
Connection between Periodontitis-Induced Low-Grade Endotoxemia and Systemic Diseases: Neutrophils as Protagonists and Targets

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
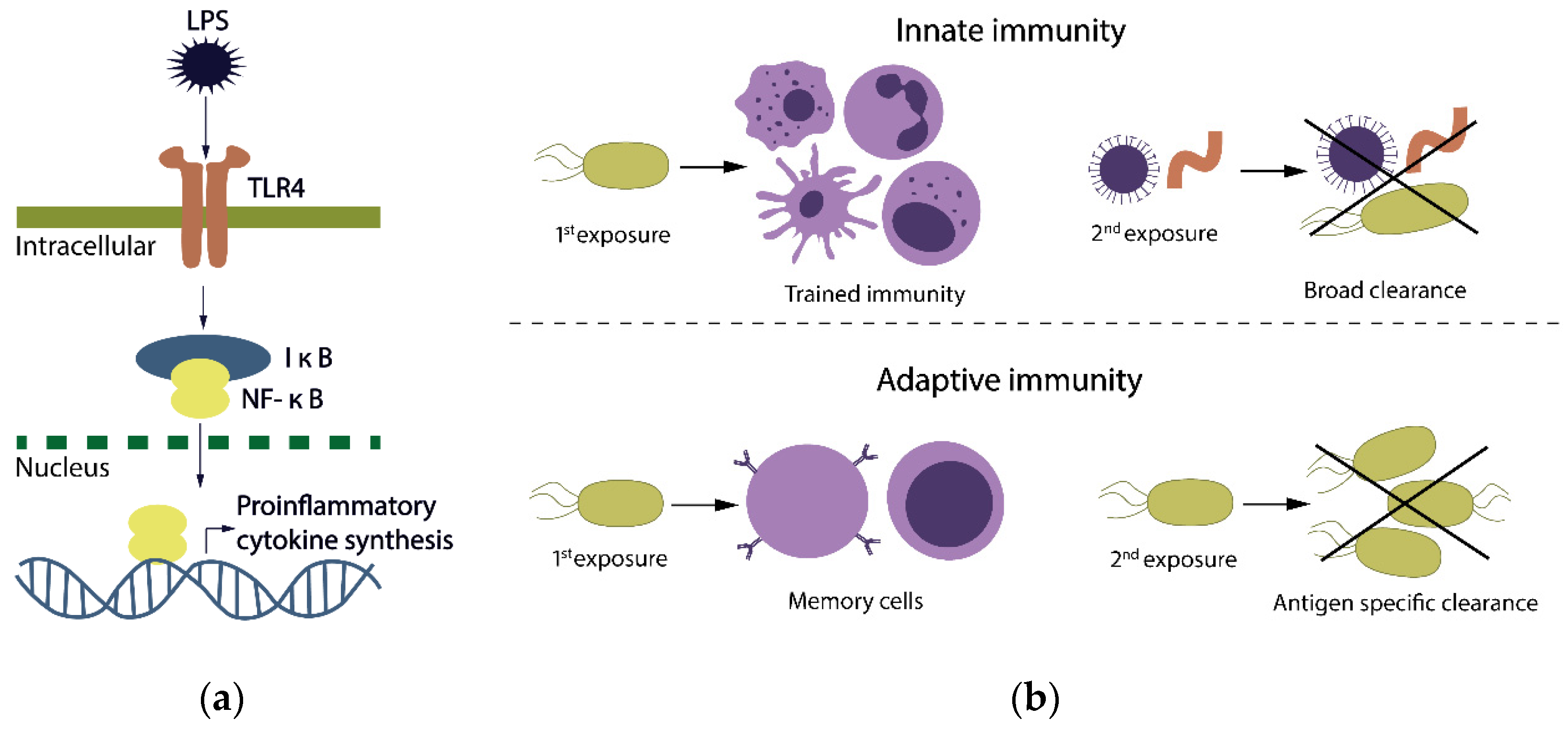
2. Development of Trained Immunity
3. LGE Induces Systemic Diseases
4. Trained Immunity and Neutrophil Hyper-Responsiveness
5. Correlation between Periodontitis and LGE-Related SDs, the Role of Periodontal Pathogens
- (i)
- (ii)
- Trained immunity marked by periodontitis-induced neutrophil hyper-responsiveness [47,48,49,50]. This aspect might be responsible for the gingiva ulceration, and hence might act as a promoter of both LGE and bacteremia (see Section 6.2.);
- (iii)
6. Endotoxemia and LPS Metabolism
- LPS penetration occurs via the inflamed periodontal pocket.
- LPS penetration might be to, a large part, a consequence of transient bacteremia.
- Intermittent pressure on the pocket epithelium appears to be crucial for bacteremia and subsequent endotoxemia.
6.1. LPS Penetrates the Periodontal Pocket
6.2. Ulcerations Circumvent the Epithelial Barrier in Periodontitis
6.3. The Pocket Pump
7. Targeting Neutrophils in the Prophylaxis of Systemic Diseases and Periodontitis
7.1. Reverting the Polarized Neutrophils back to the Homeostatic State
7.2. Diminishing the Exaggerated Crevicular NETs
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FPN | ferroportin |
| GCF | gingival crevicular fluid |
| HDL | high density lipoprotein |
| HSPC | hematopoietic stem and progenitor cells |
| IL | interleukin |
| LBP | LPS-binding protein |
| LDL | low density lipoprotein |
| LGE | low-grade endoxemia |
| LPS | lipopolysaccharide |
| LRR | leucine-rich repeat containing |
| MDPI | multidisciplinary Digital Publishing Institute |
| MMP | matrix metalloprotease |
| NETs | neutrophil extracellular traps |
| SDs | systemic diseases |
| PADI4 | peptidylarginine deiminases 4 |
| PAMPs | pathogen associated molecular patterns |
| PMN | polymorphonuclear cells |
| SLGI | systemic low-grade inflammation |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| Treg | Regulatory T cells |
References
- Fine, N.; Chadwick, J.W.; Sun, C.; Parbhakar, K.K.; Khoury, N.; Barbour, A.; Goldberg, M.; Tenenbaum, H.C.; Glogauer, M. Periodontal Inflammation Primes the Systemic Innate Immune Response. J. Dent. Res. 2020. [Google Scholar] [CrossRef]
- Pussinen, P.; Paju, S.; Mantyla, P.; Sorsa, T. Serum Microbial- and Host-Derived Markers of Periodontal Diseases: A Review. Curr. Med. Chem. 2007, 14, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From Microbial Immune Subversion to Systemic Inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Holmstrup, P.; Damgaard, C.; Olsen, I.; Klinge, B.; Flyvbjerg, A.; Nielsen, C.H.; Hansen, P.R. Comorbidity of Periodontal Disease: Two Sides of the Same Coin? An Introduction for the Clinician. J. Oral Microbiol. 2017, 9, 1332710. [Google Scholar] [CrossRef]
- Liljestrand, J.M.; Paju, S.; Buhlin, K.; Persson, G.R.; Sarna, S.; Nieminen, M.S.; Sinisalo, J.; Mäntylä, P.; Pussinen, P.J. Lipopolysaccharide, a Possible Molecular Mediator between Periodontitis and Coronary Artery Disease. J. Clin. Periodontol. 2017, 44, 784–792. [Google Scholar] [CrossRef]
- Khumaedi, A.I.; Purnamasari, D.; Wijaya, I.P.; Soeroso, Y. The Relationship of Diabetes, Periodontitis and Cardiovascular Disease. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1675–1678. [Google Scholar] [CrossRef]
- Jepsen, S.; Suvan, J.; Deschner, J. The Association of Periodontal Diseases with Metabolic Syndrome and Obesity. Periodontology 2000 2020, 83, 125–153. [Google Scholar] [CrossRef]
- Munford, R.S. Endotoxemia-Menace, Marker, or Mistake? J. Leukoc. Biol. 2016, 100, 687–698. [Google Scholar] [CrossRef]
- Nicu, E.A.; Laine, M.L.; Morré, S.A.; Van der Velden, U.; Loos, B.G. Soluble CD14 in Periodontitis. Innate Immun. 2009, 15, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Shaddox, L.M.; Wiedey, J.; Calderon, N.L.; Magnusson, I.; Bimstein, E.; Bidwell, J.A.; Zapert, E.F.; Aukhil, I.; Wallet, S.M. Local Inflammatory Markers and Systemic Endotoxin in Aggressive Periodontitis. J. Dent. Res. 2011, 90, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Eberhard, J.; Grote, K.; Luchtefeld, M.; Heuer, W.; Schuett, H.; Divchev, D.; Scherer, R.; Schmitz-Streit, R.; Langfeldt, D.; Stumpp, N.; et al. Experimental Gingivitis Induces Systemic Inflammatory Markers in Young Healthy Individuals: A Single-Subject Interventional Study. PLoS ONE 2013, 8, e55265. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Diao, N.; Yuan, R.; Chen, K.; Geng, S.; Li, M.; Li, L. Subclinical-Dose Endotoxin Sustains Low-Grade Inflammation and Exacerbates Steatohepatitis in High-Fat Diet–Fed Mice. J. Immunol. 2016, 196, 2300–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.W.; Moorlag, S.J.C.F.M.; Novakovic, B.; Li, Y.; Wang, S.-Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.-S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef] [Green Version]
- de Laval, B.; Maurizio, J.; Kandalla, P.K.; Brisou, G.; Simonnet, L.; Huber, C.; Gimenez, G.; Matcovitch-Natan, O.; Reinhardt, S.; David, E.; et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 2020, 26, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S.J.C.F.M.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Ahmed, S.S.; Curtis, N.; Kollmann, T.R.; Levy, O.; Netea, M.G.; Pollard, A.J.; van Crevel, R.; Wilson, C.B. Harnessing the Beneficial Heterologous Effects of Vaccination. Nat. Rev. Immunol. 2016, 16, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; ONeill, L.A.J.; Xavier, R.J. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Giamarellos-Bourboulis, E.J.; Domínguez-Andrés, J.; Curtis, N.; van Crevel, R.; van de Veerdonk, F.L.; Bonten, M. Trained Immunity: A Tool for Reducing Susceptibility to and the Severity of SARS-CoV-2 Infection. Cell 2020, 181, 969–977. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.-D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240. [Google Scholar] [CrossRef]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic Progenitor Cells as Integrative Hubs for Adaptation to and Fine-Tuning of Inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef]
- Plachetka, A.; Chayka, O.; Wilczek, C.; Melnik, S.; Bonifer, C.; Klempnauer, K.-H. C/EBPβ Induces Chromatin Opening at a Cell-Type-Specific Enhancer. Mol. Cell. Biol. 2008, 28, 2102–2112. [Google Scholar] [CrossRef] [Green Version]
- Grøntved, L.; John, S.; Baek, S.; Liu, Y.; Buckley, J.R.; Vinson, C.; Aguilera, G.; Hager, G.L. C/EBP Maintains Chromatin Accessibility in Liver and Facilitates Glucocorticoid Receptor Recruitment to Steroid Response Elements. EMBO J. 2013, 32, 1568–1583. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin Contributions to the Regulation of Innate Immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef]
- Ghisletti, S.; Barozzi, I.; Mietton, F.; Polletti, S.; De Santa, F.; Venturini, E.; Gregory, L.; Lonie, L.; Chew, A.; Wei, C.-L.; et al. Identification and Characterization of Enhancers Controlling the Inflammatory Gene Expression Program in Macrophages. Immunity 2010, 32, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Natoli, G.; Ostuni, R. Adaptation and Memory in Immune Responses. Nat. Immunol. 2019, 20, 783–792. [Google Scholar] [CrossRef]
- Stoll, L.L.; Denning, G.M.; Weintraub, N.L. Potential Role of Endotoxin as a Proinflammatory Mediator of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2227–2236. [Google Scholar] [CrossRef]
- Smith, B.J.; Lightfoot, S.A.; Lerner, M.R.; Denson, K.D.; Morgan, D.L.; Hanas, J.S.; Bronze, M.S.; Postier, R.G.; Brackett, D.J. Induction of Cardiovascular Pathology in a Novel Model of Low-Grade Chronic Inflammation. Cardiovasc. Pathol. 2009, 18, 1–10. [Google Scholar] [CrossRef]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut Microbiota, Lipopolysaccharides, and Innate Immunity in the Pathogenesis of Obesity and Cardiovascular Risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [Green Version]
- van der Crabben, S.N.; Blümer, R.M.E.; Stegenga, M.E.; Ackermans, M.T.; Endert, E.; Tanck, M.W.T.; Serlie, M.J.; van der Poll, T.; Sauerwein, H.P. Early Endotoxemia Increases Peripheral and Hepatic Insulin Sensitivity in Healthy Humans. J. Clin. Endocrinol. Metab. 2009, 94, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.D.; Mehta, N.N.; Wolfe, M.L.; Hinkle, C.C.; Pruscino, L.; Comiskey, L.L.; Tabita-Martinez, J.; Sellers, K.F.; Rickels, M.R.; Ahima, R.S.; et al. Innate Immunity Modulates Adipokines in Humans. J. Clin. Endocrinol. Metab. 2007, 92, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.W.; Harrison, L.E.A.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.-C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating Endotoxemia: A Novel Factor in Systemic Inflammation and Cardiovascular Disease in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Zhong, C.; Yang, X.; Feng, Y.; Yu, J. Trained Immunity: An Underlying Driver of Inflammatory Atherosclerosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.; Anderson, A.; Appella, E.; Oppenheim, J.; Matsushima, K. The Neutrophil-Activating Protein (NAP-1) Is Also Chemotactic for T Lymphocytes. Science 1989, 243, 1464–1466. [Google Scholar] [CrossRef]
- Barcia, A.M.; Harris, H.W. Triglyceride-Rich Lipoproteins as Agents of Innate Immunity. Clin. Infect. Dis. 2005, 41, S498–S503. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Nelson, A.J. HDL and Cardiovascular Disease. Pathology 2019, 51, 142–147. [Google Scholar] [CrossRef]
- Geng, S.; Chen, K.; Yuan, R.; Peng, L.; Maitra, U.; Diao, N.; Chen, C.; Zhang, Y.; Hu, Y.; Qi, C.-F.; et al. The Persistence of Low-Grade Inflammatory Monocytes Contributes to Aggravated Atherosclerosis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Geng, S.; Zhang, Y.; Lee, C.; Li, L. Novel Reprogramming of Neutrophils Modulates Inflammation Resolution during Atherosclerosis. Sci. Adv. 2019, 5, eaav2309. [Google Scholar] [CrossRef] [Green Version]
- Hamam, H.; Khan, M.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, H.J.; Palaniyar, N. Histone Deacetylase Inhibitors Dose-Dependently Switch Neutrophil Death from NETosis to Apoptosis. Biomolecules 2019, 9, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.-H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [Green Version]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Maitra, U.; Deng, H.; Glaros, T.; Baker, B.; Capelluto, D.G.S.; Li, Z.; Li, L. Molecular Mechanisms Responsible for the Selective and Low-Grade Induction of Proinflammatory Mediators in Murine Macrophages by Lipopolysaccharide. J. Immunol. 2012, 189, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Maitra, U.; Morris, M.; Li, L. Molecular Mechanism Responsible for the Priming of Macrophage Activation. J. Biol. Chem. 2013, 288, 3897–3906. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, M.I.; Gustafsson, A.K.; Bergström, K.G.; Åsman, B.E. Constitutionally Hyperreactive Neutrophils in Periodontitis. J. Periodontol. 2003, 74, 219–224. [Google Scholar] [CrossRef]
- Gustafsson, A.; Ito, H.; Asman, B.; Bergstrom, K. Hyper-Reactive Mononuclear Cells and Neutrophils in Chronic Periodontitis. J. Clin. Periodontol. 2006, 33, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.B.; Wright, H.J.; Roberts, A.; Cooper, P.R.; Chapple, I.L.C. Hyperactivity and Reactivity of Peripheral Blood Neutrophils in Chronic Periodontitis: Neutrophil Hyperactivity and Reactivity in Chronic Periodontitis. Clin. Exp. Immunol. 2006, 147, 255–264. [Google Scholar] [CrossRef]
- Johnstone, A.M.; Koh, A.; Goldberg, M.B.; Glogauer, M. A Hyperactive Neutrophil Phenotype in Patients With Refractory Periodontitis. J. Periodontol. 2007, 78, 1788–1794. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida Albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Boeltz, S.; Amini, P.; Anders, H.-J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or Not to NET:Current Opinions and State of the Science Regarding the Formation of Neutrophil Extracellular Traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Polak, D.; Sanui, T.; Nishimura, F.; Shapira, L. Diabetes as a Risk Factor for Periodontal Disease—Plausible Mechanisms. Periodontology 2000 2020, 83, 46–58. [Google Scholar] [CrossRef]
- Genco, R.J.; Borgnakke, W.S. Diabetes as a Potential Risk for Periodontitis: Association Studies. Periodontology 2000 2020, 83, 40–45. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakschevitz, F.S.; Aboodi, G.M.; Glogauer, M. Oral Neutrophil Transcriptome Changes Result in a Pro-Survival Phenotype in Periodontal Diseases. PLoS ONE 2013, 8, e68983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host Defense against Oral Microbiota by Bone-Damaging T Cells. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Hassanpour, S.; Borenstein, A.; Sima, C.; Oveisi, M.; Scholey, J.; Cherney, D.; Glogauer, M. Distinct Oral Neutrophil Subsets Define Health and Periodontal Disease States. J. Dent. Res. 2016, 95, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Ukai, T.; Kuramoto, A.; Yoshinaga, Y.; Nakamura, H.; Takamori, Y.; Yamashita, Y.; Hara, Y. The Histopathological Comparison on the Destruction of the Periodontal Tissue between Normal Junctional Epithelium and Long Junctional Epithelium. J. Periodontal Res. 2017, 52, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Gunti, S.; Notkins, A.L. Polyreactive Antibodies: Function and Quantification. J. Infect. Dis. 2015, 212, S42–S46. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, J.L.; Cappelli, D.; Steffen, M.J. Characteristics and Utilization of Antibody Measurements in Clinical Studies of Periodontal Disease. J. Periodontol. 1992, 63, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.J.; King, W.J.; Radford, D.J.; Adu, D.; McGRATH, M.; Savage, C.O.S. IL-1beta Production by Human Polymorphonuclear Leucocytes Stimulated by Anti-Neutrophil Cytoplasmic Autoantibodies: Relevance to Systemic Vasculitis. Clin. Exp. Immunol. 1996, 106, 273–279. [Google Scholar] [CrossRef]
- Edwards, S.W.; Hallett, M.B. Seeing the Wood for the Trees: The Forgotten Role of Neutrophils in Rheumatoid Arthritis. Immunol. Today 1997, 18, 320–324. [Google Scholar] [CrossRef]
- Brusko, T.M.; Putnam, A.L.; Bluestone, J.A. Human Regulatory T Cells: Role in Autoimmune Disease and Therapeutic Opportunities. Immunol. Rev. 2008, 223, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Mou, Z.; Onyilagha, N.; Jia, P.; Gounni, A.S.; Uzonna, J.E. Deficiency of Phosphatidylinositol 3-Kinase δ Signaling Leads to Diminished Numbers of Regulatory T Cells and Increased Neutrophil Activity Resulting in Mortality Due to Endotoxic Shock. J. Immunol. 2017, 199, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Lewkowicz, P.; Lewkowicz, N.; Sasiak, A.; Tchórzewski, H. Lipopolysaccharide-Activated CD4 + CD25 + T Regulatory Cells Inhibit Neutrophil Function and Promote Their Apoptosis and Death. J. Immunol. 2006, 177, 7155–7163. [Google Scholar] [CrossRef] [Green Version]
- Lewkowicz, N.; Mycko, M.P.; Przygodzka, P.; Ćwiklińska, H.; Cichalewska, M.; Matysiak, M.; Selmaj, K.; Lewkowicz, P. Induction of Human IL-10-Producing Neutrophils by LPS-Stimulated Treg Cells and IL-10. Mucosal Immunol. 2016, 9, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Lewkowicz, N.; Klink, M.; Mycko, M.P.; Lewkowicz, P. Neutrophil—CD4+CD25+ T Regulatory Cell Interactions: A Possible New Mechanism of Infectious Tolerance. Immunobiology 2013, 218, 455–464. [Google Scholar] [CrossRef]
- Zardawi, F.; Gul, S.; Abdulkareem, A.; Sha, A.; Yates, J. Association Between Periodontal Disease and Atherosclerotic Cardiovascular Diseases: Revisited. Front. Cardiovasc. Med. 2021, 7, 625579. [Google Scholar] [CrossRef]
- Pressman, G.S.; Qasim, A.; Verma, N.; Miyamae, M.; Arishiro, K.; Notohara, Y.; Crudu, V.; Figueredo, V.M. Periodontal Disease Is an Independent Predictor of Intracardiac Calcification. BioMed Res. Int. 2013, 2013, 854340. [Google Scholar] [CrossRef]
- Ishai, A.; Osborne, M.T.; El Kholy, K.; Takx, R.A.P.; Ali, A.; Yuan, N.; Hsue, P.; Van Dyke, T.E.; Tawakol, A. Periodontal Disease Associates With Arterial Inflammation Via Potentiation of a Hematopoietic-Arterial Axis. JACC Cardiovasc. Imaging 2019, 12, 2271–2273. [Google Scholar] [CrossRef]
- Reyes, L.; Herrera, D.; Kozarov, E.; Roldán, S.; Progulske-Fox, A. Periodontal Bacterial Invasion and Infection: Contribution to Atherosclerotic Pathology. J. Clin. Periodontol. 2013, 40, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Herrera, D.; Molina, A.; Buhlin, K.; Klinge, B. Periodontal Diseases and Association with Atherosclerotic Disease. Periodontology 2000 2020, 83, 66–89. [Google Scholar] [CrossRef]
- Velsko, I.M.; Chukkapalli, S.S.; Rivera, M.F.; Lee, J.-Y.; Chen, H.; Zheng, D.; Bhattacharyya, I.; Gangula, P.R.; Lucas, A.R.; Kesavalu, L. Active Invasion of Oral and Aortic Tissues by Porphyromonas Gingivalis in Mice Causally Links Periodontitis and Atherosclerosis. PLoS ONE 2014, 9, e97811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velsko, I.M.; Chukkapalli, S.S.; Rivera-Kweh, M.F.; Zheng, D.; Aukhil, I.; Lucas, A.R.; Larjava, H.; Kesavalu, L. Periodontal Pathogens Invade Gingiva and Aortic Adventitia and Elicit Inflammasome Activation in Avβ6 Integrin-Deficient Mice. Infect. Immun. 2015, 83, 4582–4593. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, S.S.; Velsko, I.M.; Rivera-Kweh, M.F.; Zheng, D.; Lucas, A.R.; Kesavalu, L. Polymicrobial Oral Infection with Four Periodontal Bacteria Orchestrates a Distinct Inflammatory Response and Atherosclerosis in ApoEnull Mice. PLoS ONE 2015, 10, e0143291. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, Q. Periodontitis Aggravated Pancreatic Β-cell Dysfunction in Diabetic Mice through Interleukin-12 Regulation on Klotho. J. Diabetes Investig. 2016, 7, 303–311. [Google Scholar] [CrossRef]
- Blasco-Baque, V.; Garidou, L.; Pomié, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis Induced by Porphyromonas Gingivalis Drives Periodontal Microbiota Dysbiosis and Insulin Resistance via an Impaired Adaptive Immune Response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [Green Version]
- Artese, H.P.C.; Foz, A.M.; de S. Rabelo, M.; Gomes, G.H.; Orlandi, M.; Suvan, J.; D’Aiuto, F.; Romito, G.A. Periodontal Therapy and Systemic Inflammation in Type 2 Diabetes Mellitus: A Meta-Analysis. PLoS ONE 2015, 10, e0128344. [Google Scholar] [CrossRef] [Green Version]
- Faggion, C.M.; Cullinan, M.P.; Atieh, M. An Overview of Systematic Reviews on the Effectiveness of Periodontal Treatment to Improve Glycaemic Control. J. Periodontal Res. 2016, 51, 716–725. [Google Scholar] [CrossRef]
- Teshome, A.; Yitayeh, A. The Effect of Periodontal Therapy on Glycemic Control and Fasting Plasma Glucose Level in Type 2 Diabetic Patients: Systematic Review and Meta-Analysis. BMC Oral Health 2017, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Geerts, S.O.; Nys, M.; Mol, P.D.; Charpentier, J.; Albert, A.; Legrand, V.; Rompen, E.H. Systemic Release of Endotoxins Induced by Gentle Mastication: Association With Periodontitis Severity. J. Periodontol. 2002, 73, 73–78. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia Associated With Toothbrushing and Dental Extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef] [Green Version]
- Crasta, K.; Daly, C.G.; Mitchell, D.; Curtis, B.; Stewart, D.; Heitz-Mayfield, L.J.A. Bacteraemia Due to Dental Flossing. J. Clin. Periodontol. 2009, 36, 323–332. [Google Scholar] [CrossRef]
- Tomás, I.; Diz, P.; Tobías, A.; Scully, C.; Donos, N. Periodontal Health Status and Bacteraemia from Daily Oral Activities: Systematic Review/Meta-Analysis. J. Clin. Periodontol. 2012, 39, 213–228. [Google Scholar] [CrossRef]
- Chukkapalli, S.S.; Easwaran, M.; Rivera-Kweh, M.F.; Velsko, I.M.; Ambadapadi, S.; Dai, J.; Larjava, H.; Lucas, A.R.; Kesavalu, L. Sequential Colonization of Periodontal Pathogens in Induction of Periodontal Disease and Atherosclerosis in LDLR null Mice. Pathog. Dis. 2017, ftx003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra e Silva Filho, W.; Casarin, R.C.V.; Nicolela Junior, E.L.; Passos, H.M.; Sallum, A.W.; Gonçalves, R.B. Microbial Diversity Similarities in Periodontal Pockets and Atheromatous Plaques of Cardiovascular Disease Patients. PLoS ONE 2014, 9, e109761. [Google Scholar] [CrossRef]
- Fernandes, C.P.; Oliveira, F.A.F.; de Barros Silva, P.G.; Alves, A.P.N.N.; Mota, M.R.L.; Montenegro, R.C.; Burbano, R.M.R.; Seabra, A.D.; Lobo Filho, J.G.; Lima, D.L.F.; et al. Molecular Analysis of Oral Bacteria in Dental Biofilm and Atherosclerotic Plaques of Patients with Vascular Disease. Int. J. Cardiol. 2014, 174, 710–712. [Google Scholar] [CrossRef] [Green Version]
- Guerville, M.; Boudry, G. Gastrointestinal and Hepatic Mechanisms Limiting Entry and Dissemination of Lipopolysaccharide into the Systemic Circulation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G1–G15. [Google Scholar] [CrossRef] [Green Version]
- Bevins, C.L.; Salzman, N.H. Paneth Cells, Antimicrobial Peptides and Maintenance of Intestinal Homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Tuin, A.; Huizinga-Van der Vlag, A.; van Loenen-Weemaes, A.-M.M.A.; Meijer, D.K.F.; Poelstra, K. On the Role and Fate of LPS-Dephosphorylating Activity in the Rat Liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G377–G385. [Google Scholar] [CrossRef]
- Schumann, R.R.; Latz, E. Lipopolysaccharide-Binding Protein. In Chemical Immunology and Allergy; Jack, R.S., Ed.; KARGER: Basel, Switzerland, 1999; Volume 74, pp. 42–60. ISBN 978-3-8055-6917-0. [Google Scholar]
- Moludi, J.; Maleki, V.; Jafari-Vayghyan, H.; Vaghef-Mehrabany, E.; Alizadeh, M. Metabolic Endotoxemia and Cardiovascular Disease: A Systematic Review about Potential Roles of Prebiotics and Probiotics. Clin. Exp. Pharmacol. Physiol. 2020, 47, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.; Stinson, F.L.; Parker, R.B. The Passage of Tritiated Bacterial Endotoxin across Intact Gingival Crevicular Epithelium. J. Periodontol. 1972, 43, 270–276. [Google Scholar] [CrossRef]
- Pillay, J.; Ramakers, B.P.; Kamp, V.M.; Loi, A.L.T.; Lam, S.W.; Hietbrink, F.; Leenen, L.P.; Tool, A.T.; Pickkers, P.; Koenderman, L. Functional Heterogeneity and Differential Priming of Circulating Neutrophils in Human Experimental Endotoxemia. J. Leukoc. Biol. 2010, 88, 211–220. [Google Scholar] [CrossRef]
- Ryder, M.P.; Wu, X.; McKelvey, G.R.; McGuire, J.; Schilke, K.F. Binding Interactions of Bacterial Lipopolysaccharide and the Cationic Amphiphilic Peptides Polymyxin B and WLBU2. Colloids Surf. B Biointerfaces 2014, 120, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtchen, A.; Malmsten, M. (Lipo)Polysaccharide Interactions of Antimicrobial Peptides. J. Colloid Interface Sci. 2015, 449, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munford, R.S. Detoxifying Endotoxin: Time, Place and Person. J. Endotoxin Res. 2005, 11, 69–84. [Google Scholar] [CrossRef]
- Hayashi, J.; Masaka, T.; Ishikawa, I. Increased Levels of Soluble CD14 in Sera of Periodontitis Patients. Infect. Immun. 1999, 67, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Ding, P.-H.; Wang, C.-Y.; Darveau, R.P.; Jin, L. Porphyromonas Gingivalis LPS Stimulates the Expression of LPS-Binding Protein in Human Oral Keratinocytes in Vitro. Innate Immun. 2013, 19, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Sasaki, N.; Yamaga, S.; Kuboniwa, M.; Matsusaki, M.; Amano, A. Porphyromonas Gingivalis Induces Penetration of Lipopolysaccharide and Peptidoglycan through the Gingival Epithelium via Degradation of Junctional Adhesion Molecule 1. PLOS Pathog. 2019, 15, e1008124. [Google Scholar] [CrossRef]
- Shands, J.W.; Chun, P.W. The Dispersion of Gram-Negative Lipopolysaccharide by Deoxycholate. Subunit Molecular Weight. J. Biol. Chem. 1980, 255, 1221–1226. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-Membrane Vesicles from Gram-Negative Bacteria: Biogenesis and Functions. Nat. Rev. Microbiol. 2015, 13, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodson, J.M. Gingival Crevice Fluid Flow. Periodontology 2000 2003, 31, 43–54. [Google Scholar] [CrossRef]
- Kayar, N.A.; Oduncuoğlu, B.F.; Haliloğlu, S.; Serpek, B.; Ataoğlu, T.; Alptekin, N.Ö. Methodological Evaluation of Gingival Crevicular Fluid Volume and Neutrophil Elastase Levels: Sequential Sampling, Length of Sampling Time and Two Different Sampling Methods. Acta Odontol. Scand. 2020, 78, 290–296. [Google Scholar] [CrossRef]
- Mahajan, A.; Grüneboom, A.; Petru, L.; Podolska, M.J.; Kling, L.; Maueröder, C.; Dahms, F.; Christiansen, S.; Günter, L.; Krenn, V.; et al. Frontline Science: Aggregated Neutrophil Extracellular Traps Prevent Inflammation on the Neutrophil-rich Ocular Surface. J. Leukoc. Biol. 2019, 105, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, B.; Zhu, D.; Yan, F. Biomarker Levels in Gingival Crevicular Fluid of Subjects with Different Periodontal Conditions: A Cross-Sectional Study. Arch. Oral Biol. 2016, 72, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Di Lenardo, D.; da Silva, F.R.P.; de Carvalho França, L.F.; Carvalho, J. dos S.; Alves, E.H.P.; Vasconcelos, D.F.P. Evaluation of Biochemical Parameters Present in the Saliva of Patients with Chronic Periodontitis: Results from a Meta-Analysis. Genet. Test. Mol. Biomark. 2019, 23, 255–263. [Google Scholar] [CrossRef]
- Chen, K.T.; Malo, M.S.; Beasley-Topliffe, L.K.; Poelstra, K.; Millan, J.L.; Mostafa, G.; Alam, S.N.; Ramasamy, S.; Warren, H.S.; Hohmann, E.L.; et al. A Role for Intestinal Alkaline Phosphatase in the Maintenance of Local Gut Immunity. Dig. Dis. Sci. 2011, 56, 1020–1027. [Google Scholar] [CrossRef]
- Shao, B.; Lu, M.; Katz, S.C.; Varley, A.W.; Hardwick, J.; Rogers, T.E.; Ojogun, N.; Rockey, D.C.; DeMatteo, R.P.; Munford, R.S. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J. Biol. Chem. 2007, 282, 13726–13735. [Google Scholar] [CrossRef] [Green Version]
- Roth, R.I.; Levin, F.C.; Levin, J. Distribution of Bacterial Endotoxin in Human and Rabbit Blood and Effects of Stroma-Free Hemoglobin. Infect. Immun. 1993, 61, 3209–3215. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Demacker, P.N.M.; Kullberg, B.J.; Jacobs, L.E.H.; Verver-Jansen, T.J.G.; Boerman, O.C.; Stalenhoef, A.F.H.; Van der Meer, J.W.M. Bacterial lipopolysaccharide binds and stimulates cytokine-producing cells before neutralization by endogenous lipoproteins can occur. Cytokine 1998, 10, 766–772. [Google Scholar] [CrossRef]
- Kitchens, R.L.; Thompson, P.A.; Viriyakosol, S.; O’Keefe, G.E.; Munford, R.S. Plasma CD14 Decreases Monocyte Responses to LPS by Transferring Cell-Bound LPS to Plasma Lipoproteins. J. Clin. Investig. 2001, 108, 485–493. [Google Scholar] [CrossRef]
- Kitchens, R.L.; Wolfbauer, G.; Albers, J.J.; Munford, R.S. Plasma Lipoproteins Promote the Release of Bacterial Lipopolysaccharide from the Monocyte Cell Surface. J. Biol. Chem. 1999, 274, 34116–34122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsbach, P.; Weiss, J. The Bactericidal/Permeability-Increasing Protein (BPI), a Potent Element in Host-Defense Against Gram-Negative Bacteria and Lipopolysaccharide. Immunobiology 1993, 187, 417–429. [Google Scholar] [CrossRef]
- Elsbach, P. Mechanisms of Disposal of Bacterial Lipopolysaccharides by Animal Hosts. Microbes Infect. 2000, 2, 1171–1180. [Google Scholar] [CrossRef]
- Bucki, R.; Georges, P.C.; Espinassous, Q.; Funaki, M.; Pastore, J.J.; Chaby, R.; Janmey, P.A. Inactivation of Endotoxin by Human Plasma Gelsolin †. Biochemistry 2005, 44, 9590–9597. [Google Scholar] [CrossRef]
- Bosshardt, D.D. The Periodontal Pocket: Pathogenesis, Histopathology and Consequences. Periodontology 2000 2018, 76, 43–50. [Google Scholar] [CrossRef]
- Vitkov, L.; Klappacher, M.; Hannig, M.; Krautgartner, W.D. Extracellular Neutrophil Traps in Periodontitis. J. Periodontal Res. 2009, 44, 664–672. [Google Scholar] [CrossRef]
- Vitkov, L.; Klappacher, M.; Hannig, M.; Krautgartner, W.D. Neutrophil Fate in Gingival Crevicular Fluid. Ultrastruct. Pathol. 2010, 34, 25–30. [Google Scholar] [CrossRef] [PubMed]
- WHO. Epidemiology, Etiology, and Prevention of Periodontal Diseases. Report of a WHO Scientific Group. World Health Organ. Tech. Rep. Ser. 1978, 1–60. [Google Scholar]
- Kozarov, E.V.; Dorn, B.R.; Shelburne, C.E.; Dunn, W.A.; Progulske-Fox, A. Human Atherosclerotic Plaque Contains Viable Invasive Actinobacillus Actinomycetemcomitans and Porphyromonas Gingivalis. Arterioscler. Thromb. Vasc. Biol. 2005, 25. [Google Scholar] [CrossRef] [Green Version]
- Blanc, V.; O’Valle, F.; Pozo, E.; Puertas, A.; León, R.; Mesa, F. Oral Bacteria in Placental Tissues: Increased Molecular Detection in Pregnant Periodontitis Patients. Oral Dis. 2015, 21, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.G.; Martin, A.W.; McBride, B.C. Experimental Transient Bacteraemias in Human Subjects with Varying Degrees of Plaque Accumulation and Gingival Inflammation. J. Clin. Periodontol. 1977, 4, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, H.; Moradi, A.; Herbort, M. The Effect of Gentamicin in Irrigating Solutions on Articular Infection Prophylaxis during Arthroscopic ACL Reconstruction. Arch. Orthop. Trauma Surg. 2014, 134, 257–261. [Google Scholar] [CrossRef]
- Crispian, S. Aphthous Ulceration. N. Engl. J. Med. 2006, 8, 165–172. [Google Scholar]
- Crispian, S. Oral and Maxillofacial Medicine: The Basis of Diagnosis and Treatment. Oral Surg. 2008, 1, 236–237. [Google Scholar] [CrossRef]
- Vitkov, L.; Krautgartner, W.D.; Hannig, M. Bacterial Internalization in Periodontitis. Oral Microbiol. Immunol. 2005, 20, 317–321. [Google Scholar] [CrossRef]
- Vitkov, L.; Krautgartner, W.D.; Hannig, M.; Weitgasser, R.; Stoiber, W. Candida Attachment to Oral Epithelium: Candida Attachment to Oral Epithelium. Oral Microbiol. Immunol. 2002, 17, 60–64. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in Modulating Acute and Chronic Inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Mäntylä, P.; Stenman, M.; Kinane, D.F.; Tikanoja, S.; Luoto, H.; Salo, T.; Sorsa, T. Gingival Crevicular Fluid Collagenase-2 (MMP-8) Test Stick for Chair-Side Monitoring of Periodontitis: MMP-8 Test in Monitoring Periodontitis. J. Periodontal Res. 2003, 38, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, C.M.S.; Fischer, R.G.; Gustafsson, A. Aberrant Neutrophil Reactions in Periodontitis. J. Periodontol. 2005, 76, 951–955. [Google Scholar] [CrossRef]
- Sorsa, T.; Gursoy, U.K.; Nwhator, S.; Hernandez, M.; Tervahartiala, T.; Leppilahti, J.; Gursoy, M.; Könönen, E.; Emingil, G.; Pussinen, P.J.; et al. Analysis of Matrix Metalloproteinases, Especially MMP-8, in Gingival Crevicular Fluid, Mouthrinse and Saliva for Monitoring Periodontal Diseases. Periodontology 2000 2016, 70, 142–163. [Google Scholar] [CrossRef]
- Podolska, M.J.; Mahajan, A.; Hahn, J.; Knopf, J.; Maueröder, C.; Petru, L.; Ullmann, M.; Schett, G.; Leppkes, M.; Herrmann, M.; et al. Treatment with DNases Rescues Hidden Neutrophil Elastase from Aggregated NETs. J. Leukoc. Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil Extracellular Traps Produced during Inflammation Awaken Dormant Cancer Cells in Mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hägele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from Dying Renal Cells Aggravate Kidney Injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 2012, 23, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, J.; Wang, Y.; Wang, C.; Liu, X.; Han, Z.; Fu, Y.; Yang, Z. Effects of Neutrophil Extracellular Traps on Bovine Mammary Epithelial Cells in Vitro. Front. Immunol. 2019, 10, 1003. [Google Scholar] [CrossRef] [Green Version]
- Knopf, J.; Leppkes, M.; Schett, G.; Herrmann, M.; Muñoz, L.E. Aggregated NETs Sequester and Detoxify Extracellular Histones. Front. Immunol. 2019, 10, 2176. [Google Scholar] [CrossRef] [PubMed]
- Hiyoshi, T.; Domon, H.; Maekawa, T.; Nagai, K.; Tamura, H.; Takahashi, N.; Yonezawa, D.; Miyoshi, T.; Yoshida, A.; Tabeta, K.; et al. Aggregatibacter Actinomycetemcomitans Induces Detachment and Death of Human Gingival Epithelial Cells and Fibroblasts via Elastase Release Following Leukotoxin-dependent Neutrophil Lysis. Microbiol. Immunol. 2019, 63, 100–110. [Google Scholar] [CrossRef]
- Figueredo, C.M.S.; Gustafsson, A. Increased Amounts of Laminin in GCF from Untreated Patients with Periodontitis. J. Clin. Periodontol. 2000, 27, 313–318. [Google Scholar] [CrossRef]
- Emingil, G.; Atilla, G.; Sorsa, T.; Savolainen, P.; Baylas, H. Effectiveness of Adjunctive Low-Dose Doxycycline Therapy on Clinical Parameters and Gingival Crevicular Fluid Laminin-5 Γ2 Chain Levels in Chronic Periodontitis. J. Periodontol. 2004, 75, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Emingil, G.; Kuula, H.; Pirila, E.; Atilla, G.; Sorsa, T. Gingival Crevicular Fluid Laminin-5 Gamma2-Chain Levels in Periodontal Disease. J. Clin. Periodontol. 2006, 33, 462–468. [Google Scholar] [CrossRef]
- An, S.; Raju, I.; Surenkhuu, B.; Kwon, J.-E.; Gulati, S.; Karaman, M.; Pradeep, A.; Sinha, S.; Mun, C.; Jain, S. Neutrophil Extracellular Traps (NETs) Contribute to Pathological Changes of Ocular Graft-vs.-Host Disease (OGVHD) Dry Eye: Implications for Novel Biomarkers and Therapeutic Strategies. Ocul. Surf. 2019. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.I.; Akhter, M.P.; Neuville, K.G.; Trcalek, C.R.; Leeper, A.M.; Williams, A.A.; Rivera, M.; Kesavalu, L.; Ke, H.Z.; Liu, M.; et al. Age-Related Periodontitis and Alveolar Bone Loss in Rice Rats. Arch. Oral Biol. 2017, 73, 193–205. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, P.A.; de Pizzol-Júnior, J.P.; Longhini, R.; Sasso-Cerri, E.; Cerri, P.S. Cimetidine Reduces Interleukin-6, Matrix Metalloproteinases-1 and -9 Immunoexpression in the Gingival Mucosa of Rat Molars With Induced Periodontal Disease. J. Periodontol. 2017, 88, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low Concentrations of Neutrophil Extracellular Traps Induce Proliferation in Human Keratinocytes via NF-KB Activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, G.C. Clinical Evaluation of Periodontal Diseases. Periodontology 2000 1995, 7, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, A.R.; Thorat, M.S. Clinical Effect of Subgingivally Delivered Simvastatin in the Treatment of Patients With Chronic Periodontitis: A Randomized Clinical Trial. J. Periodontol. 2010, 81, 214–222. [Google Scholar] [CrossRef]
- Hall, J.A.G.; Payne, A.G.T.; Purton, D.G.; Torr, B.; Duncan, W.J.; De Silva, R.K. Immediately Restored, Single-Tapered Implants in the Anterior Maxilla: Prosthodontic and Aesthetic Outcomes After 1 Year. Clin. Implant Dent. Relat. Res. 2007, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Teeuw, W.J.; Slot, D.E.; Susanto, H.; Gerdes, V.E.A.; Abbas, F.; D’Aiuto, F.; Kastelein, J.J.P.; Loos, B.G. Treatment of Periodontitis Improves the Atherosclerotic Profile: A Systematic Review and Meta-Analysis. J. Clin. Periodontol. 2014, 41, 70–79. [Google Scholar] [CrossRef]
- Liu, N.; Qiang, W.; Kuang, X.; Thuillier, P.; Lynn, W.S.; Wong, P.K.Y. The Peroxisome Proliferator Phenylbutyric Acid (PBA) Protects Astrocytes from Ts 1 MoMuLV-Induced Oxidative Cell Death. J. Neurovirol. 2002, 8, 318–325. [Google Scholar] [CrossRef]
- Li, X.; Baumgart, E.; Dong, G.-X.; Morrell, J.C.; Jimenez-Sanchez, G.; Valle, D.; Smith, K.D.; Gould, S.J. PEX11α Is Required for Peroxisome Proliferation in Response to 4-Phenylbutyrate but Is Dispensable for Peroxisome Proliferator-Activated Receptor Alpha-Mediated Peroxisome Proliferation. Mol. Cell. Biol. 2002, 22, 8226–8240. [Google Scholar] [CrossRef] [Green Version]
- Gondcaille, C.; Depreter, M.; Fourcade, S.; Lecca, M.R.; Leclercq, S.; Martin, P.G.P.; Pineau, T.; Cadepond, F.; ElEtr, M.; Bertrand, N.; et al. Phenylbutyrate Up-Regulates the Adrenoleukodystrophy-Related Gene as a Nonclassical Peroxisome Proliferator. J. Cell Biol. 2005, 169, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α Promotes Degradation of Mammalian Peroxisomes by Selective Autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated Neutrophil Extracellular Traps Resolve Inflammation by Proteolysis of Cytokines and Chemokines and Protection from Antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The Antidiabetic Drug Metformin Blunts NETosis in Vitro and Reduces Circulating NETosis Biomarkers in Vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitkov, L.; Muñoz, L.E.; Knopf, J.; Schauer, C.; Oberthaler, H.; Minnich, B.; Hannig, M.; Herrmann, M. Connection between Periodontitis-Induced Low-Grade Endotoxemia and Systemic Diseases: Neutrophils as Protagonists and Targets. Int. J. Mol. Sci. 2021, 22, 4647. https://doi.org/10.3390/ijms22094647
Vitkov L, Muñoz LE, Knopf J, Schauer C, Oberthaler H, Minnich B, Hannig M, Herrmann M. Connection between Periodontitis-Induced Low-Grade Endotoxemia and Systemic Diseases: Neutrophils as Protagonists and Targets. International Journal of Molecular Sciences. 2021; 22(9):4647. https://doi.org/10.3390/ijms22094647
Chicago/Turabian StyleVitkov, Ljubomir, Luis E. Muñoz, Jasmin Knopf, Christine Schauer, Hannah Oberthaler, Bernd Minnich, Matthias Hannig, and Martin Herrmann. 2021. "Connection between Periodontitis-Induced Low-Grade Endotoxemia and Systemic Diseases: Neutrophils as Protagonists and Targets" International Journal of Molecular Sciences 22, no. 9: 4647. https://doi.org/10.3390/ijms22094647