
Radiation- and Photo-Induced Oxidation Pathways of Methionine in Model Peptide Backbone under Anoxic Conditions

 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Pulse Radiolysis Studies
2.2. γ-Radiolysis and Product Analysis
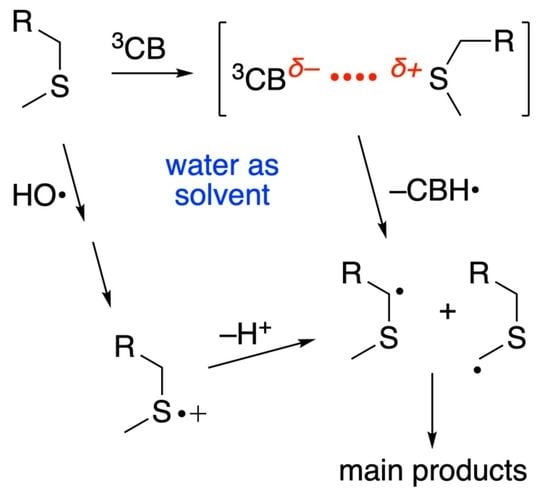
2.3. Photosensitized Oxidation by 3CB
- (i)
- In the interval of 17.5–19.5 min there are three major peaks that correspond to compounds 8, 9 and 10, and 2 minor peaks that correspond to compounds 7 and 11, which all are the dimers of αS• radicals observed in γ-radiolysis experiments (cf. Scheme 3). It is worth underlining that the accurate masses of these products and the fragmentation patterns are identical in all sets of experiments (Figure S10). We assigned the structures 8, 9 and 10 to the 3 diastereoisomers of the αS(2)–αS(2) dimers and 7 and 11 (minor peaks) to the 2 diastereoisomers of αS(2)–αS(1) reported in the previous radiolysis section (see Scheme 5).
- (ii)
- In the interval of 32–34 min there are two peaks that are individuated as compounds 18 and 19 (Figure 6). Their accurate masses (m/z 455.1527, and 455.1522) correspond to the MH+ of the dimer CBH–CBH (Figure S11). Figure 7 shows that CBH–CBH has two stereocenters and a plane of symmetry that correspond to erythro and threo diastereoisomers.
- (iii)
- In the interval of 25–29 min there is the major peak that corresponds to CB, with two doublets on the right and left sides, respectively, and one singlet in the shoulder of CB (Figure 6). In this area of HPLC run there are the cross-coupling products of αS• and CBH• radicals. The accurate masses of two couples of compounds named 12, 13 (m/z 414.1391, 414.1393) and 16, 17 (m/z 414.1390, 414.1393), as well as their fragmentation patterns, are identical and assigned to αS(2)–CBH (Scheme 5 and Figure S12). Figure 7 shows that αS(2)–CBH has three stereocenters, one is from the starting material fixed at the S configuration and two stereocenters are generated from the cross-termination of the two radicals, producing the diastereoisomers SSS, SRS, SSR and SRR. Regarding the singlet in the shoulder of CB, having also m/z 414.1392, but different fragmentation patterns, it is assigned to αS(1)–CBH (Figure S13). As shown in Figure 7, this compound has two stereocenters, the usual S configuration from the starting material and a new one generated from the cross-termination of the two radicals, producing the diastereoisomers SS and SR. It is likely that, under our HPLC conditions, the two diastereoisomers be under the same peak, or one of them overlap with CB.
3. Materials and Methods
3.1. Pulse Radiolysis
3.2. Spectral Resolutions of Transient Absorption Spectra
3.3. Steady-State γ-Radiolysis
3.4. Laser Flash Photolysis
3.5. Steady-State Photolysis
3.6. LC-MS/MS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Levine, R.L. Methionine in proteins defends against oxidative stress. FASEB J. 2009, 23, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.M.; Kim, G.; Levine, R.L. Methionine in Proteins: It’s Not Just for Protein Initiation Anymore. Neurochem. Res. 2019, 44, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Javitt, G.; Cao, Z.; Resnick, E.; Gabizon, R.; Bulleid, N.J.; Fass, D. Structure and Electron-Transfer Pathway of the Human Methionine Sulfoxide Reductase MsrB3. Antioxid. Redox Signal. 2020, 33, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (●OH/●O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Schöneich, C. Radical-Based Damage of Sulfur-Containing Amino Acid Residues. In Encyclopedia of Radical in Chemistry, Biology and Materials; Studer, A.S., Ed.; John Wiley & Sons: Chichester, UK, 2012; Volume 3, pp. 1459–1474. [Google Scholar]
- Schöneich, C. Sulfur Radical-Induced Redox Modifications in Proteins: Analysis and Mechanistic Aspects. Antioxid. Redox Signal. 2017, 26, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C.; Bobrowski, K. Intramolecular hydrogen transfer as the key step in the dissociation of hydroxyl radical adducts of (alkylthio)ethanol derivatives. J. Am. Chem. Soc. 1993, 115, 6538–6547. [Google Scholar] [CrossRef]
- Houée-Levin, C.; Bobrowski, K. The use of methods of radiolysis to explore the mechanisms of free radical modifications in proteins. J. Proteom. 2013, 92, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.S.; Hug, G.L.; Schöneich, C.; Wilson, G.S.; Kuznetsova, L.; Lee, T.; Ammam, M.; Lorance, E.; Nauser, T.; Nichol, G.S.; et al. Neighboring Amide Participation in Thioether Oxidation: Relevance to Biological Oxidation. J. Am. Chem. Soc. 2009, 131, 13791–13805. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Wisniowski, P.; Hug, G.L.; Bobrowski, K. Intramolecular Sulfur-Oxygen Bond Formation in Radical Cations of N-Acetylmethionine Amide. J. Am. Chem. Soc. 2000, 122, 10224–10225. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free radical reactions of methionine in peptides: Mechanisms relevant to b-amyloid oxidation and Alzheimer’s disease. J. Am. Chem. Soc. 2003, 125, 13700–13713. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schöneich, C. Stabilization of sulfide radical cations through complexation with the peptide bond: Mechanisms relevant to oxidation of proteins containing multiple methionine residues. J. Phys. Chem. B 2007, 111, 9608–9620. [Google Scholar] [CrossRef] [PubMed]
- Hug, G.L.; Bobrowski, K.; Pogocki, D.; Hörner, G.; Marciniak, B. Conformational influence on the type of stabilization of sulfur radical cations in cyclic peptides. ChemPhysChem 2007, 8, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Ferreri, C.; Zhang, T.; Permentier, H.; Bischoff, R.; Bobrowski, K.; Chatgilialoglu, C. Radiation chemical studies of Gly-Met-Gly in aqueous solution. Free Radic. Res. 2016, 50, S24–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schöneich, C. Sulfur radical cation peptide bond complex in the one-electron oxidation of S-methylglutathione. J. Am. Chem. Soc. 2007, 129, 9236–9245. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Ferreri, C.; Chatgilialoglu, C. Radiation chemical studies of methionine in aqueous solution: Understanding the role of molecular oxygen. Chem. Res. Toxicol. 2010, 23, 258–263. [Google Scholar] [CrossRef]
- Torreggiani, A.; Barata-Vallejo, S.; Chatgilialoglu, C. Combined Raman and IR spectroscopic studies on the radical-based modifications of methionine. Anal. Bioanal. Chem. 2011, 401, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Houée-Levin, C.; Marciniak, B. Stabilization and Reactions of Sulfur Radical Cations: Relevance to One-Electron Oxidation of Methionine in Peptides and Proteins. Chimia 2008, 62, 728–734. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Schöneich, C.; Marciniak, B. N-Terminal Decarboxylation as a Probe for Intramolecular Contact Formation in γ-Glu-(Pro)n-Met Peptides. J. Phys. Chem. B 2020, 124, 8082–8098. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Kozubek, H.; Marciniak, B. Photo-oxidation of Methionine-containing Peptides by the 4-Carboxybenzophenone Triplet State in Aqueous Solution. Competition between Intramolecular Two-centered Three-electron Bonded (S\S)+ and (S\N)+ Formation. Photochem. Photobiol. 2000, 72, 1–9. [Google Scholar] [CrossRef]
- Ignasiak, M.T.; Marciniak, B.; Houée-Levin, C. A Long Story of Sensitized One-Electron Photo-oxidation of Methionine. Isr. J. Chem. 2014, 54, 248–253. [Google Scholar] [CrossRef]
- Pędzinski, T.; Grzyb, K.; Kaźmierczak, F.; Frański, R.; Filipiak, P.; Marciniak, B. Early Events of Photosensitized Oxidation of Sulfur-Containing Amino Acids Studied by Laser Flash Photolysis and Mass Spectrometry. J. Phys. Chem. B 2020, 124, 7564–7573. [Google Scholar] [CrossRef]
- Mönig, J.; Goslich, R.; Asmus, K.-D. Thermodynamics of S\S 2σ/1σ* three-electron bonds and deprotonation kinetics of thioether radical cations in aqueous solution. Ber. Bunsenges. Phys. Chem. 1986, 90, 115–121. [Google Scholar] [CrossRef]
- Rauk, A.; Yu, D.; Taylor, J.; Shustov, G.V.; Block, D.A.; Armstrong, D.A. Effects of Structure on aC-H Bond Enthalpies of Amino Acid residues: Relevance to H-Transfers in Enzyme mechanisms and in Protein Oxidation. Biochemistry 1999, 38, 9089–9096. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chemistry of Acyl Radicals. Chem. Rev. 1999, 114, 1991–2069. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Melchiore, M.; Sansone, A.; Torreggiani, A. Lipid geometrical isomerism: From chemistry to biology and diagnostics. Chem. Rev. 2014, 114, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Torreggiani, A.; Salzano, A.M.; Renzone, G.; Scaloni, A. Radiation-induced reductive modifications of sulfur-containing amino acids within peptides and proteins. J. Proteom. 2011, 74, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Pędzinski, T.; Markiewicz, A.; Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Res. Chem. Intermed. 2009, 35, 497–506. [Google Scholar] [CrossRef]
- Ignasiak, M.T.; Pędzinski, T.; Rusconi, F.; Filipiak, P.; Bobrowski, K.; Houée-Levin, C.; Marciniak, B. Photosensitized Oxidation of Methionine-Containing Dipeptides. From the Transients to the Final Products. J. Phys. Chem. B 2014, 118, 8549–8558. [Google Scholar] [CrossRef]
- Bobrowski, K. Free radicals in chemistry, biology and medicine: Contribution of radiation chemistry. Nukleonika 2005, 50 (Suppl. 3), S67–S76. [Google Scholar]
- Mirkowski, J.; Wiśniowski, P.; Bobrowski, K. INCT Annual Report 2000; INCT: Warsaw, Poland, 2001. [Google Scholar]
- Schuler, R.H.; Hartzell, A.L.; Behar, B. Track effects in radiation chemistry. Concentration dependence for the scavenging of OH by ferrocyanide in N2O-saturated aqueous solutions. J. Phys. Chem. 1981, 85, 192–199. [Google Scholar] [CrossRef]
- Janata, E.; Schuler, R.H. Rate constant for scavenging e−aq in N2O-saturated solutions. J. Phys. Chem. 1982, 86, 2078–2084. [Google Scholar] [CrossRef]
- Klassen, N.V.; Shortt, K.R.; Seuntjens, J.; Ross, C.K. Fricke dosimetry: The difference between G(Fe3+) for 60Co gamma-rays and high-energy X-rays. Phys. Med. Biol. 1999, 44, 1609–1624. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (µs) | HOS● | αC● | αS(1)● + αS(2)● | SS●+ | SN● | Total R● |
|---|---|---|---|---|---|---|
| 1.1 | 0.39 (73.6%) | 0.06 (11.3%) | 0.01 (1.9%) | 0.04 (7.5%) | 0.03 (5.7%) | 0.53 |
| 3 | 0.16 (28.6%) | 0.02 (3.6%) | 0.17 (30.3%) | 0.11 (19.6%) | 0.10 (17.9%) | 0.56 |
| 6 | 0.03 (5.3%) | 0.02 (3.5%) | 0.26 (45.6%) | 0.13 (22.8%) | 0.13 (22.8%) | 0.57 |
| k, s−1 | HOS● | αC● | αS● | SS●+ | SN● |
|---|---|---|---|---|---|
| kgrowth | 2.1 × 106 1.1 × 1010 a | 1.5 × 106 7.5 × 109 a | 4.1 × 105 b 3.6 × 104 c | 4.7 × 105 2.2 × 109 d | 3.7 × 105 |
| kdecay | 5.6 × 105 | 1.4 × 106 | 4.0 × 103 | 3.6 × 104 | 8.2 × 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pędzinski, T.; Grzyb, K.; Skotnicki, K.; Filipiak, P.; Bobrowski, K.; Chatgilialoglu, C.; Marciniak, B. Radiation- and Photo-Induced Oxidation Pathways of Methionine in Model Peptide Backbone under Anoxic Conditions. Int. J. Mol. Sci. 2021, 22, 4773. https://doi.org/10.3390/ijms22094773
Pędzinski T, Grzyb K, Skotnicki K, Filipiak P, Bobrowski K, Chatgilialoglu C, Marciniak B. Radiation- and Photo-Induced Oxidation Pathways of Methionine in Model Peptide Backbone under Anoxic Conditions. International Journal of Molecular Sciences. 2021; 22(9):4773. https://doi.org/10.3390/ijms22094773
Chicago/Turabian StylePędzinski, Tomasz, Katarzyna Grzyb, Konrad Skotnicki, Piotr Filipiak, Krzysztof Bobrowski, Chryssostomos Chatgilialoglu, and Bronislaw Marciniak. 2021. "Radiation- and Photo-Induced Oxidation Pathways of Methionine in Model Peptide Backbone under Anoxic Conditions" International Journal of Molecular Sciences 22, no. 9: 4773. https://doi.org/10.3390/ijms22094773