L.V.; Goldberg, S.R.; Ciccocioppo, R. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists as Promising New Medications for Drug Addiction: Preclinical Evidence. Curr. Drug Targets 2013, 14, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domi, E.; Caputi, F.F.; Romualdi, P.; Domi, A.; Scuppa, G.; Candeletti, S.; Atkins, A.; Heilig, M.; Demopulos, G.; Gaitanaris, G.; et al. Activation of PPARγ Attenuates the Expression of Physical and Affective Nicotine Withdrawal Symptoms through Mechanisms Involving Amygdala and Hippocampus Neurotransmission. J. Neurosci. 2019, 39, 9864–9875. [Google Scholar] [CrossRef]
- Schmitz, J.M.; Green, C.E.; Hasan, K.M.; Vincent, J.; Suchting, R.; Weaver, M.F.; Moeller, F.G.; Narayana, P.A.; Cunningham, K.A.; Dineley, K.T.; et al. PPAR-gamma agonist pioglitazone modifies craving intensity and brain white matter integrity in patients with primary cocaine use disorder: A double-blind randomized controlled pilot trial. Addiction 2017, 112, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Comer, S.D.; Metz, V.E.; Manubay, J.M.; Mogali, S.; Ciccocioppo, R.; Martinez, S.; Mumtaz, M.; Bisaga, A. Pioglitazone, a PPARγ agonist, reduces nicotine craving in humans, with marginal effects on abuse potential. Pharmacol. Biochem. Behav. 2017, 163, 90–100. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2009, 152, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Hudson, R.; Rushlow, W.; Laviolette, S.R. Functional interactions between cannabinoids, omega-3 fatty acids, and peroxisome proliferator-activated receptors: Implications for mental health pharmacotherapies. Eur. J. Neurosci. 2020, ejn.15023. [Google Scholar] [CrossRef] [PubMed]
- Renard, J.; Norris, C.; Rushlow, W.; Laviolette, S.R. Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: Implications for novel schizophrenia treatments. Neurosci. Biobehav. Rev. 2017, 75, 157–165. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, R.M.; Iannotti, F.A.; Amodeo, P. The (Poly)Pharmacology of Cannabidiol in Neurological and Neuropsychiatric Disorders: Molecular Mechanisms and Targets. Int. J. Mol. Sci. 2021, 22, 4876. https://doi.org/10.3390/ijms22094876
Vitale RM, Iannotti FA, Amodeo P. The (Poly)Pharmacology of Cannabidiol in Neurological and Neuropsychiatric Disorders: Molecular Mechanisms and Targets. International Journal of Molecular Sciences. 2021; 22(9):4876. https://doi.org/10.3390/ijms22094876
Chicago/Turabian StyleVitale, Rosa Maria, Fabio Arturo Iannotti, and Pietro Amodeo. 2021. "The (Poly)Pharmacology of Cannabidiol in Neurological and Neuropsychiatric Disorders: Molecular Mechanisms and Targets" International Journal of Molecular Sciences 22, no. 9: 4876. https://doi.org/10.3390/ijms22094876